NARBreakthroughArticle Formation and persistence of polyglutamine aggregates in mistranslating cells - Oxford Academic
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Published online 28 October 2021 Nucleic Acids Research, 2021, Vol. 49, No. 20 11883–11899
https://doi.org/10.1093/nar/gkab898
NAR Breakthrough Article
Formation and persistence of polyglutamine
aggregates in mistranslating cells
Jeremy T. Lant1 , Rashmi Kiri1 , Martin L. Duennwald3 and Patrick O’Donoghue 1,2,*
1
Department of Biochemistry, The University of Western Ontario, London, Ontario N6A 5C1, Canada, 2 Department
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
of Chemistry, The University of Western Ontario, London, Ontario N6A 5C1, Canada and 3 Department of Anatomy &
Cell Biology, The University of Western Ontario, London, Ontario N6A 5C1, Canada
Received August 11, 2021; Revised September 03, 2021; Editorial Decision September 10, 2021; Accepted September 20, 2021
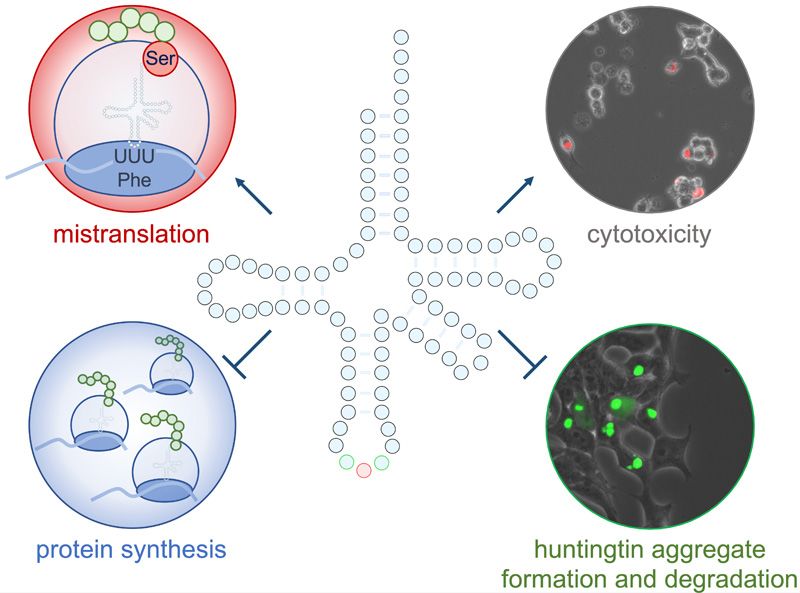
ABSTRACT GRAPHICAL ABSTRACT
In neurodegenerative diseases, including patholo-
gies with well-known causative alleles, genetic fac-
tors that modify severity or age of onset are not
entirely understood. We recently documented the
unexpected prevalence of transfer RNA (tRNA) mu-
tants in the human population, including variants
that cause amino acid mis-incorporation. We hy-
pothesized that a mistranslating tRNA will exacer-
bate toxicity and modify the molecular pathology
of Huntington’s disease-causing alleles. We charac-
terized a tRNAPro mutant that mistranslates proline INTRODUCTION
codons with alanine, and tRNASer mutants, including
High-fidelity translation of messenger RNAs (mRNAs) was
a tRNASer AGA G35A variant with a phenylalanine anti-
considered essential for life by assuring the functional re-
codon (tRNASer AAA ) found in ∼2% of the population. production of ‘so many highly evolved protein molecules’ (1).
The tRNAPro mutant caused synthetic toxicity with In fact, translation of mRNAs is the most erroneous step
a deleterious huntingtin poly-glutamine (polyQ) al- on the path from gene expression to protein synthesis in
lele in neuronal cells. The tRNASer AAA variant showed cells (2–5). Errors in protein synthesis can result from ri-
synthetic toxicity with proteasome inhibition but did bosome stalling or pausing, frameshifting, and amino acid
not enhance toxicity of the huntingtin allele. Cells mis-incorporation. Error rates in cells are normally con-
mistranslating phenylalanine or proline codons with sidered low, with 1 mis-incorporation event occurring for
serine had significantly reduced rates of protein syn- every 1000–10 000 codons translated (6,7). Cells can toler-
thesis. Mistranslating cells were slow but effective ate or even derive a selective advantage from elevated mis-
in forming insoluble polyQ aggregates, defective in translation rates as a result of stress (8–11), chemical treat-
ment (12–14) or mutations in the protein synthesis machin-
protein and aggregate degradation, and resistant to
ery (15–18).
the neuroprotective integrated stress response in- The conserved sequence and structure of transfer RNAs
hibitor (ISRIB). Our findings identify mistranslating (tRNAs) are a major determinant of proteome fidelity.
tRNA variants as genetic factors that slow protein ag- Consequently, single nucleotide substitutions in individ-
gregation kinetics, inhibit aggregate clearance, and ual tRNA genes can lead to proteome-wide mistransla-
increase drug resistance in cellular models of neu- tion in bacteria (19,20), yeast (18), and mammalian cells
rodegenerative disease. (17,21,22). The relevance of tRNA mutations to human dis-
ease is becoming more evident since the discovery that cyto-
plasmic tRNA variants, including those likely to cause er-
rors in translation, are more common in the human popu-
* To whom correspondence should be addressed. Tel: +1 519 850 2373; Fax: +1 519 661 3175; Email: patrick.odonoghue@uwo.ca
C The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which
permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.
11884 Nucleic Acids Research, 2021, Vol. 49, No. 20
lation than previously recognized (23). Sequencing efforts, eases of the nervous system, including Charcot-Marie-
such as the 1000 Genomes Project and our own work (24), Tooth (CMT) disease (36) and early-onset epileptic en-
confirm that mistranslating tRNA mutants occur in indi- cephalopathy (37).
viduals as both rare and common variants with some found Inspired by these studies, we hypothesized that a tRNA
in 1–5% of the population. variant that also caused mistranslation will act to modify
Mutations in protein coding genes that cause mistrans- the progression of neurodegenerative disease at the molec-
lation, such as aminoacyl-tRNA synthetases, are associ- ular level by affecting protein homeostasis and protein ag-
ated with neurodegeneration in mice (25) and Drosophila gregation. We investigated the disease-modifying potential
(26). Mutant tRNAs that lead to a loss of tRNA func- of mistranslating tRNA variants in combination with mul-
tion were linked to neurodegenerative disease and neuronal tiple mammalian cellular models of Huntington’s disease
phenotypic defects in mice (27,28) and a genetic disor- (HD). HD, like many other neurodegenerative diseases (38),
der in humans (29). Mitochondrial tRNA variants have is characterized by the misfolding and aggregation of spe-
long been associated with human diseases, including neu- cific proteins. Protein misfolding typically has the great-
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
rodegeneration (30). MELAS (mitochondrial myopathy, est impact on post-mitotic cells such as those found in the
encephalopathy, lactic acidosis, and stroke-like episodes) heart, brain, and eye (25,39). Since these cells cannot read-
(31) and MERRF (myoclonus epilepsy associated with ily divide or undergo apoptosis, misfolded proteins and pro-
ragged red fibers) (32) are two major neurodegenerative tein aggregates accumulate over time, leading to dysregula-
diseases caused by mutations in mitochondrial tRNAs. tion of the proteome, cytotoxicity, and eventually cell death
In both cases, the mutant tRNAs, which occur at differ- (39).
