Recent Progress in the Asymmetric Syntheses of α-Heterofunctionalized (Masked) α- and -Amino Acid Derivatives - JKU ePUB
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
Asymmetric Synthesis | Very Important Paper |
Recent Progress in the Asymmetric Syntheses of
α-Heterofunctionalized (Masked) α- and β-Amino Acid
Derivatives
Isabella Eder,[a][‡] Victoria Haider,[a][‡] Paul Zebrowski,[a][‡] and Mario Waser*[a]
Abstract: The asymmetric synthesis of α-heterofunctionalized to give an illustrative overview of the most successfully applied
α- and β-amino acid derivatives has been a heavily investigated concepts to access these targets, like asymmetric α-heterofunc-
topic over the last years, benefiting from the development of tionalizations or asymmetric C–C-bond forming reactions of al-
novel catalysis concepts as well as from the introduction of ready heterofunctionalized precursors.
suited new precursor entities. Within this short review, we wish
1. Introduction that contain other, non-halogen, heteroatom groups (e.g. OR,
SR, NR2, ...) in the α-position. Given the increasing number of
Asymmetric α-heterofunctionalization reactions of prochiral
recent publications dealing with the development of novel syn-
pronucleophiles are important transformations to access valu-
thesis and catalysis concepts to access α-heterofunctionalized
able chiral target molecules.[1] The hereby obtained products
α- and β-AA derivatives stereoselectively, it is the intention of
may possess promising (biological) properties themselves or
this short review to provide an illustrative (but not encyclope-
can serve as versatile building blocks for further manipulations.
dic) overview about some of the most significant (recent)
Therefore, it comes as no surprise that the development of
achievements in the field (primarily covering developments
stereoselective (catalytic) methods for the introduction of
from the last 10–15 years but also highlighting selected pio-
heteroatoms in the α-position of carbonyl compounds (or
neering older studies). It should be noted, that the majority of
analogous derivatives) has been heavily investigated in the
the reports covered herein focused primarily on the develop-
past.[1] Among the different classes of frequently investigated
ment of stereoselective methods for the synthesis of masked/
α-substituted chiral carbonyl derivatives, α-functionalized α-
protected AA-derivatives, while full deprotection to the free
and β-amino acids (α-AA, β-AA) have been amongst the most
amino acids or incorporation into peptides was in a lot of cases
prominent compounds of interest.[2–4] The importance of intro-
either not attempted or, if tried, sometimes also not possible
ducing robust and powerful methods to access (non-natural)
(i.e. for the case of α-F-α-AA).
amino acid derivatives stereoselectively cannot be overesti-
mated, given the value of amino acids, peptides, and proteins
for numerous applications.[2,5,6,7] Among the broad variety of
different amino acid derivatives that have been investigated
over the last decades, halogenated ones have received consid-
erable attention,[5] especially fluorine-containing ones.[6] Here it
can be distinguished between those derivatives that contain
the halogen (i.e. fluorine) somewhere in a side chain, and those
which possess an α-halogenated stereogenic center.[5,6] In addi-
tion, the last years have also witnessed an increasing interest in
the asymmetric synthesis of α- and β-amino acid derivatives
[a] I. Eder, V. Haider, P. Zebrowski, Prof. Dr. M. Waser
Institute of Organic Chemistry, Johannes Kepler University Linz,
Altenbergerstr. 69, 4040 Linz, Austria
E-mail: mario.waser@jku.at
[‡] Equal contribution by these three authors (alphabetical order)
ORCID(s) from the author(s) for this article is/are available on the WWW
under https://doi.org/10.1002/ejoc.202001077.
© 2020 The Authors. Published by Wiley-VCH GmbH · This is an open access
article under the terms of the Creative Commons Attribution License, which
permits use, distribution and reproduction in any medium, provided the Scheme 1. Most commonly described strategies to access α-heterofunctional-
original work is properly cited. ized α- and β-amino acid derivatives.
