Neonatal Guidelines Chapter 10: Metabolic - WISDOM
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Neonatal Guidelines
Chapter 10: Metabolic
V2017.1
Specialty: Neonatal Medicine
Revised by: Jean Matthes
Date Revised : January 2017
Ratified 6th February 2017
Approved by: ABMU Joint Perinatal Forum
Date for Review: 1st of March 2021
Neonatal Guidelines Valid until 1st March 2021 1
Chapter 10: Metabolic v2017.1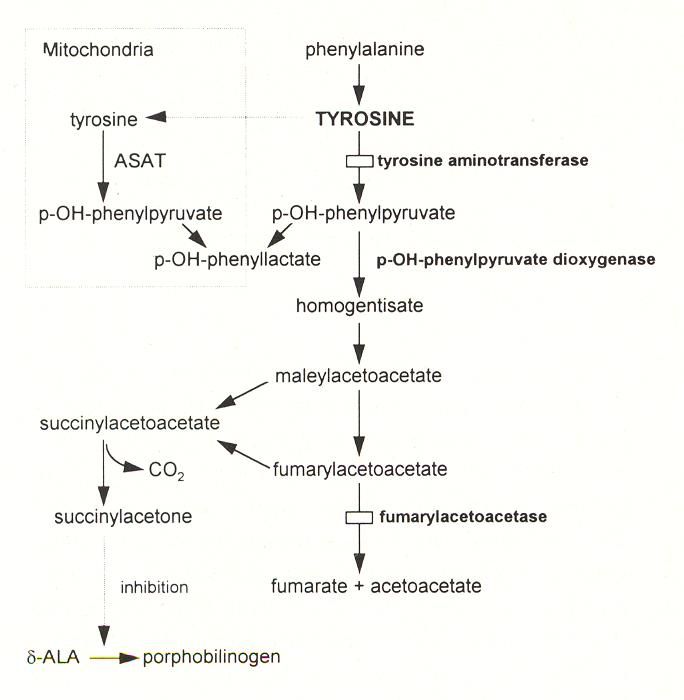
Directorate of Child Health
Checklist for Clinical Guidelines being submitted for Approval by
ABMU Joint Perinatal Forum
Chapter 10: Metabolic v2017.1
Title of Guideline:
Name(s) of revising author: Jean Matthes, Edited by S. Banerjee
Chair of Group or Committee
Neonatal Guideline Group – Sujoy Banerjee
supporting submission:
Issue / Version No: Chapter 10: Metabolic v2017.1
Next Review / Guideline Expiry: Review date: 1st March 2021
Neonatal Consultants, Neonatal junior doctors,
Details of persons included in
Nursing Managers, Neonatal Pharmacist
consultation process:
Brief outline giving reasons for
document being submitted for Routine Revision
ratification
Name of Pharmacist
Katherine Willson
(mandatory if drugs involved):
Please list any policies/guidelines
Chapter 10:Metabolic v2013
this document will supercede:
Keywords linked to document: Metabolic , neonate
Date approved by ABMU Joint
6th February 2017
Perinatal Forum:
File Name: Used to locate where
file is stores on hard drive
Neonatal Guidelines Valid until 1st March 2021 2
Chapter 10: Metabolic v2017.1CONTENTS Topics Page number ------------------------------------------------------------------------------------------------------------------ Introduction to IEM 4 Clinical presentation 4 Clinical Examination 6 Initial screen for metabolic disease 9 Further investigations for specific conditions 11 Treatment – general principles 13 Treatment of hyperammonaemia 13 Some specific disorders 15 Notes on specimens 22 Neonatal Guidelines Valid until 1st March 2021 3 Chapter 10: Metabolic v2017.1
Inborn errors of metabolism
Introduction:
Inborn errors of metabolism (IEM) are individually rare. Some conditions may only be seen
once in a professional’s working life. But there are very many different conditions so their
combined incidence is significant. Usually a high index of suspicion is required to make the
diagnosis. Early detection and treatment may prevent lifelong neurological damage. Early
discussions with the newborn screening laboratory in UHW may ensure that the correct
investigations are taken and processed urgently rather than routinely, saving valuable time.
Likewise a discussion with our biochemistry laboratory in ABMU is required to ensure that
samples are sent to Cardiff expeditiously. These discussions are best undertaken consultant
to consultant.