ent locations in the tRNA body, cannot be properly post- Disorders characterized by protein folding stress or by
transcriptionally modified at position 34 of the anticodon. impaired protein quality control may be particularly sus-
Single mitochondrial tRNA mutants lacking the base 34 ceptible to the effects of mistranslating tRNAs, since mis-
taurine modification are less efficient in decoding their cog- translation increases the synthesis of misfolded proteins to
nate codons, resulting in reduced protein synthesis in the further burden the cellular protein folding stress responses
mitochondria. The tRNALeu variants lacking U34 modifi- (17,21,40). In the case of HD, proteinopathy is triggered by
cation can read UUA but not UUG Leu codons (33). an expanded CAG (Gln) codon repeat in exon1 of the HTT
Cytoplasmic tRNAs that specifically cause amino acid gene encoding the huntingtin protein. Pathogenicity results
mis-incorporation, however, have not been assessed for their primarily from a region corresponding to the first exon of
contribution to neurodegenerative disease. Recently, loss- the HTT gene (mHTTexon1), which can generate a polyQ
of-function cytoplasmic tRNA variants emerged with con- expanded Htt protein either by mRNA splicing or prote-
nections to neurodegeneration (23). The n-Tr20 mutant is olysis (41,42). CAG repeats of >38 glutamine residues are
a tRNAArg CTC with a C50T substitution in the T-arm of associated with disease risk (43). CAG repeat length there-
the tRNA (27). The mutation leads to a processing defect after correlates with age of onset and severity, but the rela-
and the mature tRNA is not produced. Although there are tionship is highly variable (44). Indeed, some patients with
five nearly identical tRNAArg CTC genes in the genome, the the same CAG repeat length differ by over 20 years in age
affected tRNA gene normally shows high expression in the of onset (44). Further, the severity of symptoms can differ
cerebellum where the n-Tr20 gene product accounts for 60% greatly between individuals with a similar age of onset (45).
of the tRNAArg CUC pool (27). In a screen for neurodegener- The discrepancies imply the existence of genetic modifiers
ation in mice, this tRNA mutant was found to be causative of Huntington’s disease, and several protein-coding genes
when co-incident with a mutation in the ribosome recycling have been proposed (44). Searches for genetic modifiers,
factor GTPBP2. Together both mutants act to stall the ri- thus far, have relied on whole exome sequencing and single
bosome and reduce the rate of protein synthesis. Mice with nucleotide polymorphism (SNP) arrays which do not cap-
these mutations displayed multiple neurodegenerative phe- ture tRNA or other non-protein coding RNA gene variants.
notypes, locomotor defects, and died at 8–9 weeks (27). Fur- In addition, whole genome sequencing approaches lack the
ther studies of this tRNAArg mutant found altered synaptic depth of coverage, read length, and mapping strategy re-
transmission and increased susceptibility to seizures in mice quired to confidently identify all tRNA variants in a human
(28). genome (24). In this study, we demonstrate that a naturally
Mistranslation is also associated with proteotoxic stress occurring tRNA variant has significant potential to act as
and neurodegeneration in mice (25) and patient-derived a genetic modifier to Huntington’s disease and conceivably
cell lines (34). Many studies have focused on tRNA syn- other forms of neurodegenerative disease.
thetase mutants, particularly those defective in editing mis-
charged amino acids. One example involves an editing de- MATERIALS AND METHODS
fective AlaRS that mis-charges tRNAAla with Ser, causing
Plasmids and strains
increased levels of misfolded proteins in neurons. Mice ex-
pressing the mutant AlaRS displayed reduced body weight Expression constructs, cloning procedures and primers
and a neurodegenerative phenotype resulting from cell loss (Supplementary Table S1) are described in supplemental
and ataxia of Purkinje cells in the cerebellum (25). An- methods. Plasmid DNA for transfection in mammalian cells
other AlaRS editing-defective mutation caused cardiopro- was purified by Midi-Prep (GeneAid, New Taipei City, Tai-
teinopathy in mice, characterized by protein aggregation wan) from 100 ml Escherichia coli DH5␣ cultures accord-
in cardiomyocytes, cardiac fibrosis and dysfunction (35). ing to manufacturers’ instructions. For all tRNA genes used
AlaRS mutants are also associated with additional dis- in our study, we collected data on the folding predictions
Nucleic Acids Research, 2021, Vol. 49, No. 20 11885
of the wild-type and mutant tRNAs (gtRNAdb (46)), and 2332; ␣-Phospho-eEF2 (Thr56), Cell Signaling Technology,
available expression data from human cells (gtRNAdb (46), 2331.
UCSC genome browser (47)). All tRNAs used were pre-
dicted to be expressed and fold into a canonical tRNA
Mass spectrometry
structure in human cells (Table 1).
Detailed mass spectrometry protocols are described in the
Cell culture and transfection supplemental methods. Briefly, mCherry protein and wild-
type or mistranslating tRNASer were co-expressed in N2a
Experiments were performed in murine Neuro2a Neu- cells and mCherry protein was purified using RFP-trap
roblastoma (N2a; ATCC #CCL-131), human SH-SY5Y agarose bead immunoprecipitation (Chromotek, Munich,
neuroblastoma (SH-SY5Y; ATCC #CRL-226), or rat Germany). Immunoprecipitated mCherry was visualized
pheochromocytoma (PC12; parent cells: ATCC #CRL- on SDS-PAGE and bands at the correct molecular weight
1721)-derived cells. PC12-derived cell lines containing were excised from the gel for MS/MS following tryptic di-
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
HTT-exon1 fused to EGFP with 23Q or 74Q polyQ un- gest of the protein samples. Hits representing Ser misincor-
der doxycycline promoter (48) were a gift from David Ru- poration at Phe codons were curated to include only pep-
binsztein (University of Cambridge, UK). All cell lines tides with a peptide score (–10log P) of >50.
were grown at 37◦ C with humidity and 5% CO2 . N2a and
SHSY5Y cells were cultured in high glucose Dulbecco’s
modified Eagle medium (DMEM, 4.5 g/l glucose; Gibco by Fluorescence microscopy
Life Technologies, Carlsbad, CA) containing penicillin (100 Detailed fluorescence microscopy methods are described in
IU/ml), streptomycin (100 g/ml; P/S; Wisent Bioprod- supplemental methods. Briefly, Fluorescent microscopy im-
ucts, Montreal, QC, Canada) and 10% fetal bovine serum ages were captured on an EVOS FL auto fluorescent micro-
(FBS; Gibco). PC12-derived cell lines were cultured in high scope (Thermo Fisher Scientific). GFP (470 ± 22 nm exci-
glucose DMEM containing P/S, 10% horse serum (Gibco), tation, 510 ± 42 nm emission) and RFP (531 ± 40 nm exci-
5% FBS (Gibco), 50 g/ml G418 (Gibco), and 150 g/ml tation, 593 ± 40 nm emission) filter cubes were used to cap-
hygromycin B (Invitrogen, Carlsbad, CA, USA). All trans- ture green or red fluorescence. An EVOS onstage incubator
fections were performed using Lipofectamine 2000 trans- was used for live cell experiments and images were quanti-
fection reagent (Invitrogen) with 2 g/ml plasmid DNA, tated using ImageJ/Fiji (49,50) (see supplemental methods,
following the manufacturer’s instructions. supplemental appendix).
Small molecules and peptides
Cycloheximide chase protein degradation assays
Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132;
N2a cells were transfected for 48 hr before the experiment in
Sigma-Aldrich 474790, Darmstadt, Germany) and inte-
96-well plates with three biological and six technical repli-
grated stress response inhibitor (ISRIB; Sigma-Aldrich
cates. Cells were washed once with Hank’s buffered salt so-
SML0843) were dissolved in DMSO and cells were treated
lution (HBSS; Gibco by Life Technologies), then media was
with final concentrations as described.
replaced with DMEM (10% FBS, P/S) containing 50 g/ml
cycloheximide (Sigma-Aldrich) and either 10 M MG132
Cellular viability and toxicity assays or equivalent concentration of DMSO. The plate was im-
Cellular viability was assessed using a CelltitreGlo 2.0 Lu- mediately transferred to the EVOS FL environment cham-
minescent Viability Assay (Promega, Madison, WI) in at ber pre-heated to 37◦ C with 5% CO2 and humidity. After
least three biological replicates following the manufacturer’s acclimatizing the plate for 1 h, images were captured every
instructions. Cytotoxicity was assayed with a CytotoxGlo 30 min for 12 h. Fluorescence was quantitated using a semi-
luminescent cytotoxicity assay (Promega) in at least four automated approach in ImageJ (see supplemental informa-
biological replicates, following the manufacturer’s instruc- tion). Initially, increasing concentrations of cycloheximide
tions. Cells were assayed 48 h post-transfection. For assays (0, 50, 250 and 500 g/ml) were also assessed with single
containing the proteasome inhibitor, cells were treated with fluorescent images with the same approach after 24 h incu-
MG132 (0.1–10 M as indicated) or vehicle (DMSO) for 4 bation (see supplemental information). All concentrations
h immediately before assay. resulted in a similar reduction in fluorescence after 24 h, so
the lowest concentration (50 g/ml) was selected for the ki-
netic assay.