Eur. J. Org. Chem. 2021, 202–219 202 © 2020 The Authors. Published by Wiley-VCH GmbH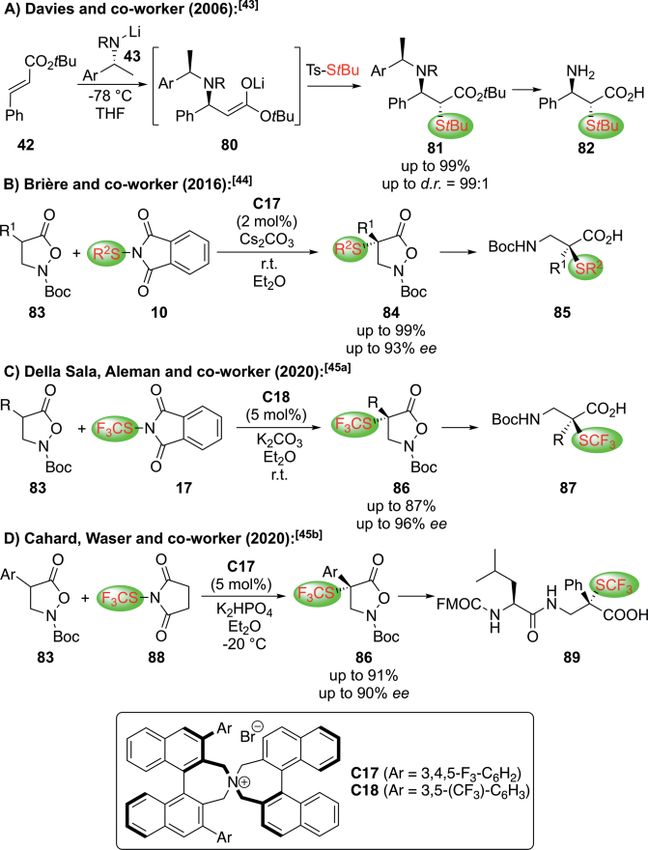
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
The first part of this review will focus on α-heterofunctional- far, the main focus was on the development of methods to
ized α-AA (Scheme 1A). Here the most commonly reported facilitate the stereoselective introduction of the heteroatom in
strategies rely on the use of masked (protected) α-AA deriva- the masked surrogate, while further manipulations to the free
tives that undergo stereoselective α-heterofunctionalization re- α-AA have rarely been described.
actions with suited electrophilic heteroatom transfer reagents.
The second part will cover methods for the asymmetric synthe-
sis of α-heterofunctionalized β-AA (Scheme 1B). Here several
fundamentally different strategies have emerged. On the one 2.1. α-F-α-AA Derivatives
hand, suitable masked β-AA derivatives can undergo asymmet-
The synthesis of fluorine-containing amino acids is one of the
ric electrophilic or nucleophilic α-heterofunctionalization reac-
most prominent tasks in amino acid/peptide chemistry nowa-
tions. In addition, the use of α-heterofunctionalized pronucleo-
days.[6] While the introduction of fluorine or a fluorine-contain-
philes in asymmetric C–C-bond forming reactions (i.e. Mannich
ing group in the amino acid side chain is well established,[8]
type approaches), or asymmetric hydrogenation reactions of
the stereoselective syntheses of α-F-α-AA derivatives are rather
appropriately substituted alkenes represent complementary
challenging targets transformations.[9–12] First, only a handful
powerful methods to access masked α-heterofunctionalized
of asymmetric approaches for the α-introduction of fluorine in
β-AA derivatives as well.
masked α-AA derivatives have been developed so far (vide in-
fra).[11] In addition, the H–N–C–F structural motive easily under-
goes HF-elimination, especially when the N-lone pair is not de-
2. α-Amino Acid Derivatives
localized in e.g. a (sulfon)-amide bond.[9] As a consequence, and
The asymmetric synthesis of chiral α-heterofunctionalized β-AA despite the successful development of methods for the α-fluor-
derivatives has been thoroughly investigated over the last ination of N-protected α-AA derivatives,[11,12] further manipula-
years. Hereby the choice of appropriately masked or protected tions and N-deprotection to the free α-F-α-AA are very difficult
α-AA analogs is of crucial importance, as a direct functionaliza- or almost impossible. Nevertheless, interesting stereoselective
tion of the free α-AA is usually not possible. In addition, it approaches towards masked α-F-α-AA have been reported
should be emphasized that in most of the cases reported so (Scheme 2),[11] and the development of novel strategies to ac-
Isabella Eder born in Linz, Austria, in 1996. She started to study chemistry at the Johannes Kepler University in Linz in the year 2014 and
graduated in 2019. Her master thesis was supervised by Mario Waser focusing on the enantioselective synthesis of α-fluorinated β-amino
acids. At the moment she is carrying out Ph.D. studies within the same group focusing on chiral isothiourea catalysis.