A very useful Website for investigation and treatment of metabolic conditions is BIMDG –
the British inherited metabolic diseases group. This also covers what interim treatment
may be given to a newborn infant who may have a disorder, whilst awaiting the results of
the diagnostic tests. It also gives treatment regimens for specific conditions. Please refer to
this site when implementing emergency treatment.
Newborn screening is undertaken in Wales for glutaric aciduria, homocysteinuria, isovaleric
acidaemia, maple syrup urine disease, phenylketonuria and medium chain acyl CoA
dehydrogenase deficiency. These tests are done on the 5 day heel prick Guthrie test but the
result may not routinely be available until the child is several weeks old. Earlier diagnosis is
definitely beneficial. So if you suspect one of these disorders, do not wait for the routine
Guthrie to be reported. Contact the laboratory and expedite the test!
Metabolic conditions may present in the following ways
Antenatally suspected: babies born to mothers with acute fatty liver of pregnancy
[AFLP], or to mothers with recurrent HELLP syndrome [haemolysis, elevated liver
enzymes and low platelets] are at increased risk of LCHAD.
Abnormalities evident at birth: dysmorphic features, severe hypotonia, seizures and
apnoea, hydrops and ascites.
Neonatal Guidelines Valid until 1st March 2021 4
Chapter 10: Metabolic v2017.1 Presentation following an asymptomatic interval [of varying lengths – hours to
weeks]. Presentation may be non-specific, and may mimic sepsis.
a. Encephalopathy: poor feeding, persistent vomiting with no anatomical cause,
persistent hiccups, seizures, coma.
b. Acid-base disturbance
c. Liver impairment
d. Cardiac impairment
e. Unexplained hypoglycaemia
f. Hyperammonaemia
g. Others: Unusual odours, Cataract, Neutropenia, Thrombocytopenia
Other clues from the history.
a. Parental consanguinity
b. Previous neonatal death
c. Recurrent non-immune hydrops fetalis
d. Siblings with known inborn errors of metabolism
In the work up of a baby presenting with any of the above, IEMs should be considered in the
differential diagnosis. Therefore, in addition to any other investigations requested, a set of
preliminary tests should be sent aiming to identify the presence of an IEM.
Pathogenesis:
Majority are autosomal recessive.
A few are X-linked recessive, e.g. ornithine carbamyl transferase (OCT) deficiency.
a) Problems making and breaking complex molecules.
Making: Zellweger – Inability to synthesise peroxisomes
Smith Lemli Opitz – block in cholesterol synthesis
CDG (congenital disorders of glycosylation) – block in glycosylation
Breaking: Hurler Syndrome – failure to breakdown mucopolysaccharides
Tay Sachs – failure to breakdown gangliosides
Fabry disease – failure to breakdown glycolipids
Neonatal Guidelines Valid until 1st March 2021 5
Chapter 10: Metabolic v2017.1b) Intoxication:
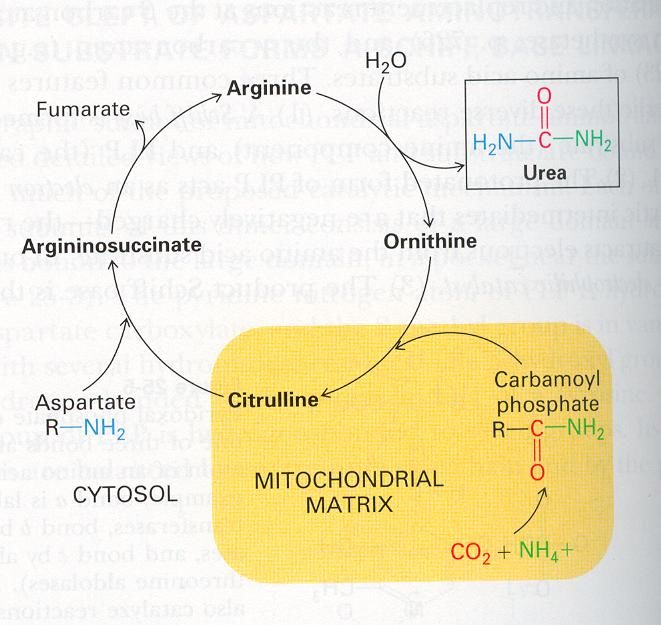
Urea cycle disorders – block in ammonia clearance
Organic acidaemias – block in amino acid breakdown
Galactosaemia – block in Galactose metabolism
c) Energy Insufficiency:
Congenital lactic acidosis –Respiratory chain disorders (mitochondrial disorders)
Pyruvate metabolism disorders
Energy supply - Fat oxidation defects (FAO)
Glycogen storage disorders (GSD)
Gluconeogenesis defect
d) Deficiencies of glucose transporter enzymes (e.g. GLUT1)
Clinical Examination:
A careful clinical examination of all the systems is required. These include:-
Dysmorphic features
Cardiovascular system, especially tachycardia, capillary refill time, signs of cardiac
failure.