Western blotting
Detailed western blotting procedures are described in
Semi-denaturing detergent agarose gel electrophoresis
supplemental methods. The following primary antibod-
(SDD-AGE)
ies were used in this work: ␣-GFP, Abcam, Cambridge,
UK, ab32146; ␣-GAPDH, Sigma-Aldrich, Darmstadt, N2a cells were transfected as above. In one experiment
Germany, MAB374m; ␣-HSP70, Invitrogen, MA3-006; (Supplementary Figure S6A), cells were incubated with
␣-HSP90, Protein Tech, Rosemont, IL, USA, 13171–1- DNA and lipofectamine 2000 for 24 h. In another exper-
AP; ␣-Phospho-eIF2␣ Ser52, ThermoFisher Scientific, 44– iment (Figure 5C–E), cells were incubated for 48 h with
728G; ␣-eIF2␣, ThermoFisher Scientific, AHO0802; ␣- lipofectamine 2000 and DNA, then cells were treated for
eEF2, Cell Signaling Technology, Danvers, MA, USA, 4 h with either 10 M MG132 dissolved in DMSO or
11886 Nucleic Acids Research, 2021, Vol. 49, No. 20
Table 1. tRNA genes and variants
Variant Documented tRNA scoref Expression
tRNA gene Variants Mistranslation description mistranslation wt;variant ARMg ;CHIPh
Ser-AGA-2–3 G35A Ser at Phe natural N2A cellsb 89.6 89.6 + +
(UUU/C), Leu MAF = 1.8%a
(UUA) codons
Ser-CGA-2–1 C34T, A36G Ser at Pro codons synthetic yeastc N2A cellsb 94.0 94.1 + +
Pro-TGG-1–1 C3G, G70T Ala at Pro codons synthetic yeastd HEK293e cells 74.9 70.8 + +
a Data from 1000 Genomes Project (46,82); b this study; c homolog of Ser-CGA C34T, A36G mistranslation documented in yeast (56,83), d homolog of
Pro-TGG-1–1 3G, 70T mistranslation documented in yeast (18), e Pro-TGG-1–1 C3G, G70T mistranslation documented in HEK293 cells (22), f tRNA
gene score calculated using tRNA-Scan SE (Infernal score) (46); g ARM = ARM-seq data suggesting expression (46,84); h CHIP = CHIP-seq hits for at
least three proteins found in RNA Polymerase III holoenzyme or initiation complex (RPC155, POLR3G, BRF1, BDP1, GTF3C2, TBP) (47,85–88).
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
an equal volume of DMSO. Cell lysates were prepared as and tRNALeu have potential to cause amino acid misincor-
above (see Western blotting) and protein concentrations poration. Seryl-tRNA synthetase is especially flexible in rec-
were measured with the BCA assay following manufac- ognizing tRNASer with different anticodons (53), since ser-
turer’s instructions. SDD-AGE assays were performed sim- ine is encoded by six codons with no single nucleotide com-
ilarly to published approaches (51). Lysate volumes con- mon to all possible sequences. Indeed, engineered tRNASer
taining 40 g protein were diluted in 3× loading dye (0.5 anticodon variants cause mistranslation in mouse and hu-
M Tris–HCl, pH 6.8; 1.12 M sucrose; 0.025% bromophenol man cells (21), but naturally occurring variants have not
blue; 3.8% SDS) with sterile milliQ H2 O, loaded and sepa- been tested.
rated on 1.5% agarose gels containing 0.1% SDS. Proteins By searching the genomic tRNA database (46), we found
were transferred to a nitrocellulose membrane by capillary an uncharacterized tRNASer anticodon mutant (tRNA-Ser-
electrophoresis overnight using Tris-acetate-EDTA buffer AGA-2–3 G35A) that occurs in 1.8% of the sequenced pop-
(40 mM Tris-base, 20 mM acetic acid, 1 mM EDTA) with ulation (Figure 1A) (23). The mutant occurs primarily in the
0.1% SDS. EGFP-tagged polyQ aggregates were visualized tRNASer -AGA-2-3 gene and is also found more rarely in
by western blotting with ␣-GFP antibody (see supplemental the identical tRNASer -AGA-2-2 gene (23). In an indepen-
information). dent targeted sequencing effort covering all human tRNA
genes, we identified the same mutant in the tRNASer -AGA-
2-3 gene at a minor allele frequency of 3% in a popula-
Protein aggregation clearance assay tion of 84 individuals (24). In eukaryotes, tRNASer AGA is
PC12-derived cells were transfected using lipofectamine a substrate for A34 -to-I34 editing (54), yielding an IGA an-
2000 and transfectants expressing wild-type or mu- ticodon which can decode UCU, UCC and to a lesser ex-
tant tRNAs were identified by red fluorescence from a tent UCA codons (55). Hence, assuming canonical A:U
plasmid-encoded mCherry protein. Forty-eight hours pairing in the second anticodon:codon position, the tRNA-
post-transfection, mHTT-exon1 74Q-EGFP expression AGA-2-3 G35A variant has potential to decode UUU
was induced with 2 g/ml doxycycline (doxycycline hy- (Phe), UUC (Phe), and UUA (Leu) codons. We also in-
drochloride; Sigma-Aldrich) as before (48). After 96 h, vestigated two additional mistranslating tRNA variants
cells were washed once with doxycycline-free medium and that we previously characterized in yeast (18,56) and mam-
thereafter maintained in doxycycline-free medium. Images malian cells (22) for their ability to direct amino acid mis-
were captured daily in RFP (531 ± 40 nm excitation, incorporation. One variant is caused by mutation of an
593 ± 40 nm emission) and GFP (470 ± 22 nm excitation, identity element such that a different AARS recognizes the
510 ± 42 nm emission) settings from the time at induction tRNA (Figure 1B). The resulting Ala-tRNAPro decodes Pro
to 72 h after doxycycline was removed. To determine the (CCA/G/U) codons with Ala (18,22). The second variant
fraction of transfected cells containing aggregates at each (tRNASer CGA-2-1 C34T, A36G) is a human homolog of a
time point, the number of transfected cells (red) containing tRNASer with a UGG (proline) anticodon that led to mis-
visible aggregates (green) in each image was counted incorporation of Ser at Pro codons (CCA/G/U) in yeast
manually by overlaying fluorescent images in ImageJ. (56). Properties of the tRNA genes and variants are noted
below (Table 1), while additional gene and SNP identifiers
as well as sequences for each tRNA gene locus are listed in
RESULTS the supplemental information (Supplementary Table S2).