Victoria Haider was born in Linz, Austria, in 1994. She started studying chemistry at the Johannes Kepler University in Linz in 2012, where
she received her master's degree in 2018 under the supervision of Mario Waser with the main focus on the synthesis and applications of
guanidine-containing chiral quaternary ammonium salt catalysts. She is currently a Ph.D. candidate in the same group and her research
project focuses on the syntheses of chiral fluorinated amino acids and peptides.
Paul Zebrowski was born in Klosterneuburg, Austria, in 1990. He studied chemistry at JKU Linz, Austria, where he graduated in January 2020.
During his master thesis in the group of Mario Waser, he investigated the enantioselective synthesis of α- and β-amino nitriles via bifunctional
ammonium salt catalysis. Since February 2020, he is working on his Ph.D. in the same group, where his main focus is currently laid on the
asymmetric α-heterofunctionalization of masked β-amino acid derivatives.
Mario Waser was born in Steyr, Austria in 1977 and studied chemistry at JKU Linz, Austria where he obtained his Ph.D. in 2005 in the group
of Prof. Heinz Falk. After a postdoctoral stay with Prof. Alois Fürstner (Max-Planck Institut für Kohlenforschung, Mülheim, Germany), he spent
two years as an R&D chemist working for DSM. In 2009 he started his independent career at JKU Linz. In 2014 he obtained his habilitation
(venia docendi) and became Associate Professor and in 2020 he became Full Professor for Organic Stereochemistry. His main research
interests are on the design and application of asymmetric organocatalysts (i.e. ion-pairing catalysts) and on the development of asymmetric
organocatalytic synthesis methods.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 203 © 2020 The Authors. Published by Wiley-VCH GmbH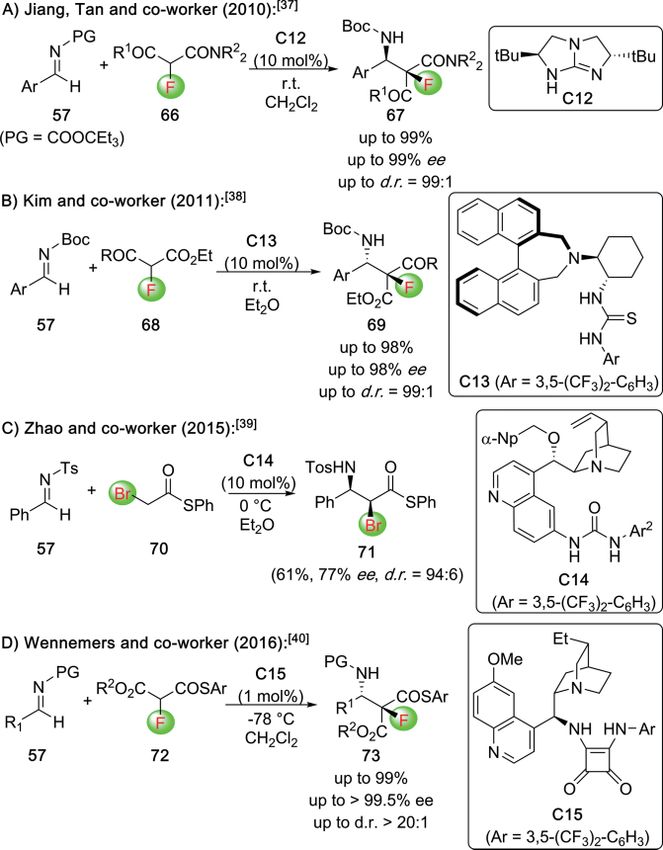
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
cess such targets even in a racemic manner is still a topic of
current interest.[12]
Scheme 3. Asymmetric organocatalytic α-sulfanylation of masked α-AA deriv-
atives.
Scheme 2. Enantioselective approaches towards masked α-F-α-AA deriva-
tives.
