Medical Apps- when are they medical devices? - Valerie Field- Head of Devices Software/apps - UK Medicines Information
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Medical Apps- when are they medical devices? Valerie Field– Head of Devices Software/apps

OVERVIEW
• Pace of new technology
• Types of app/algorithm changing
• Regulation & its challenges.
• New regulation- the differences.
• What it means for me and you?
• Guidance & Help
• Maintaining patient safety
2
PACE OF TECHNOLOGY 3

TYPE OF TECHNOLOGY 4
TYPE OF TECHNOLOGY is Changing
• Multiple Datasets- Big Data
• Exciting new methods and algorithms being developed
will be used as the basis for new software based medical
devices including;
• Bioinformatics tools used in clinical genomics
• Apps for dose calculation linked to big data
• Symptom checkers-Triage apps
• Machine Learning/AI- Already becoming a reality
• element of ‘learning from errors’.
• tolerance for errors in healthcare low
• important emphasise transparency and clinical
validation for AI applications designed for
healthcare.
5
TYPE OF TECHNOLOGY is Changing
• AI has great potential to transform the UK health and
social care sectors.
• already in use and will continue to grow
• ability to reduce face-to-face time with clinicians
• can be an important part of a care pathway and
patient self-management
• but not without risk
• Transparency on
• How algorithms are continually tested, verified and
clinically validated
• How processes are made understandable to
clinicians to gain support and use.
6
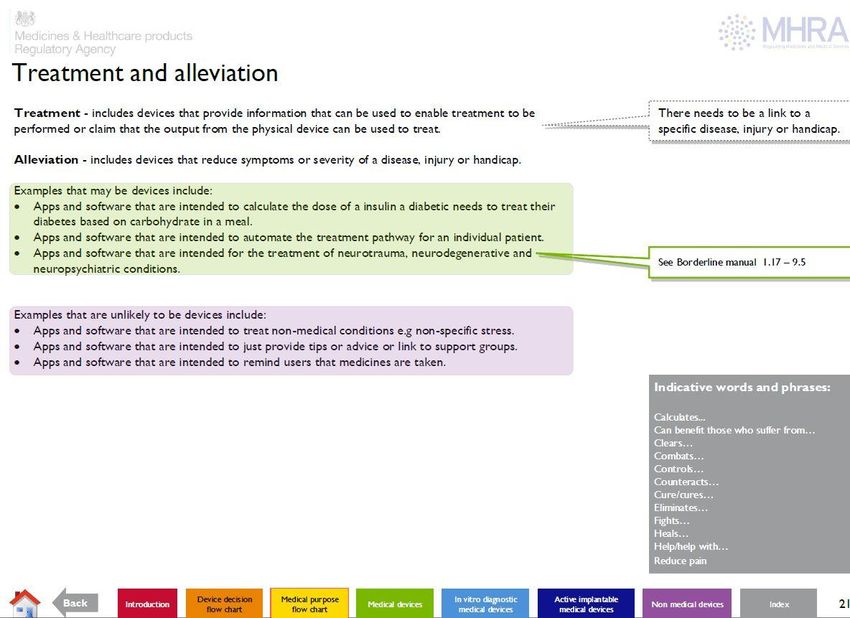
WHEN IS SOFTWARE A MEDICAL DEVICE?
• What does the manufacturer claim used for?
• Is this use considered to be a ‘medical purpose’?
• prevent disease/illness
• used for diagnosis
• monitoring
• treatment*
• compensation for an injury or handicap
• Investigation, replacement or modification
• prediction, prognosis
• software intended for life-style and well-being purposes
is not a medical device.
http://www.legislation.gov.uk/uksi/2002/618/regulation/2/made
7
Current Regulatory framework
In Vitro
Diagnostics
Medical Device Active
Medical Device Directive Implantable
Directive 98/79/EC Medical Device
93/42/EEC Directive
90/385/EEC
UK Medical
Device
Regulations
2002
8
NEW Regulatory framework
General and
In Vitro
Active
Diagnostics
Implantable
Medical Device
Medical Device
Regulation
Regulations
3 Years 5 Years
9
NEW MDR/ IVDR TIMETABLE
EUDAMED??
2017 2018 2019 2020 2021 2022
The Regulations
entered into force in
May 2017.
3 year transition
period However, not
EU MDR EU MDR
Formal Full application everything will fully
publication Implementation
enters into
apply until MAY 2020
force Medical Devices, and
5 year transition
MAY 2022 for In Vitro
EU IVDR
period EU IVDR Diagnostic Medical
Full application
Formal
implementation
Devices.
publication
enters into
force
10New Regs-SOFTWARE SPECIFIC CHANGES
14.2(d)…remove as far as possible risks associated with
the possible negative interaction between s/w and the IT
environment within which it operates & interacts.
14.5 Devices that are intended to be operated together
with other devices shall be designed and manufactured in
such a way that the interoperability and compatibility are
reliable and safe
17.1…or software that are devices themselves shall be
designed to ensure repeatability, reliability and
performance in line with their intended use.
11New Regs-SOFTWARE SPECIFIC CHANGES.
17.2 For devices that incorporate software or for software
that are devices in themselves, the software shall be
developed and manufactured in accordance with the state
of the art taking into account the principles of development
life cycle, risk management, including information security,
verification and validation
17.3. Software referred to in this Section that is intended
to be used in combination with mobile computing
platforms shall be designed and manufactured taking into
account the specific features of the mobile platform (e.g.
size and contrast ratio of the screen) and the external
factors related to their use (varying environment as
regards level of light or noise).