Transfer RNA variants used in this study
tRNASer AAA -dependent amino acid misincorporation
Transfer RNA variants can elicit several different types of
errors in protein synthesis (23). Here, we focused on tRNA To confirm that the tRNASer AAA variant causes the ex-
variants that specifically cause amino acid misincorpora- pected Ser incorporation at Phe codons in cells, we im-
tion. The cognate aminoacyl-tRNA synthetases (AARS) munoprecipitated mCherry protein from cells expressing
for Ser, Ala, and to a lesser extent for Leu, do not use the tRNASer AGA or tRNASer AAA and identified multiple pep-
anticodon as an identity or recognition element (52). Thus, tides corresponding to Ser misincorporation by mass spec-
nonsynonymous anticodon variants of tRNASer , tRNAAla trometry. The mCherry coding sequence contains ten UUC
Nucleic Acids Research, 2021, Vol. 49, No. 20 11887
A
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
B
Figure 1. Mechanisms of tRNA-dependent mistranslation. Anticodon (A) or identity element (B) mutations in tRNAs can lead to mistranslation. Anti-
codon mutations in tRNASer genes (A) lead to a mutant tRNA that still accepts serine and now decodes other codons. The tRNASer G35A mutant decodes
phenylalanine or leucine (UUN) codons with serine. We also characterized a tRNASer UGG variant (not shown) that decodes proline codons with serine.
Mutations in human tRNAs can lead to the acquisition of a G3:U70 base pair, which is a critical identity element for AlaRS (B). The resulting tRNAPro
(G3:U70) is an efficient alanine but not proline acceptor that retains the ability to decode Pro codons.
Table 2. Selected observed peptides showing Ser mis-incorporation at Phe codons in mCherry
Supplementary tRNASer Phe codon AA change
Figure S1 panel anticodon mCherry mCherry Peptide sequence -10logP Area
A AGA UUC F70S GGPLPFAWDILSPQ(+.98)SMYGSKA 58.75 1.7 × 109
B AAA UUC F70S GGPLPFAWDILSPQSM(+15.99)YGSK 94.78 3.8 × 107
C AAA UUC F70S GGPLPFAWDILSPQ(+.98)SMYGSKA 48.50 2.3 × 107
D AAA UUC F104S VM(+15.99)NSEDGGVVTVTQDSSLQDGEFIYK 88.19 2.0 × 107
E AAA UUC F123S VM(+15.99)NFEDGGVVTVTQDSSLQDGESIYK 92.40 2.0 × 107
S indicates Ser misincorporation at Phe codons. Spectra for these peptides are shown in Supplementary Figure S1.
codons that may be mistranslated by tRNASer AAA . We peptide hits with –10log P > 50 for Phe or Ser incorpora-
detected Ser misincorporation at multiple Phe codons in tion at each Phe codon in mCherry (Table 3). The spectral
mCherry from cells expressing tRNASer AAA. In addition, we counts indicate a greater level of mis-incorporation of Ser at
observed potential mistranslated peptide hits, i.e. probabil- Phe codons in mistranslating cells (26 Ser / 266 Phe) com-
ity of random hit score (–10log P) ∼40–60, in both nor- pared to the background level (6 Ser / 304 Phe) observed
mal (Supplementary Figure S1A) and mistranslating cells in normal cells. Most of the hits in wild-type cells occur at
(Supplementary Figure S1C, Tables 2 and 3). While these Phe70 (Supplementary Figure S1A), yet these spectra lack
lower scoring hits match the full peptide mass, following sufficient y and b ion support to confirm these peptides as
fragmentation, both hits contain significantly more uniden- evidence that mis-incorporation occurs in normal cells.
tified peaks and lack multiple y and b ions that cover the site
of interest. This is in contrast to the higher scoring hits doc-
umenting mistranslation in cells expressing tRNASer AAA , Reduced protein levels in mistranslating cells
where the spectra have multiple ions with a much higher Previous reports established a translation-inhibition re-
signal to noise ratio confirming misincorporation (Supple- sponse to mistranslating tRNAs expressed in mammalian
mentary Figure S1B, D, E, Table 2). For an overview of mis- cells (21,40). Through inhibition of mRNA translation initi-
incorporation in the mCherry reporter, we summarized all ation or elongation, general protein synthesis can be down-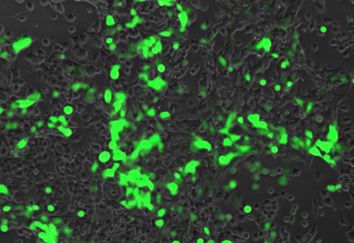
11888 Nucleic Acids Research, 2021, Vol. 49, No. 20
Table 3. Observed spectral counts for Ser or Phe incorporation at Phe the same result in both mouse (Figure 3A) and human cells
codons in mCherry (Figure 3B).
tRNASer AGA tRNASer AAA
Phe codon Interactions of polyQ HTT alleles with tRNASer mutants
position in No. Phe No. Ser No. Phe No. Ser
mCherry peptides peptides peptides peptides We cloned the tRNA-Ser-AGA-2–3 wild type and G35A
variant and tRNA-Ser-CGA-2–1 wild type and C34G,
32 122 1 86 5 A36G double mutant with native genomic context
61 18 0 15 1
70 14 4 9 9 (±300 bp) into plasmids containing mCherry fused to a
92 5 0 5 2 fluorescence-inactivated GFP variant (see supplemental
96 0 0 0 0 methods). In N2a cells, we co-transfected plasmids with
104 54 1 36 4 tRNASer AAA , tRNASer UGG or the respective WT tRNA
123 55 0 36 5
controls along with the 23Q or 46Q HTT-exon1 allele.
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
134 36 0 42 0
Total: 304 6 229 26 Unlike our studies with tRNAPro G3:U70, the tRNASer
anticodon variants showed no significant loss of cellular
Peptides hits with –10log P score >50 are shown. viability on their own or in combination with the 46Q
HTT-exon1 (Supplementary Figure S3A, B).
For the following experiments, we focused our investi-
regulated in response to mistranslation or tRNA dysfunc- gations on tRNASer AGA and the G35A variant, since the
tion (21,57). Using fluorescence microscopy, we measured variant occurs naturally in the human population and leads
red fluorescence (ex. 542 ± 20 nm, em. 593 ± 40 nm) of N2a to phenotypic defects, including inhibited protein synthe-
cells expressing tRNASer AGA , tRNASer AAA , tRNASer CGA or sis. We cloned the tRNASer AGA and the tRNASer AAA vari-
tRNASer UGG and mCherry. We observed a significant re- ant genes into plasmids expressing 23Q or 74Q HTT-exon1
duction in fluorescence of cells expressing either tRNASer allele fused to EGFP. We used a CytotoxGlo assay to mea-
mutant compared to wild-type tRNA (Figure 2A, B). The sure cellular toxicity. In this assay, the tRNASer AAA vari-
transfections were repeated with plasmids expressing wild- ant was significantly cytotoxic compared to the wild-type
type tRNASer AGA or the tRNASer AAA variant for western tRNASer AGA , but the mutant tRNA showed no apparent
blotting analysis of GFP (S65F)-mCherry expression (see additional toxic effect in combination with 74Q HTT-exon1
supplementary methods). Compared to a GAPDH con- (Supplementary Figure S3C, D).
trol, the GFP-mCherry protein levels were reduced > 3.6- We hypothesized that inhibition of the proteasome would
fold in mistranslating cells (Figure 2C). We captured images increase the toxicity of mistranslating cells because of the
of HEK239 cells expressing wild-type and mistranslating accumulation of mistranslated and misfolded proteins. We
tRNAPro G3:U70 with an EGFP reporter as before (22). used increasing concentrations of MG132 in cells express-
Analysis of these images confirmed that tRNAPro G3:U70 ing 23Q HTT-exon1-EGFP and either tRNASer AGA (Fig-
does not elicit the translation suppression response (Supple- ure 3C) or the tRNASer AAA variant (Figure 3D) and mea-
mentary Figure S2A) that we observed with the anticodon sured cell viability. We observed a MG132 concentration-
variants of tRNASer . dependent decrease in cell viability only in cells express-
ing the mistranslating tRNA variant, demonstrating a syn-
thetic toxic interaction between the naturally occurring
tRNASer AAA mutant and proteasome inhibition. MG132
tRNA-dependent toxicity in human and mouse cellular mod-
treatments at the same concentrations had no effect on the
els of HD
viability of cells expressing wild-type tRNA (Figure 3C).