Recently, the (asymmetric) di- or trifluoromethylthiolation of
(prochiral) nucleophiles became an important and heavily in-
One classical approach to carry out asymmetric α-fluorin-
vestigated topic.[16] Owing to the value of the incorporation of
ation reactions of prochiral nucleophiles relies on the use of N-
fluorinated Cinchona derivatives F as chiral electrophilic these F-containing groups in potentially biologically relevant
F-transfer reagents.[10,11] Cahard and co-worker,[11a,11b] as well molecules,[16] it comes as no surprise that the development of
as Togni's group[11c] have shown that this methodology also methods for the asymmetric synthesis of α-di-/trifluoromethyl-
thiolated-α-AA has attracted the attention of the synthesis com-
allows accessing the masked enantioenriched α-F-α-AA deriva-
munity lately (Scheme 4).[17] Shen and co-workers succeeded
tives 2, 4, and 6 as outlined in Scheme 2. Unfortunately, how-
ever, as already stated above, further manipulations were found in carrying out the asymmetric α-difluoromethylthiolation of
to be rather difficult and the synthesis of free α-F-α-AA or pept- azlactones 12 with excellent enantioselectivities by using the
ides thereof still represents one of the major challenges in the chiral CF2HS-reagent 15 (Scheme 4A).[17a] On the other hand,
field of amino acid chemistry. the analogous α-CF3S-derivatives 18 could be accessed with
very high selectivities under asymmetric phase-transfer cataly-
sis conditions by using the established Cinchona alkaloid
ammonium salt C4, as reported by the groups of Della Sala and
2.2. α-SR-α-AA Derivatives Aleman recently (Scheme 4B).[17b]
The stereoselective synthesis of α-sulfanylated-α-AA derivatives
has attracted considerable interest over the last decade.[13–15]
Hereby, especially the use of asymmetric organocatalysis turned
out to be rather promising to access a variety of different
masked α-SR-α-AA derivatives straightforwardly with good to
excellent selectivities (Scheme 3). In 2009, Olenyuk and
co-workers first reported the Cinchona alkaloid C1-catalyzed
α-sulfanylation of the cyclic α-AA derivatives 7 with reasonable
enantioselectivities (Scheme 3A).[13]
A few years later, Fang, Zhu, and co-workers developed a
prolinol C2-controlled α-sulfanylation of the α-nitroester 5
(Scheme 3B)[14] while Jiang's group introduced the highly
enantioselective α-sulfanylation of azlactones 12 using
the squaramide-containing Cinchona alkaloid catalyst C3
(Scheme 3C).[15] The latter group also demonstrated the
successful nucleophilic ring-opening of products 14 to access Scheme 4. Recently reported methods for the asymmetric syntheses of
differently protected acyclic α-SR-α-AA derivatives. α-CF2HS- and α-CF3S-α-AA derivatives.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 204 © 2020 The Authors. Published by Wiley-VCH GmbH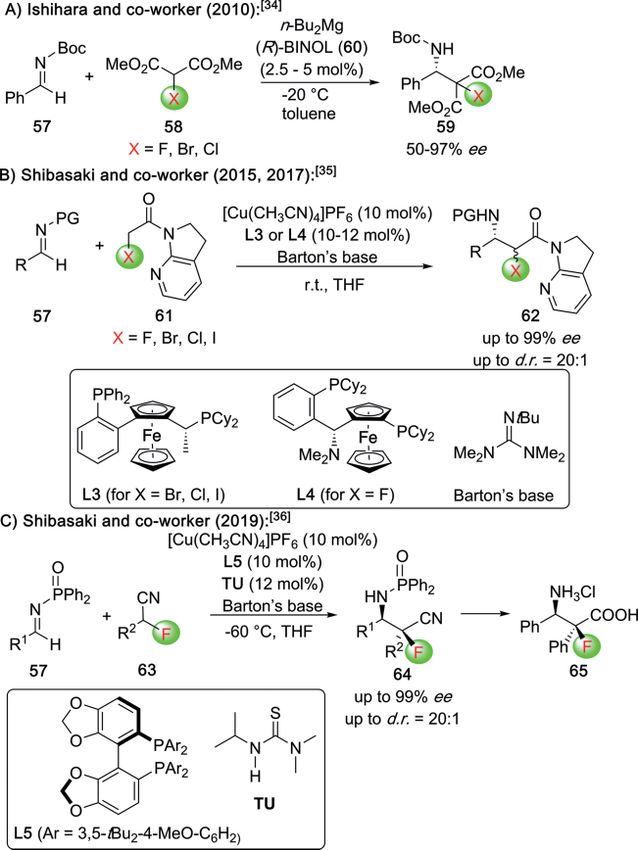
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
2.3. α-NR2-α-AA Derivatives ther the syn or the anti diastereomer could be perfectly con-
trolled by variation of the reduction conditions.[19a] In 2011,
The direct asymmetric α-amination of α-AA derivatives has so
Shibatomi and co-workers developed a practical one-pot di-
far received very little attention only, but in general, such an
halogenation protocol to access the α-Cl-α-F-β-AA derivatives
approach provides an interesting entry towards quaternary
26 from simple β-keto esters 21 (Scheme 6B).[19b] Hereby the
stereogenic centers containing two (orthogonal) nitrogen-
starting material 21 was first α-chlorinated, followed by an
based functional groups. In 2012, the groups of Guo and Zhou
asymmetric Cu-catalyzed α-fluorination with NFSI, giving the
showed that the addition of α-nitroesters 5 to diazocarboxylate
highly functionalized β-keto ester 25 (which could be trans-
19 can be rendered enantioselective by using the Cinchona-
ferred into 26 by established means).