12New regs-SOFTWARE SPECIFIC CHANGES.
17.4 Manufacturers shall set out minimum requirements
regarding hardware, IT networks, characteristics and Its
security measures, including protection against
unauthorised access, necessary to run the software as
intended
13New regs-
SOFTWARE NEW CLASSIFICATION RULE 11
• Software intended to provide information which is used
to take decisions with diagnosis or therapeutic
purposes, is in class IIa, except if such decisions have
an impact that may directly or indirectly cause:
• the death or an irreversible deterioration of the state
of health, in which case it is in class III;
• a serious deterioration of the state of health or a
surgical intervention, in which case it is in class IIb.
• Software intended to monitor physiological processes is
in class IIa except if it is……..
• All other software is in class I.
14New Regs-TRACEABILITY – UDI requirements
• Device software
• The UDI shall be assigned at system level of the s/w.
• Only s/w which is commercially available on its own
and s/w which constitutes a device shall be subject
to that requirement.
• Developers
• Allocate UDI – and update for every modification
• Provide data for European UDI database
• Record of who they supplied it to
• Economic operators – (importers, distributors etc.)
• Record of who supplied it and who they supplied it to.
15The Health Institution Exemption
• Regulatory exemption for devices that are manufactured and
used within a single health institution
• Health institutions may manufacture, modify and use a
device ‘in-house’, in order to target a specific need
• Health institution must meet all safety and performance
requirements of the regulations – but will not need notified
body scrutiny or a CE marking
• Will continue in the revised EU regulations
• MHRA continue to partner with stakeholders to update the
guidance on exemptions for health institutions – for greater
reassurance re. safety and performance whilst applying the
exemption in as simple a way as possible
16OTHER KEY CHANGES
Distributors/Importers- to ensure;
• CE marked
• EU declaration of conformity of the device completed
• A manufacturer is identified and that an authorised
representative identified;
• Appropriately labelled
• Where applicable, a UDI has been assigned by the
manufacturer
• Inform the competent authority of any serious risk
All
• Establish and update a clinical evaluation plan
• Conduct Post-Market clinical follow-up
• Centralised European database (EUDAMED) to capture data
on CE certificates, incidents, clinical investigations, market
surveillance etc.
1718
19
20
21
22
Registration of Class 1 device manufacturers
• Registration only – Not approval
• £100.00
• Self-declare conformity
• Details appear on the publicly accessible database
23
2324
General guidance already issued 25
26
So what are your challenges? • Recognising medical device apps/software • Changes to in-house exemption rules • Current class 1 software developers to check if their devices are going to be ‘up classified’ in the MDR /IVDR such that they will need a Notified Body. • Engaging Notified Bodies • UDI requirements • Increased post market surveillance requirements • Reporting requirements https://yellowcard.mhra.gov.uk/ 27
CHALLENGES – for MHRA
• MDR/IVDR- Work with EU on agreeing interpretation and
implementation of the changes, update of MEDDEV 2.1/6
• International approaches- IMDRF, FDA & others
• Informing developers/ manufacturers of what these changes mean
for them
• Working with Notified Bodies on being ready for the changes
• Engage with the major app stores to let them know what the
requirements will be for importers and distributors.
• How do we ensure verification/clinical validation especially for
Machine learning algorithms.
• Update our software guidance in appropriate timescale
• Ensuring those messages relayed and understood by all involved.
28Example—-Social media signal
MHRA made aware that a company was allegedly
contacting ambulatory insulin pump users with type 1
diabetes through social media and asking them to switch
to their product!!
Issued a generic Medical Device Alert 2016/020 and an
associated press release
“Managing diabetes: patients should not change their
insulin delivery device without checking with their
healthcare specialist and should report such approaches”
29Example app calculation
Report of an app associated with a blood glucose meter used to
determine insulin dose
Issue
• When used with a secondary mobile device [phone or tablet] it was
not downloading information correctly from the cloud database.
• Data on blood glucose meter and primary mobile device not
affected.
• Implications could be extremely serious
Action
• Reported to MHRA
• Company issued Field Safety Notice to customers
• Cloud synchronisation disabled and new software version
developed
30Example
Article published in health publication undertaken by a UK university
Issue
• Article claimed the majority of 40+ ‘stand alone’ insulin dosing
apps they tested were unreliable and put patients at risk of
incorrect doses of insulin
Action
• MHRA liaised with authors of study at university
• All university findings supplied to us
• All issues identified were passed to our compliance unit
• All cases not CE marked or evidence to suggest error investigated
• All UK based apps brought into compliance or removed from
market
• Liaison with organisations to be aware.
31How can we help each other?
MHRA Innovation Office;
Innovationoffice@mhra.gsi.gov.uk
Devices Regulatory Enquiries;
Devices.Regulatory@mhra.gsi.gov.uk
Register your devices;
https://www.gov.uk/guidance/medical-devices-how-to-comply-with-
the-legal-requirements
Report issues to us;
https://yellowcard.mhra.gov.uk
32Please note that whilst we are willing to give any help and advice we can, any views given by us on the interpretation of the Regulations represent our best judgement at the time, based on the information available. Such views are not meant to be a definitive statement of law, which may only be given by the Courts. Accordingly we would always advise you to seek the views of your own professional advisors.
Any Questions?
.
34You can also read

















