To investigate the viability of cells expressing a known mis- Anticipating that proteasome inhibition would exacer-
translating tRNA combined with an aggregating polyQ al- bate toxicity of both the mistranslating tRNA and the
lele, we co-transfected plasmids encoding a wild-type or 74Q allele, we measured the toxicity of cells with or with-
G3:U70 human tRNAPro with a non-pathogenic (25Q) or out treatment of MG132. We again confirmed cytotoxicity
mildly pathogenic (46Q) version of HTT-exon1 (58). The resulting from the tRNASer AAA variant in comparison to
experiments were performed in murine N2a and human wild-type tRNA, however, we observed no additional toxi-
SHSY5Y cells. Both are neuroblastoma-derived lines that city with 74Q compared to the 23Q HTT-allele in mistrans-
are routinely used as a model for protein misfolding dis- lating cells (Supplementary Figure S3D). Compared to nor-
ease, including HD (48,59). Using a luminescent assay for mal conditions (Supplementary Figure S3C), we note that
cell viability (Celltitre Glo 2.0), we observed no significant the addition of MG132 (Supplementary Figure S3D) re-
loss of viability from the mutant tRNA alone or from the sulted in a greater and more significant increase in cytotox-
mildly deleterious HTT-allele co-expressed with a wild-type icity for mistranslating cells compare with wild type cells.
tRNA. Only the combination of HTT-exon1 46Q expres-
sion in mistranslating cells resulted in a significant reduc-
Kinetics of polyQ aggregate formation in mistranslating cells
tion (1.3 ± 0.05-fold in SHSY5Y; 1.2 ± 0.06-fold in N2a)
in cellular viability compared to cells expressing wild-type Using live cell fluorescence microscopy, we captured the
tRNA and 25Q (Figure 3A, B). The data demonstrate a syn- formation of EGFP-tagged polyQ aggregates in N2a cells
thetic toxic interaction between the Ala accepting tRNAPro expressing either tRNASer AGA or the tRNASer AAA variant
G3:U70 mutant and a deleterious HTT allele. We observed over an 18 h time-course. We quantified the fluorescence
Nucleic Acids Research, 2021, Vol. 49, No. 20 11889
A B
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
C
Figure 2. Fluorescence and expression of mCherry protein in mistranslating cells. N2a cells were transfected with a plasmid encoding human tRNASer AGA
or G35A variant tRNASer AAA ; or tRNASer CGA or the C34G, A36G variant tRNASer UGG and fluorescently dead GFP (S65F or P) fused to an active
mCherry transfection marker. Fluorescence of cells was measured by fluorescence microscopy (RFP, ex 531 nm, em 593 nm). Each box represents data from
135 cells from three biological and nine technical replicates (A). Midline represents the median, boxes represent quartiles, and whiskers represent 1.5× the
interquartile range. Representative images were captured under 20× magnification with phase or RFP settings (B). Cell lysates were harvested and western
blotted for fluorescent protein expression with anti-GFP and anti-GAPDH antibodies as a loading control (C). Anti-GFP blots were quantitated in three
biological replicates by densitometry normalized to GAPDH. Stars indicate P-values from independent sample t-tests (n.s. = no significant difference, *
P < 0.05, ** P < 0.01, *** P < 0.001) and letters indicate significantly different groups determined by Tukey’s Honestly Significant Different (HSD) test,
where groups sharing a letter are not significantly different and groups not sharing a letter are significantly different (␣ = 0.05).
and number of aggregates in each image series using a semi- Mistranslating cells accumulate smaller and fewer polyQ ag-
automated approach in ImageJ (Supplementary Figure S4). gregates
Mistranslating cells accumulated fewer 74Q aggregates and
To assess the effects of a mistranslating tRNA on insolu-
less overall fluorescence signal over the time-course (Figure
ble polyQ aggregate formation, we performed a membrane
4A, Supplementary Figure S5, Data File S1). We normal-
detergent assay to quantify insoluble EGFP-HTT-exon1
ized the number of aggregates in each image to total fluores-
polyQ aggregates in cells expressing either tRNASer AGA
cence of the image to account for the reduced fluorescence
or the tRNASer AAA variant. Aggregates were allowed to
in mistranslating cells and variability in the number of flu-
from over a 48 h transfection period, after which cells
orescing cells between images. In cells expressing the wild-
were treated with Triton X-100 to permeate the cell mem-
type tRNA, the number of aggregates per unit fluorescence
brane. Large, insoluble fluorescent aggregates are retained
increased over time. In mistranslating cells, the appearance
in the cell, whereas soluble polyQ or small oligomers
of aggregates proceeded at a slower rate and plateaued ear-
defuse into the media (Figure 5A) (60). We used thresh-
lier at the 10-h time point (Figure 4B). To further validate
olding analysis to assess the number and size of aggre-
that the observed reduction in fluorescence was due to ex-
gates in images captured before and after Triton X-100
pression of the mutant tRNA, we co-transfected plasmids
treatment. All fluorescent foci disappeared from cells ex-
expressing mCherry and the 23Q HTT-exon1 allele fused
pressing the non-aggregating 23Q-EGFP following Tri-
to EGFP, with the mutant tRNASer AAA encoded on either
ton X-100 treatment, while the 74Q foci were clearly
one plasmid or the other. Regardless of which plasmid the
visible (Figure 5A). Foci remaining in the mistranslat-
tRNA was expressed from, 23Q-EGFP fluorescence was
ing cells were significantly smaller than cells expressing
significantly and equivalently reduced compared to cells ex-
wild-type tRNA, with a median area of 257 m2 com-
pressing no additional tRNA, and the effect was maintained
pared to 315 m2 in the wild-type tRNA-expressing cells
for at least 48 hrs post-transfection (Supplementary Figure
(Figure 5B).
S2B).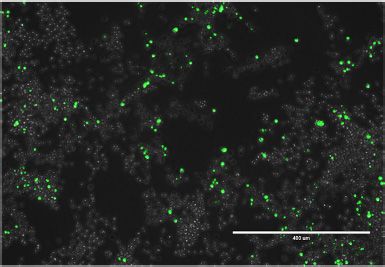
11890 Nucleic Acids Research, 2021, Vol. 49, No. 20
A B
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
C D
Figure 3. Toxic interactions of tRNA variants with a deleterious polyQ allele or proteosome inhibition. N2a (A) or SHSY5Y cells (B) were co-transfected
with two plasmids encoding human tRNAPro UGG or the G3:U70 variant and HTT-exon1 containing 25 (25Q) or 46 (46Q) CAG/CAA mixed codon repeats
encoding polyQ. Cellular viability was measured 24 h post-transfection with CellTitreGlo 2.0. Luminescence readings were normalized to the ‘25Q WT’
control. N2a cells were transfected with a plasmid encoding human tRNASer AGA (C) or G35A variant tRNASer AAA (D) and HTT-exon1 containing 23Q
fused to EGFP as a transfection marker. Cellular viability was assayed 48 h post-transfection with the CellTitreGlo 2.0 assay following 4 hr treatment with
increasing concentrations of MG132 or equal concentration of DMSO. Luminescence readings were normalized to the DMSO controls. Letters indicate
significantly different groups determined by Tukey’s HSD test (␣ = 0.05). Error bars represent the mean ± 1 standard deviation of at least three biological
replicates.