Respiratory - especially respiratory rate ↑ in metabolic acidosis, apnoea in altered
conscious state.
GIT - jaundice, hepatomegaly, splenomegaly.
Neurological - conscious level, seizures, abnormal movements, tone, posture,
irritability, feel the fontanel, measure the head circumference.
Urinalysis – glucose and ketones. Smell.
Eyes – look carefully for cataracts, corneal clouding. Fundus for pigment or cherry
red spot.
Neonatal Guidelines Valid until 1st March 2021 6
Chapter 10: Metabolic v2017.1Clinical Presentation and Differential Diagnosis
Seizures:
When the usual causes of HIE, infection and biochemical causes have been excluded or seem
unlikely, metabolic causes need to be considered.
a) Isolated Seizures
- Pyridoxine dependent seizures
- Folinic acid responsive seizures
- Biotin responsive multicarboxylase deficiency
- Congenital malabsorption of magnesium
b) Seizures with other Severe Neurological Signs
- Non-ketotic hyperglycinaemia
- Sulfite oxidase deficiency
- Peroxisomal disorders
- GLUT1 deficiency
c) Seizures with pre-existing stupor, coma or hypoglycaemia
- MSUD
- Organic acidaemia
- Urea cycle disorder
Hypotonia:
The most severe metabolic causes of hypotonia are:-
- Congenital lactic acidosis
- Respiratory chain disorders
- Urea cycle disorders
- Non ketotic hyperglycinaemia (NKH)
- Sulphite oxidase (SO) Deficiency
- Peroxisomal disorders
Hepatic Presentation:
a) Hepatomegaly and seizures suggest:-
- Glycogen storage Type I or III
Neonatal Guidelines Valid until 1st March 2021 7
Chapter 10: Metabolic v2017.1- Gluconeogenesis defects
- Severe hyperinsulinism
b) Liver Failure
- Galactosaemia
- Tyrosinaemia type 1
- Neonatal haemochromatosis
- Respiratory chain disorders
c) Cholestatic jaundice and failure to thrive
- Alpha-1-antitrypsin deficiency
- Inborn errors of bile acid metabolism
- Peroxisomal disorders
- Niemann Pick Type C disease
- CDG syndrome
- Cholesterol Biosynthesis Defects
d) Hepatosplenomegaly
- Lysosomal storage disorder
Cardiac Presentation:
a) Cardiac failure, Cardiomyopathy, Hypotonia and muscle weakness suggests:-
- Respiratory chain disorders
- Pompe disease
- Fatty acid oxidation disorders
b) Cardiac failure, pericardial effusions, cardiac tamponade, Cardiomyopathy
- CDG syndrome
c) Cardiomyopathy and conduction defects
- Long chain fatty acid disorders
Neonatal Guidelines Valid until 1st March 2021 8
Chapter 10: Metabolic v2017.1Initial screen for metabolic diseases:
Unless one is looking for a specific condition (for example when there is a known positive
family history), the following test must ALL be undertaken in an initial screen for metabolic
disease.
Blood gas
Metabolic acidosis: organic acidemias, primary lactic acidosis, following collapse in
any IEM (even urea cycle disorders!)
Respiratory alkalosis: seen early in urea cycle defects
Electrolytes, urea & creatinine
Low urea in relation to creatinine early in urea cycle defects
Calculate anion gap [Na+ + K+] – [Cl- + HCO3-]: increased > 20 in organic acidemias,
primary lactic acidosis, some fatty acid oxidation defects
Laboratory blood glucose (Unexplained hypoglycaemia):
Hypoglycaemia in babies may accompany the following disorders
fatty acid oxidation defects
organic acidemias
primary lactic acidosis
glycogen storage disorders
Endocrine causes eg cortisol deficiency, growth hormone
deficiency, hyperinsulinaemia
Liver enzymes, including albumin, and split bilirubin
Elevated liver enzymes and bilirubin are found in :
Galactosemia,
Tyrosinaemia, peroxisomal disorders eg refsum’s disease
Full blood count and film
Pancytopaenia: in secondary infection or overwhelming disease, which may
complicate IEM.