derived organocatalyst C5 under cryogenic conditions
(Scheme 5).[18] To the best of our knowledge, this report repre-
sents the only highly enantioselective example to access
masked or protected α-AA derivatives with an additional α-N-
functional group. However, no comprehensive studies concern-
ing further manipulations of these interesting compounds were
reported so far.
Scheme 5. Asymmetric α-amination of α-nitroesters 5.
3. β-Amino Acid Derivatives Scheme 6. Asymmetric syntheses of α-halogenated β-AA starting from β-keto
esters.
Owing to the higher structural diversity of β-amino acids com-
pared to α-amino acids a much broader variety of complemen- Isoxazolinones 27 can be considered as versatile masked β-
tary asymmetric syntheses protocols to access α-heterofunc- AA derivatives as well and the asymmetric α-fluorination of
tionalized β-AA derivatives have been developed so far. These those compounds has been reported by the groups of Ma and
methods differ fundamentally, by either carrying out the heter- Wang (Scheme 7).[20] First, Ma and co-workers reported the chi-
ofunctionalization in the stereodefining step or by using al- ral thiourea C7-catalyzed addition of 27 to nitroalkenes 28 fol-
ready heterofunctionalized starting materials e.g. asymmetric lowed by α-fluorination (Scheme 7A).[20a] Two years later, Wang
C–C bond forming reactions. et al. developed a direct Cinchona dimer C8-catalyzed α-fluorin-
ation of α-substituted isoxazolinones 27 (Scheme 7B).[20b] Both
protocols allowed for high enantioselectivities for a broader
3.1. α-Halogen-β-AA Derivatives via Asymmetric
substrate scope, but unfortunately, to the best of our knowl-
Electrophilic α-Halogenations
edge, no further manipulations towards e.g. free β-amino acids
A variety of different approaches for electrophilic asymmetric were reported so far.
α-halogenation reactions using differently decorated β-AA de-
rivatives or precursors have been reported so far, and in the
following chapter some of the most versatile concepts shall be
discussed.
Asymmetric electrophilic α-halogenation reactions of β-keto
esters are very commonly investigated target transforma-
tions.[19] The hereby accessed α-halogenated 1,3-dicarbonyl
compounds can be utilized for a variety of further manipula-
tions, among them also conversions to the corresponding α-
halogenated β-AA derivatives (Scheme 6). As an impressive
early example in the field (2002), Sodeoka's group has shown
that the α-fluorinated β-keto esters 23 can be accessed with
high selectivities under asymmetric Pd-catalysis using NFSI as
the electrophilic F-transfer reagent (Scheme 6A).[19a] Com-
pounds 6 were then successfully converted into the α-F-β-AA
derivatives 24 via reduction to the alcohol, followed by a stereo-
specific Mitsunobu-type inversion. Hereby, the formation of ei- Scheme 7. Asymmetric α-fluorination of isoxazolinones 27.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 205 © 2020 The Authors. Published by Wiley-VCH GmbH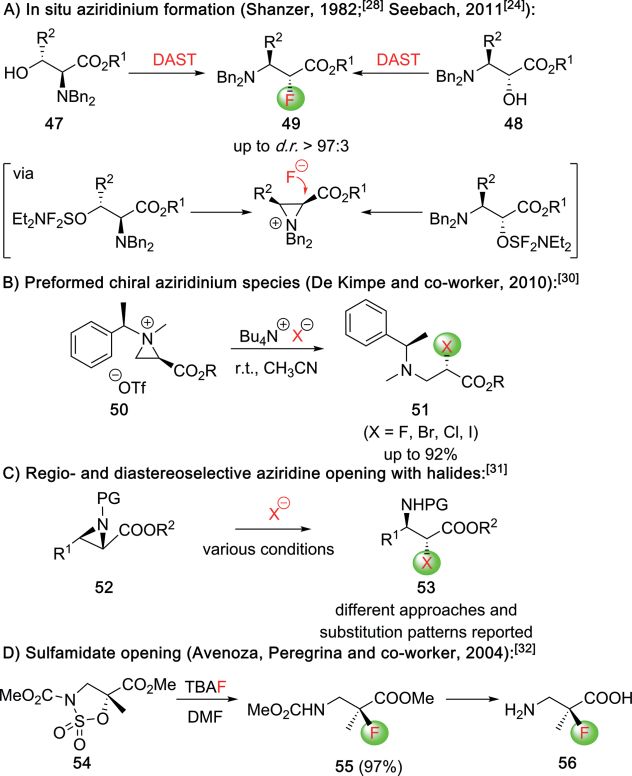