A B
Figure 4. Formation of polyQ aggregates in mistranslating cells. N2a cells were transfected with a plasmid encoding human tRNASer AGA or G35A variant
tRNASer AAA and HTT-exon1 containing 74Q-EGFP. Images were captured beginning 24 h post-transfection by fluorescence microscopy in GFP settings
at 10× magnification every 30 min for an 18 h time course. (A) Representative images from the beginning (t = 0 h) and end (t = 18 h) of the time course are
shown. (B) The number of aggregates in each image was quantitated in ImageJ (see supplemental information). The number of aggregates in each image of
the series was then normalized to total fluorescence of the same image (Naggregates /total fluorescence), and initial values were subtracted (Naggregates /total
fluorescence). Error bars represent the mean ± 1 standard deviation of six biological replicates.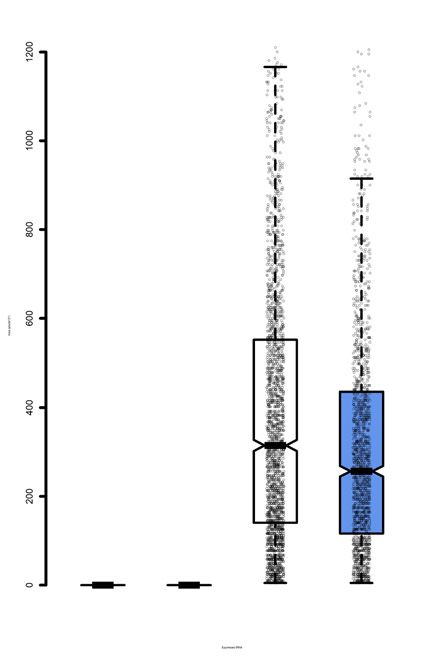
Nucleic Acids Research, 2021, Vol. 49, No. 20 11891
A B
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
C D E
Figure 5. Insoluble PolyQ aggregate size and levels in mistranslating cells. N2a cells were transfected with a plasmid encoding human tRNASer AGA or
G35A variant tRNASer AAA and HTT-exon1 containing 23Q or 74Q fused to EGFP. (A) Representative images were captured by fluorescence microscopy
before and after Triton X-100 treatment at 10× magnification. Images are overlaid from GFP and phase settings. (B) The area of fluorescent bodies
remaining after detergent was measured in ImageJ (see supplemental methods). Midline represents the median, boxes represent quartiles, and whiskers
represent 1.5× the interquartile range. (C) Cell lysates were harvested from N2a cells transfected with the same plasmids and analyzed by SDD-AGE
and western blotting (␣-GFP). Cells were treated for 4 h with 10 M MG132 or an equivalent volume of DMSO. Higher molecular weight smears in the
74Q lanes indicate the presence of aggregated proteins. The SDD-AGE images were quantified for (D) 23Q and (E) 74Q aggregates by densitometry (see
Supplementary Figure S6). Intensity of the high molecular weight aggregates was normalized to total intensity and expressed as a percentage. Error bars
represent mean ± 1 standard deviation from 3 biological replicates. Stars indicate P-values from independent sample t-tests (n.s. = no significant difference,
* P < 0.05, ** P < 0.01, *** P < 0.001). Letters indicate significantly different groups determined by Tukey’s HSD test, where groups sharing a letter are
not significantly different and groups not sharing a letter are significantly different (␣ = 0.05).
We further investigated the effect of the tRNASer AAA vari- ior. Cells expressing 23Q with the wild-type tRNA showed
ant on aggregate formation using semi-denaturing deter- a small fraction of aggregated polyQ in the SDD-AGE as-
gent agarose gel electrophoresis (SDD-AGE) (51). SDD- say with 13% of the HTT-23Q protein aggregated. In 23Q-
AGE is a semi-quantitative method to visualize insoluble expressing cells with the mistranslating tRNA, however, we
protein aggregates as high molecular weight products after found a significant increase in the proportion of aggregated
agarose gel electrophoresis and western blotting. We used protein (33%) compared to cells expressing the wild-type
an ␣-GFP antibody to detect the EGFP-tagged polyQ pro- tRNA (Figure 5C, D). The data suggest that mistranslation
teins. At 24 h post-transfection, we observed less aggregated of the HTT-23Q allele contributes to an increase in aggre-
74Q protein in cells expressing the mistranslating tRNA gation of the non-deleterious HTT allele.
compared to wild type, and no evidence of aggregated 23Q- Cells expressing either wild type or mutant tRNA both
EGFP protein in cells expressing either tRNA (Supplemen- showed aggregation of the 74Q protein, but mistranslat-
tary Figure S6). ing cells displayed a smaller fraction of high molecular
To further promote formation of protein aggregates, we weight 74Q aggregates (Figure 5C, E) in agreement with
transfected cells for 48 hrs and then treated them for 4 h with our Triton-X100 treatments (Figure 5A, B). Proteasome in-
MG132 or DMSO as a control. With 48 h transfections, hibition with MG132 had no significant effect on aggrega-
high molecular weight aggregates were observed even in tion of either 23Q or 74Q in mistranslating cells. We ob-
23Q-expressing cells (Figure 5C, D). Comparing the wild- served a greater accumulation of 23Q aggregates in MG132-
type tRNASer AGA and mistranslating tRNASer AAA , we ob- treated cells expressing wild-type tRNA, which was still sig-
served different effects on 23Q and 74Q aggregation behav- nificantly less than the level of 23Q aggregates seen in mis-11892 Nucleic Acids Research, 2021, Vol. 49, No. 20
translating cells (Figure 5D). MG132-treated cells express- p-eEF2 and eEF2 levels from cells expressing an mCherry
ing the wild type tRNA and 74Q showed an intermediate transfection marker and tRNASer AGA or the tRNASer AAA
level of protein aggregation, which was not significantly dif- variant with or without treatment with 10 M MG132
ferent from either untreated wild-type cells or from cells ex- (Supplementary Figure S8B). MG132 treatment stimulated
pressing the mistranslating tRNA (Figure 5E). The absence a significant increase in p-eEF2 levels in both cell lines (Sup-
of any change in protein aggregates in mistranslating cells plementary Figure S8), but we did not observe any differ-
with or without MG132 treatment suggests that the mis- ence in p-eEF2 levels in the mistranslating cells compared
translating tRNA has a dominant effect on huntingtin pro- to wild type. In normal cells, MG132 inhibits both protein
tein aggregation that is independent of proteasome activity. degradation (see Figure 7C) and protein synthesis, leading
to no change in the steady state protein levels. Despite the
clear induction of p-eEF2 in MG132-treated cells, mistrans-
Heat shock protein levels in mistranslating cells
lating cells show a severely reduced level of protein synthesis
Heat shock protein production is a common cellular stress in conditions that have high or low p-eEF2 (Supplementary
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
response mounted to mitigate the toxic effects of misfolded Figure S8C).
proteins in cells and is known to be activated by mistrans-
lating tRNAs in yeast (18,56). Increased heat shock pro-
Mistranslating cells are resistant to the integrated stress re-
tein levels were also observed in mice expressing editing-
sponse inhibitor
defective aminoacyl-tRNA synthetases (25), and mam-
malian cells expressing mistranslating tRNAs after ex- To further probe the integrated stress response in cells ex-
tended transfection periods of up to 72 h (21). In a pre- pressing tRNASer AAA , we tested whether the reduction in
vious study, we observed no change in HSP70 or HSP90 protein levels could be reversed with a p-eIF2␣ antagonist.
levels in HEK293 cells expressing tRNAPro G3:U70 (22). The integrated stress response inhibitor, ISRIB, relieves
Here, we measured the level of heat shock response factors translation suppression caused by eIF2␣ phosphorylation.
HSP70 and HSP90 24 h after transfection in cells express- ISRIB promotes the formation of active heterodecameric
ing tRNASer AAA or wild-type tRNASer AAA . Consistent with eIF2B complexes to stimulate eIF2B-dependent transla-
our previous study on tRNAPro G3:U70, we did not ob- tion initiation (63). Cells transfected with plasmids express-
serve any evidence of increased HSP levels at 24 h (Supple- ing EGFP-fused HTT-exon1 23Q and no tRNA, wild-type
mentary Figure S7A). We also measured HSP70 levels after tRNASer AGA , or the tRNASer AAA variant were treated with
more extended transfections periods (24, 48 and 72 h) and 500 nM ISRIB over an 18 h time-course. Fluorescent images
observed no significant differences between wild-type and were captured throughout the time course, and a cytotoxic-
mistranslating cells (Supplementary Figure S7B). ity assay was completed at the final timepoint.