Ammonia
>200mmol/L: IEM until proven otherwise: urea cycle disorders, organic acidemias,
fatty acid oxidation defects, transient hyperammonemia of the newborn.
NB emergency treatment of hyperammonemia in section
Lactate
Persisting >3mmol/L: primary lactic acidosis
Beware of sampling errors – best if free flowing arterial sample
Exclude tissue hypoxia, congestive heart failure, sepsis, post convulsion
CPK
Urine
Ketones: elevated in maple syrup urine disease (MSUD), absent in fatty acid oxidation
defects (especially important when absent in presence of hypoglycaemia),
Galactosemia, fructose 1,6 biphosphate aldolase deficiency
Reducing substances
Organic & amino acid profile:
Further investigations
Recurrent hypoglycaemia
See Endocrine Chapter (Special investigations in hypoglycaemia)
Do not delay correcting the low blood glucose if the blood is difficult to obtain and do not
wait for the urine to be collected. Give 10% glucose 3mls / kg and increase the infusion rate
from the baseline. Recheck the glucose within 10-20 minutes and again at 1 hour
Further investigations of raised ammonia (Discuss with lab urgently)
U and E, Clotting, Glucose, lactate, blood gas
Plasma amino acids requested .
Blood spot acyl carnitine profile
Urine amino acids
Urine organic acids including orotic acid
Ammonia >250 micromol/litre - start immediate treatment and arrange transfer to a
specialist centre
Monitor neurological status (Glasgow coma score or similar for infant)
Neonatal Guidelines Valid until 1st March 2021 10
Chapter 10: Metabolic v2017.1Further Investigations according to clinical presentation.
(Many of these tests will be done only in specialist centres)
Presentation Condition Specific Investigation
Encephalopathy Urea cycle defects Urine amino acids
Organic acidemias & MSUD Urine organic acids
Plasma amino acids
Blood carnitine & acylcarnitines
Fatty acid oxidation defects As above, plus:
Blood for DNA mutation analysis
Primary lactic acidosis (e.g. CSF Lactate
puruvate dehydrogenase Mitochondrial DNA (blood & muscle)
deficiency, respiratory chain Muscle biopsy (histology & electron
defects) microscopy)
Skin biopsy (enzymes of pyruvate
metabolism)
Non-ketotic hyperglycinaemia Plasma & CSF amino acids
CSF: plasma glycine levels
USS brain +/-MRI (agenesis corpus callosum,
cerebellar abnormalities
Molybdenum cofactor deficiency ↓Urine sulphite
Urine amino acids (↑ hypoxanthine and
taurine)
↓ Plasma and urine uric acid
Pyridoxine dependent seizures Pipecolic acid in plasma and CSF
Trial of pyridoxine under EEG
Liver Disorders Peroxisomal disorders Blood VLCFA
Glycogen storage disease (types I, Leucocyte / liver biopsy (enzyme assay)
III, VI, IXX) ↑Plasma lactate, CPK, uric acid
Most diagnosed now on a specific genetic
panel
↓Red cell galactose-1-phosphate uridyl
Galactosaemia transferase activity
If recently transfused, above test unreliable,
so measure RBC galactose 1 phosphate as
alternative (↑)
Ophthalmology: for cataracts
Neonatal Guidelines Valid until 1st March 2021 11
Chapter 10: Metabolic v2017.1Plasma amino acids
Tyrosinaemia type I Urine organic acids
Coagulation
Investigate for renal Fanconi’s syndrome
α1 Antitrypsin deficiency Serum α1 antitrypsin
genotype
Niemann-Pick A Acid sphingomyelinase in leukocytes (↓)
(↓) HDL cholesterol and ↑cholesterol
and TG
Foam cells on histiocytes in bone
marrow
Cardiomyopathy
Mitochondrial defects (respiratory ↑CSF lactate
chain) Mitochondrial DNA (blood & muscle)
Muscle biopsy (histology & electron
Pompe’s disease (GSD type II) microscopy)
Fatty oxidation defects Lymphocyte / skin fibroblast (enzyme
assay)
Mitochondrial defects As stated under encephalopathy
CDG 1a syndrome As stated under liver disease
Lysosomal storage disorders Serum transferrin isoelectric focusing
Dysmorphism Lysosomal storage disorders Skin biopsy (enzyme assay)
White cell enzymes
Disorders of sterol synthesis
Urine oligosaccharides and
CDG (Congenital disorders of mucopolysaccharides
glycosylation) syndrome as in cardiomyopathy
Urine organic acids
Glutaric aciduria type II Plasma 7-dehydrocholesterol
Skin biopsy
Peroxisomal disorders (e.g. Serum transferrin isoelectric focusing
Zellweger) Skin biopsy (enzyme assay)
Urine organic acids
Blood carnitine & acylcarnitine, VLFA
Neonatal Guidelines Valid until 1st March 2021 12
Chapter 10: Metabolic v2017.1**There are also a variety of specialist investigations done, e.g., specific enzyme studies on
blood or skin fibroblast, muscle biopsies, etc
It is good practice to save and freeze all urine passed for future analysis, and to save a
heparinised blood sample before the first blood transfusion.