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
Cyanoacetates 30 can be considered as oxidized β-AA deriv-
atives as well, and these commonly employed nucleophiles
have been subjected to several α-fluorination attempts with
different catalysis strategies in the past (Scheme 8).[21] While
Shibata's group utilized the simple Cinchona derivative C9 in
combination with Selectfluor,[21a] Kim et al. reported methods
for the α-fluorination of 30 with NFSI either using the chiral
ammonium salt catalyst C10,[21b] or the Pd complex C11.[21c]
Unfortunately, and to the best of our knowledge, in neither of
these cases any reductions of the cyano group towards the
corresponding β-AA were described.
Scheme 9. Diastereoselective α-halogenation reactions of chiral β-AA precur-
sors.
Scheme 8. Asymmetric α-fluorination of cyanoacetates 30.
Besides the enantioselective electrophilic approaches relying as outlined in Scheme 10.[26,27] Here, two fundamentally differ-
on asymmetric catalysis modes discussed so far, also diastereo- ent approaches to control the absolute configuration were de-
selective α-halogenations of chiral (auxiliary containing) β-AA veloped. First, Duggan and co-workers showed that the use of
precursors emerged as promising strategies to access α-halo- the chiral Li-base 43 in combination with NFSI allows for the
genated-β-AA with high stereoselectivities (Scheme 9).[22–25] highly stereoselective synthesis of 44, which could then be con-
For example, around 20 years ago Tomasini and co-workers verted into the free α-F-β-AA 45 directly (Scheme 10A).[26]
showed that the LiHMDS-mediated α-halogenation of cyclic More, recently, Liu, Feng, and co-workers developed a chiral
(compounds 32) and acyclic (compounds 35) chiral β-AA deriv- Fe-catalyzed protocol for the aza-Michael addition of TMSN3
atives allows for the synthesis of α-Cl, α-Br, and α-I-derivatives with direct bromination of the α-position, giving the versatile
33 and 36 with high diastereoselectivities under operationally products 46 with excellent diastereo- and enantioselectivities
simple conditions (Scheme 9A, 9B).[22] These compounds then (Scheme 10B).[27]
served as valuable building blocks for further transformations,
like e.g. the synthesis of chiral aziridines from acyclic derivatives
34.[22a]
Abell and co-workers investigated the α-fluorination of chiral
auxiliary-based carboxylic acid derivatives like compounds 37,
which were α-fluorinated with high diastereoselectivities using
NFSI. The resulting products 38 could then be converted into
the corresponding α-alkylated α-F-β-AA 39 straightforwardly by
established means (Scheme 9C).[23] Alternatively, Seebach[24]
and Abell[25] also showed that the β-substituted chiral β-amino
esters 40 can be α-fluorinated with high diastereoselectivities
with NFSI and using LDA as the base (Scheme 4D). Accordingly,
all these reports shown in Scheme 9 clearly demonstrate the
potential and simplicity of diastereoselective approaches for
cases where the required chiral precursors are as easily available
as shown in these examples.