Unlike normal cells, mistranslating cells were unable
to increase protein synthesis levels in response to ISRIB.
Regulation of translation initiation and elongation in mis-
In cells expressing no ectopic tRNA, fluorescence of the
translating cells
EGFP-fused HTT-allele increased significantly from the be-
Previous studies established that certain tRNA anticodon ginning to end of the time course by 25% in DMSO con-
variants expressed in mammalian cells lead to increased trol and 40% in 500 nM ISRIB. In cells expressing wild-
phosphorylation of eIF2␣ at Ser52 (21,28,40). Phospho- type tRNASer AGA , fluorescence increased by 26% in DMSO
Ser52 in eIF2␣ prevents translation initiation of most cellu- control and 34% in 500 nM ISRIB. In cells expressing
lar mRNAs and is a converging point of the integrated stress tRNASer AAA , ISRIB did not significantly increase protein
response stimulated by numerous cellular stresses. However, levels, with only a 6% increase of 23Q-EGFP production in
p-eIF2␣ levels vary substantially depending on the tRNA the DMSO control, and a similar 7% increase in 500 nM
gene variant expressed (21), and how long cells have been ISRIB. Hence, the addition of ISRIB did not simulate syn-
expressing the tRNA variant (40). We used western blotting thesis of the 23Q-EGFP huntingtin protein in mistranslat-
to measure p-eIF2␣, eIF2␣ and GFP levels from cells ex- ing cells (Figure 6A, B). As anticipated, ISRIB also had no
pressing all combinations of EGFP-fused HTT-exon1 23Q effect on the cytotoxicity of the mistranslating tRNA (Fig-
and 74Q with wild-type tRNASer AGA or the tRNASer AAA ure 6C).
variant. Despite a reduction in the EGFP-fused huntingtin
protein in all mistranslating cells, we did not observe an in-
Defective protein turnover and aggregate clearance in mis-
crease in the level of p-eIF2␣ in mistranslating cells, even
translating cells
after longer transfection periods (Supplementary Figure
S8A). Given the synthetic toxic effect we observed in mistrans-
Cells can also down-regulate mRNA translation at the lating cells treated with MG132, and previous reports of
level of elongation. Eukaryotic elongation factor 2 (eEF2) is tRNA anticodon variants promoting increased proteasome
involved in ribosome repositioning and movement of tRNA activity (40), we assayed protein turnover in mistranslat-
from the A-site to P-site during translation (61). Phospho- ing cells. Cycloheximide chase assays (64) were performed
rylation of the eukaryotic elongation factor 2 (eEF2) by on N2a cells expressing no tRNA, wild-type tRNASer AGA ,
eEF2 kinase (eEF2K) at Thr56 reduces translation elonga- or the tRNASer AAA variant with EGFP-fused HTT-Exon1
tion rates in conditions of nutrient deprivation or various 23Q and 74Q (Figure 7A-D). Cycloheximide concentra-
other forms of cellular stress, including proteasome inhibi- tions of 50, 250 and 500 g/ml were effective in inhibit-
tion with MG132 (62). We used western blotting to measure ing translation and promoting turnover of the 23Q-EGFPNucleic Acids Research, 2021, Vol. 49, No. 20 11893
A
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
B C
Figure 6. Fluorescence and cytotoxicity of mistranslating cells treated with p-eIF2␣ antagonist ISRIB. N2a cells were transfected with a plasmid encoding
no tRNA, human tRNASer AGA or G35A variant tRNASer AAA and HTT-exon1 23Q-EGFP. Cells were treated for 18 h with 500 nM ISRIB or equivalent
concentration of DMSO. (A) Representative images were capture by fluorescence microscopy in GFP settings after 18 h ISRIB treatment. (B) Fluorescence
intensity per cell area of six technical and three biological replicates was quantitated in ImageJ (see supplemental methods) before and after treatment. Each
bar represents mean (±1 standard deviation) increase in fluorescence intensity per area as a percentage. (C) Cytotoxicity after treatment was measured
with CytotoxGlo. Luminescence readings were normalized to the ‘23Q −/−’ control. Stars indicate P-values from independent sample t-tests (n.s. = no
significant difference, * P < 0.05, ** P < 0.01, *** P < 0.001).
protein in N2a cells (Supplementary Figure S9). Treat- To assess whether 74Q aggregate clearance was reduced
ment with MG132 to inhibit the proteasome was used as in an independent polyQ model, we used a rat PC12-derived
a negative control as described (64). In cells expressing the cell line with a stable, genome-integrated HTT-exon1 74Q
tRNASer AAA variant (Figure 7B, D), we observed a signifi- fused to EGFP under control of a doxycycline inducible
cantly lower rate of protein turnover of both the HTT-exon1 promoter (48). We transfected either tRNASer AGA or the
23Q-EGFP and 74Q-EGFP alleles compared to cells ex- tRNASer AAA variant on plasmids with an mCherry trans-
pressing wild-type tRNASer AGA (Figure 7B, C). Compared fection marker and monitored the appearance and disap-
to cells expressing no plasmid-borne tRNA, turnover of pearance of aggregates by induction and removal of doxy-
the aggregating 74Q-EGFP but not 23Q-EGFP was signif- cycline. Using mCherry to identify tRNA-transfected cells,
icantly reduced in mistranslating cells (Figure 7B). In nor- we counted the number of transfected cells expressing ei-
mal cells, MG132 lead to a reduced rate or protein degrada- ther wild-type or mutant tRNAs containing at least one ag-
tion (Figure 7C), while in mistranslating cells MG132 was gregate (Supplementary Figure S10). The number of cells
not able to further slow their already defective rate of pro- containing aggregates was not significantly different dur-
tein degradation (Figure 7D). ing doxycycline treatment, but after doxycycline was re-11894 Nucleic Acids Research, 2021, Vol. 49, No. 20
A B
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
C D E
Figure 7. Protein turnover and clearance of PolyQ aggregates in mistranslating cells. N2a cells were transfected with a plasmid encoding human
tRNASer AGA or G35A variant tRNASer AAA and HTTexon1 containing 23Q or 74Q fused to EGFP for 48 h before cycloheximide chase assays (A−D).
Cells were treated with cycloheximide and/or MG132 and fluorescence was captured by live cell fluorescence microscopy. (A) Representative images of 23Q-
EGFP/tRNASer AGA and 23Q-EGFP/tRNASer AAA expressing cells at the indicated timepoints. Fluorescence was quantitated in ImageJ (see supplemental
methods). Average rate of protein decay (B) is shown as a change in fluorescence normalized to initial cell area (C, D) (see supplemental information). (E)
PC12 cells with genomically integrated HTT-exon1 74Q-EGFP under a doxycycline (Dox)-inducible promoter were transfected with a plasmid encoding
human tRNASer AGA or G35A variant tRNASer AAA and mCherry transfection marker. HTT-exon1 expression was induced with doxycycline 48 h post-
transfection and cells were imaged daily by fluorescence microscopy. After 96 h, Dox was removed and daily imaging was resumed for 72 h. Aggregate
counting is described in methods (see Supplementary Figure S10). Error bars represent the mean ±1 standard deviation of at least three biological repli-
cates and nine technical replicates each. Stars indicate P-values from independent sample t-tests (n.s. = no significant difference, * P < 0.05, ** P < 0.01,