General treatment – Acute:
This must be commenced as soon as preliminary results suggest a possibility of IEM. Please
see website of BIMDG emergency treatment guidelines for further details
ABC: basic neonatal intensive care
Discontinue all milk feeds
Protein content increases amino acid load; toxic in urea cycle defects and
organic acidemias.
Carbohydrate load contains lactose. Galactose is toxic in Galactosemia
Provide adequate calories – high calorie, protein free nutrition parenterally, or
enterally if specific diagnosis has been made and feeds are judged safe.
Correct hypoglycaemia; consider insertion of central access (UVC) early on.
Correct acidosis – beware of hypernatremia if many NaHCO3 corrections are given
Correct electrolyte disturbances
Be vigilant for sepsis; note that some IEMs predispose to sepsis, e.g. galactosemia
Insulin for reinforcement of anabolism (dose 0.02 – 0.1 u/kg/hr) may be considered
Liaise with Specialist team early: Dr Graham Shortland Consultant in Metabolic
Diseases at UHW if in doubt.
Treatment of Hyperammonaemia:
Metabolic team @ UHW, led by Dr Graham Shortland, recommend early contact for advice.
Remember NH3 is very neurotoxic and needs to be reduced quickly.
NH3 > 250 needs the following to commence ASAP, as well as contacting Metabolic
team.
NH3 > 450 should have haemofiltration - i.e. urgent transfer to PICU.
Neonatal Guidelines Valid until 1st March 2021 13
Chapter 10: Metabolic v2017.1(a) Increase glucose infusion rate (GIR) to 7 - 8mg/Kg/min, even when glucose
measurements are normal, and if necessary add insulin if hyperglycemia ensues. This serves
to inhibit catabolism of endogenous protein
(b) Treat any acidosis / electrolyte imbalance
(c) Sodium Benzoate 250mg/Kg IV bolus over 90 minutes, followed by 250mg/Kg over 24
hours as continuous IVI.
Together with:
(d) Sodium Phenylbutyrate 250mg/Kg IV bolus over 90 minutes, followed by 250mg/Kg over
24 hours as IVI.
(e) L-Arginine 200mg/Kg IV bolus over 90 minutes, followed by 8mg/Kg/hour IVI
(All the above three drugs are now stocked and available from Singleton NICU – If not in
stock or out of date contact on call pharmacist)
(f) Re-check NH3 four hours after onset of steps (c) to (e), to check for response.
Some Specific Disorders
Only a few of the more common disorders are covered here in minimal detail.
For all of these disorders please refer to the referenced texts and to the BIMDG emergency
guidelines for management
Glycine encephalopathy (non ketotic hyperglycinaemia)
- Basic defect is the glycine cleavage system
- Reduced fetal movements in utero
- Neonatal disease usually presents early, within 48 hours after birth
- Hiccups
- Hypotonia
- Depressed level of consciousness
- Seizures with burst suppression pattern on EEG
- Plasma glycine levels variable
- CSF and urine glycine are elevated
- Organic acids in urine are normal
- May be agenesis of corpus callosum
- Diagnosis confirmed on transformed lymphocytes or liver biopsy
Neonatal Guidelines Valid until 1st March 2021 14
Chapter 10: Metabolic v2017.1Maple Syrup Urine Disease:
- Presents at around 1 week of age
- Seizures
- Encephalopathy
- Vomiting
- Frequent hypoglycaemia
- Severe keto-acidosis and increased anion gap
- Typical sweet odour in urine
- Elevated leucine, isoleucine and valine in blood and urine
Tyrosinaemia Type 1:
Type 1 is due to a deficiency of fumaryl acetoacetase (FAH). This causes a build up of
fumaryl and maleyl acetoacetate, responsible for renal and hepatic damage.