Alternatively, aza-Michael-initiated approaches of simple vin-
ylogous acceptor molecules 42 also allowed for highly stereo- Scheme 10. Asymmetric aza-Michael-initiated syntheses of α-halogenated-β-
selective syntheses of α-halogenated β-amino acid derivatives AA.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 206 © 2020 The Authors. Published by Wiley-VCH GmbH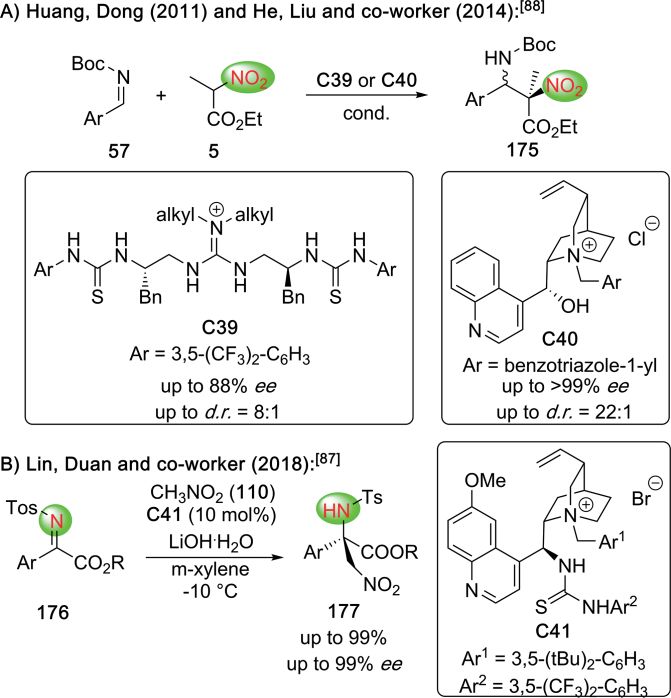
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
3.2. α-Halogen-β-AA Derivatives via Asymmetric halide species has also been explored to large extent, highlight-
Nucleophilic α-Halogenations ing the value of chiral aziridines for further manipulations
(Scheme 11C).[31] Conceptually similarly, chiral sulfamidates 54
Besides the introduction of the halogen via an electrophilic
show analogous reactivities and can undergo ring-opening to-
α-halogenation approach, also the addition of nucleophilic hal-
wards products 55 upon treatment with TBAF as a nucleophilic
ide-sources to appropriately substituted starting materials has
F-source as well (Scheme 11D).[32]
been successfully utilized to access enantioenriched α-halogen-
β-AA derivatives (Scheme 11).[28–32] In this context, the majority
of the reported methods rely on stereospecific addition reac- 3.3. α-Halogen-β-AA Derivatives via Asymmetric C–C-Bond
tions of the halide nucleophile to chiral, enantioenriched start- Formations
ing materials. Aziridines or (in situ formed) aziridinium species
are amongst the most versatile substrates for such approaches Besides incorporating the heteroatom via electrophilic or nu-
and in general, this concept is very well-established, with first cleophilic approaches on an already appropriately constituted
reports dating back to the 1980s when Shanzer's[28] and later β-AA-skeleton, asymmetric C–C-bond forming reactions, i.e.
on Seebach's[24] groups showed that the chiral amines 47 and Mannich-type reactions, are obviously the most versatile strate-
48 can both be converted into the α-F-β-AA derivatives 49 by gies for the stereoselective synthesis of α-heterofunctionalized-
using DAST. This reagent fulfills two functions, first, it activates β-AA. Given the power of asymmetric organo- and transition
the alcohol for in situ aziridinium formation and in addition it metal-catalysis to accomplish a multitude of asymmetric C–C-
serves as a nucleophilic F-source for the ring-opening again bond forming reactions, it comes as no surprise that such meth-
(Scheme 11A). The intermediate ring formation via this neigh- ods have also been heavily exploited to access chiral β-AA de-
boring group effect of the amine group also provides a ration- rivatives.[33–42]
ale for the observed stereochemistry in these reactions (reten- (Transition) Metal-catalyzed Mannich-type reactions of
tion of configuration starting from 48 and inversion from 47). α-halogenated enolate precursors represent a powerful way to
Besides in situ aziridinium formation, also preformed chiral access a variety of differently functionalized α-halogenated-β-
aziridinium species like compound 50 can be directly utilized AA (Scheme 12).[34–36] In 2010, Ishihara's group reported the
for such ring-opening reactions (Scheme 11B).[30] This approach addition of α-halo malonates 58 to imines 57 using a Mg-BINOL
allowed De Kimpe's group to access a variety of differently catalyst system (Scheme 12A).[34] A few years later, Shibasaki
α-halogenated esters 51 with perfect stereocontrol and in high and co-workers introduced a broadly applicable method by us-
yields.
Scheme 11. Stereospecific regioselective ring-opening reactions with nucleo-
philic halide sources.