*** P < 0.001) and letters indicate significantly different groups determined by Tukey’s HSD test (␣ = 0.05).
moved, aggregates persisted significantly longer in cells ex- concentrations are less likely to reach the threshold required
pressing the mistranslating tRNA (Figure 7E). Indeed, cells to seed aggregates (65). Consistent with the translation
with wild-type tRNA had a significantly reduced num- suppression response in mistranslating cells, we observed
ber of polyQ aggregates at 48 and 72 h after removal fewer and smaller aggregates in cells expressing HTT-exon1
of doxycycline, yet we did not observe any decrease in 74Q and the tRNASer AAA variant compared to wild-type
polyQ aggregates in the mistranslating cells following re- tRNASer . Further, in N2a cells and in an inducible PC12
moval of doxycycline. The data suggest that mistranslating cell line of HTTexon1 74Q, we observed a greater persis-
cells are defective in clearing protein aggregates that cause tence of aggregates in cells expressing tRNASer AAA com-
disease. pared to wild-type tRNASer . We also found that mistrans-
lating cells were generally defective in their ability to de-
DISCUSSION grade proteins. While mistranslating cells synthesize aggre-
gating proteins more slowly, they are also defective in their
Huntingtin protein aggregation in mistranslating cells ability to degrade huntingtin protein aggregates. In these
Our data show that huntingtin aggregates form readily cellular models of neurodegenerative disease, the data in-
in mistranslating cells, but at a slower rate than in wild- dicate that natural mistranslating tRNA variants have the
type cells. Since cells expressing the 74Q and mistranslating potential to affect the onset, progression, and severity of
tRNA strongly suppress protein synthesis, polyQ protein HD.Nucleic Acids Research, 2021, Vol. 49, No. 20 11895
The Ser mis-incorporation at Phe codons we observed in acerbate the dysregulation of protein synthesis caused by
cells expressing tRNASer AAA led to the accumulation of mis- deleterious and longer polyQ alleles.
folded proteins. The label-free MS/MS approach we used is
admittedly not ideal for quantifying the mistranslation rate,
Compromised proteostasis in cells expressing mistranslating
which will be the subject of future work. The data suggest an
tRNAs
estimate for the rate of Ser mis-incorporation at Phe codons
could be ∼9% per UUC codon, which is 90−900 times We found that cells expressing the naturally occurring
more than the generally accepted translation error rate of tRNASer AAA variant were characterized by several phe-
1 mistake per 1000−10 000 codons (6,7). Although spec- notypic defects. Using mass spectrometry, we confirmed
tral counting may overestimate the error rate, even mod- amino acid mis-incorporation of Ser at multiple Phe
est levels of mis-incorporation can lead to the accumulation codons. Mistranslating cells were characterized by in-
of mis-made protein as we found. Although mistranslating creased cytotoxicity, general inhibition of protein synthesis,
cells were slower in forming HTT-exon1 74Q aggregates, we sensitivity to proteosome inhibition, and resistance to the
Downloaded from https://academic.oup.com/nar/article/49/20/11883/6413606 by guest on 24 December 2021
observed the normally non-aggregating 23Q protein form neuroprotective stress response inhibitor ISRIB. Together
a significant amount of protein aggregate in mistranslat- the data demonstrate that tRNASer AAA expression leads to
ing cells. While 23Q HTT-exon1 is less aggregation prone a loss of protein homoeostasis.
than 74Q, in vitro studies have demonstrated that 23Q and Despite a mistranslation rate of ∼2–3% Ala incorpora-
shorter HTT-exon1 polyQ peptides are capable of aggrega- tion at Pro codons, tRNAPro G3:U70 is not toxic when ex-
tion (66). In mistranslating cells, we observed 23Q aggre- pressed alone (22). Here, we found that toxicity of tRNAPro
gates at 48 h post-transfection. The data suggest that com- G3:U70 is evident in combination with a mildly deleteri-
pared to 74Q, higher expression levels and longer times were ous 46Q HTT-exon1 allele. The data suggest that some mis-
needed for the 23Q-EGFP protein to aggregate in mistrans- translating tRNA variants can exacerbate polyQ toxicity
lating cells. We also observed increased 23Q aggregation in in a model of neurodegenerative disease. Conversely, the
cells treated with the proteasome inhibition. Our observa- naturally occurring tRNASer AAA variant was toxic alone,
tions on huntingtin protein aggregation suggest mistrans- but did not show additional toxicity with aggregating hunt-
lating cells are compromised in their ability to both synthe- ingtin protein. The differences in toxicity may depend on
size and degrade protein aggregates. how cells respond to different types of mistranslation. We
observed a consistent reduction in protein synthesis in cells
expressing the tRNASer AAA or tRNASer UGG variants, but
Huntingtin protein aggregation and defects in protein quality not in cells expressing tRNAPro G3:U70. Furthermore, cells
control mis-incorporating Ser at Phe codons showed a stronger re-
pression of translation than cells mistranslating Pro codons
Our work is the first to explore the interaction between mis- with Ser. The severely inhibited protein synthesis observed
translation and huntingtin polyQ aggregation. Our studies, in cells with tRNASer AAA may indeed protect the cell from
focused on mistranslation resulting from a tRNA mutant, the toxicity of the aggerating huntingtin allele and amino
suggest that other routes to amino acid mis-incorporation, acid mis-incorporation.
e.g. editing-defective AARS variants, can also exacerbate A recent study in yeast demonstrated that tRNA vari-
polyQ aggregation and HD. Interactions between other fac- ants that result in different types of amino acid change
tors regulating protein homeostasis and polyQ aggrega- can elicit distinct cellular responses (72). A tRNASer mu-
tion represent a continuing theme in yeast and mammalian tant that mistranslates Arg codons and a yeast homolog
cell models of polyQ aggregation and toxicity. For exam- of the same tRNAPro G3:U70 variant we tested here were
ple, work in yeast shows that molecular chaperones (67,68) found to mistranslate at similar levels. In RNA sequencing
and protein degradation (69) are critical systems to com- experiments, however, each tRNA-expressing strain elicited
bat polyQ aggregation. In Drosophila S2 cells expressing distinct changes in the transcriptome. For example, down-
long 138Q, but not shorter polyQ alleles, protein synthesis regulation of genes involved in cell cycle, DNA replication,
is downregulated via the translation regulator Orb2 (70). transcription, and response to stress was observed in cells
Studies using N2a cells, as we used, examined production mistranslating Pro codons with Ala but not in cells mis-
and aggregate formation of HTT alleles with 18Q, 64Q incorporating Ser at Arg codons (72).
or 150Q. Aggregates were monitored over a 48 h period, Phenotypic differences observed from expressing differ-
and in agreement with our study, no toxicity from the 64Q ent tRNA variants may also depend on the compatibility
or even the 150Q HTT allele was observed (71). In HeLa of the tRNA in question with a given gain-of-function mu-
cells, the same authors used a reporter for nuclear reten- tation. For example, nucleotides in and adjacent to the an-
tion of ribosomal protein S2, which signifies inhibition of ticodon sequence are often conserved within tRNA isoac-
ribosome biogenesis. Cells expressing a 97Q allele showed a ceptor groups (46), and can be modified to ensure opti-
3-fold increase in nuclear localization of ribosomal protein mal fidelity and efficiency in recognizing certain codon sets.
S2, demonstrating defective ribosome biogenesis in cells ex- This is true for wild-type tRNAPhe , wherein modified gua-
pressing an aggregating HTT allele (71). The authors did nine bases in position 34 (73) of the anticodon and the
not directly measure the rate of protein synthesis, but their anticodon-adjacent position 37 (74) are used to efficiently
data suggest dysregulation of protein synthesis in cells ex- and accurately decode UUU and UUC codons. Conversely,
pressing long polyQ alleles. Our data indicate that tRNA the tRNASer AAA variant we investigated has adenine at 34
variants, which compromise protein homeostasis, may ex- and 37, so it is possible that the strong reduction in pro-You can also read

















