- Progressive liver disease and renal tubular dysfunction
- Hypoglycaemia due to pancreatic islet cell hyperplasia
- Plasma tyrosine and methionine are raised
- Phosphate and potassium levels are low
- Generalised aminoaciduria, glycosuria, phosphaturia, rickets
- Urinary organic acid – increase succinyl acetone
Treatment: NTBC 2 (2 – nitro – 4 – trifluoro – methylbenzoyl 1, 3 cyclohexanedione).
NTBC inhibits hydroxy phenylpyruvate dioxygenase, so reducing toxic metabolites.
Neonatal Guidelines Valid until 1st March 2021 15
Chapter 10: Metabolic v2017.1Pathway for Degradation of Tyrosine:
Hyperammonaemia – differential diagnosis:
Inherited disorders –
- Urea cycle disorders
- Organic acidemias (e.g. propionic acidaemia, methylmalonic acidaemia, etc)
- Fatty acid oxidation disorders
- Other inborn errors (ornithine amino transferase deficiency, HHH
syndrome, etc)
Acquired disorders –
- Transient Hyperammonaemia
- Perinatal asphyxia
- Herpes simplex infection
- Liver disease
- Any severe illness
Neonatal Guidelines Valid until 1st March 2021 16
Chapter 10: Metabolic v2017.1UREA CYCLE DISORDERS: The urea cycle is a metabolic pathway enabling detoxification of ammonia and producing urea. Incidence : OTC (ornithine transcarbamylase), ASS (arginosuccinic acid synthetase) deficiency, ASL (arginosuccinic acid Lyase) deficiency , ARG (arginase deficiency), and NAGs (N acetyl glutamate synthetase) CPS (carbamyl phosphate synthetase) each have incidence of 1:100,000 approx.. Symptoms: Vomiting, drowsiness becoming unconscious, seizures, shock, hepatomegaly Diagnosis: Raised ammonia (>200 µmol/l) Plasma glutamine > 800 µmol/l Normal urinary organic acid profile. Remember to check LFT’s, glucose, ammonia and clotting, frequently. Neonatal Guidelines Valid until 1st March 2021 17 Chapter 10: Metabolic v2017.1
Early metabolic alkalosis, but can have metabolic acidosis after collapse
Treatment please see page 14
Organic Acid Disorders:
a) Propionic acidaemia
Poor feeding, vomiting, drowsiness, coma
Diagnosis
Raised ammonia
Ketoacidosis
Hyperglycinaemia
Raised urine organic acid and ketones
Increased anion gap
Long Term Treatment
High carbohydrate
Protein restriction
Sodium bicarbonate
Sodium benzoate
Carnitine 250 – 500 mg/kg/day
b) Methyl Malonic Acidaemia:
Clinically similar to proprionic acidaemia.
Presents with vomiting, acidosis and neurological depression.
Treatment:
Withdraw protein, high carbohydrate ± insulin
Vitamin B12
Biotin
Galactosaemia
This presents after a few days with vomiting, failure to thrive, jaundice, neurological
sequelae and possibly a superadded septicaemia, e.g. E. coli, which often starts as a UTI.
There is usually hepatomegaly and may be cataracts. If suspected, take urine for reducing
Neonatal Guidelines Valid until 1st March 2021 18
Chapter 10: Metabolic v2017.1substances and blood (lithium heparin) for erythrocyte galactose 1 phosphate uridyl
transferase. If the baby has previously received a blood transfusion, enzyme analysis is
unreliable and diagnosis is achieved measuring Galactose 1 phosphate in blood.
Treatment:
Stop all lactose and Galactose in diet.
Give Nutramigen milk.
Lactic Acidosis:
In neonates, lactic acidosis is usually secondary to tissue hypoxia, and is not usually
associated with ketosis.
Increased Lactate is raised in a number of inborn errors
a) Respiratory Chain (mitochondrial) Disease
Encephalopathy
Hypotonia
± Hypoglycaemia
Possible Cardiomyopathy
Changes in basal ganglia and brain stem on MRI.
Also have raised CSF lactate.
b) Fructose 1-6 bisphosphatase Deficiency
Severe anion gap metabolic acidosis presenting in first week of life
A defect in gluconeogenesis pathway
Muscle weakness, hepatomegaly
Hyperventilation.
Hypoglycaemia.
Apnea and possible death.
High alanine, lactate and pyruvate
Diagnosis established by genetic testing
Avoid fasting and give dextrose infusion.
Neonatal Guidelines Valid until 1st March 2021 19
Chapter 10: Metabolic v2017.1c) Pyruvate Dehydrogenase Deficiency
Encephalopathy
Hypotonia
Severe acidosis increased pyruvate and lactate and alanine
Patients deteriorate when given a high glucose intake.
May improve if given high doses of thiamine.
Need a high lipid, high protein, low carbohydrate regime (ketogenic diet).
d) Organic acidaemia ( eg methylmalonic, proprionic, isovaleric, and glutaric type 1
acidemia
e) Glycogen Storage Disease (GSD) Type 1:
Presents with:
Profound hypoglycaemia
Lactic acidosis
Hyperuricaemia
Hyperlipidaemia
Hepatomegaly
Patients with Type 1b also have chronic Neutropenia with functional deficiencies of
neutrophils and monocytes, which required GMCSF therapy.
f) Fatty Acid Oxidation Defects:
- A number of enzyme defects have been identified – enzymes have specificity based
on length of carbon chain.
- Medium chain acyl co A dehydrogenase (MCAD) deficiency is most common in
Caucasians.
- Usually presents with hypoketotic hypoglycaemia
- Some can cause Cardiomyopathy
- May have increased CPK, and transaminases
- Characteristic pattern of urine organic acids
- Blood spots on Guthrie for acyl carnitine analysis by tandem mass spectrometry
Neonatal Guidelines Valid until 1st March 2021 20
Chapter 10: Metabolic v2017.1Peroxisomal Disorders:
- Zellweger’s Syndrome, Infantile Refsums Disease, neonatal adrenoleukodystrophy
- Raised liver enzymes
- Dysmorphic
- Severe neurological abnormalities plus punctate epiphyseal calcification, liver
fibrosis
- Request analysis of very long chain fatty acids
Further reading
1. Text book Pediatric endocrinology and inborn errors of metabolism K Sarafoglou
2. Text book Atlas of metabolic diseases WL Nyhan, BA Barshop, PT Ozand
Neonatal Guidelines Valid until 1st March 2021 21
Chapter 10: Metabolic v2017.1NOTES ON SPECIMENS
BIOCHEMISTRY LAB: @ SINGLETON: 5037 @ MORRISTON: 713046
MEDICAL BIOCHEMISTS @ MORRISTON: 713036
Any sample that needs to be processed outside Swansea, is transported by the biochemists to the Morriston
lab first. Some assays require the sample to be frozen as soon it reaches the lab. If a frozen sample needs to be
transferred to UHW, this requires special transport, which is routinely available only twice a month from
Morriston, thus if results are needed urgently, the lab personnel must be contacted, by a senior member of our
team, to arrange earlier transport.
PAEDIATRIC SPECIMEN BOTTLES USED ON NICU
Lithium heparin: green top
Fluoride oxalate: yellow top
EDTA: lilac top
Substance Sample Special measures – Lab where processed
PLEASE adhere to these
Ammonia 500 micro litres Immediately put bottle in ice Morriston
EDTA Transport to lab immediately
Lactate 500 micro litres Singleton
Fluoride oxalate
Plasma amino acids 600 micro litres Send to lab immediately UHW, Cardiff
Lithium heparin
Carnitines 3 X blood spots on UHW, Cardiff
‘Guthrie card’
Free fatty acids 500 micro litres Send to lab immediately UHW, Cardiff
Fluoride oxalate
VLCFA 600 micro litres Southmead Hospital, Bristol
Lithium heparin
Ketones (3-hydroxy 500 micro litres UHW, Cardiff
butyric acid) Fluoride oxalate
Insulin & C-peptide 600 micro litres Immediately put bottle in ice UHW, Cardiff
Lithium heparin Transport to lab immediately
ACTH 500 micro litres EDTA Immediately put bottle in ice UHW, Cardiff
Transport to lab immediately
Cortisol 600 micro litres Remember diurnal variation Morriston
Lithium heparin not well developed in
neonates
Growth hormone 600 micro litres Morriston
Lithium heparin
Neonatal Guidelines Valid until 1st March 2021 22
Chapter 10: Metabolic v2017.1You can also read