Alternatively, the regioselective stereospecific ring-opening Scheme 12. Transition metal-catalyzed Mannich-type approaches for the syn-
of chiral aziridines 52 with a variety of different nucleophilic thesis of α-halogenated-β-AA.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 207 © 2020 The Authors. Published by Wiley-VCH GmbH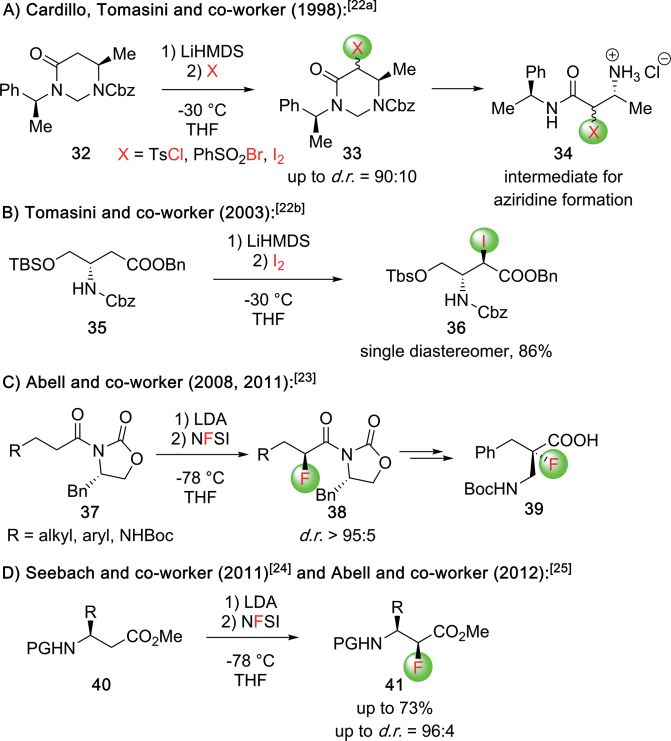
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
ing the α-halo amides 61 for Cu-catalyzed Mannich reactions, of α-F-carboxylic acids in β-lactam-forming reactions, as dem-
which yielded the amides 62 with very high levels of enantio- onstrated by Birman's group recently (Scheme 14).[41] Starting
and diastereoselectivities (Scheme 12B).[35] Very recently, the from simple starting materials 57 and 74, they developed an
same group also succeeded in engaging simple α-fluoro nitriles elegant protocol for the synthesis of the α-F-β-lactam 75 by
63 for Mannich reactions, giving the nitriles 64 which could means of an isothiourea C16-catalyzed lactamization. This β-
then be converted into the free α-F-β-AA 65 (Scheme 12C).[36] lactam then undergoes nucleophilic ring-opening reactions
Besides the above mentioned metal-catalyzed strategies, with various O-nucleophiles, resulting in a synthetically useful
also asymmetric organocatalytic Mannich-type approaches alternative as compared to the Mannich protocols shown in
have been used very successfully to access a variety of different Scheme 13.
α-halogenated-β-AA (Scheme 13).[37–40] In 2010, Jiang, Tan, and
co-workers succeeded in controlling the asymmetric addition
of the α-F-β-ketoamide 66 to aldimines 57 in the presence of
the chiral bicyclic guanidine catalyst C12 (Scheme 13A).[37] Al-
ternatively, Kim's group shortly after made use of the bifunc-
tional cyclohexanediamine-based thiourea catalyst C13, which
allowed them to achieve high levels of selectivities for the reac-
tion between the α-F-β-keto esters 68 and aldimines 57
(Scheme 13B).[38] In 2015, Zhao et al. investigated the Mannich
addition of α-Br-thioester 70 to aldimines 57 and found the
Cinchona alkaloid-based urea catalyst C14 being best suited for Scheme 14. Asymmetric synthesis of α-F-β-lactams.
this approach (Scheme 13C).[39] Expanding the applicability of
thioesters, Wennemers' group developed a highly selective pro- A conceptually different approach to access α-F-β-AA deriva-
tocol for the use of α-F-thiomalonates 72 for Mannich reactions tives by means of an asymmetric C–C-bond forming reaction
in the presence of low quantities of the squaramide catalyst was recently reported by Stoltz and co-workers (Scheme 15).[42]
C15 (Scheme 13D).[40] By carrying out a Pd-catalyzed asymmetric decarboxylative α-
allylation of allyl ester 77, the cyclic masked α-F-β-AA 78 was
obtained with high enantioselectivity. This compound could
then be converted into the free α-allyl-α-F-β-AA 79 subse-
quently, giving access to a β-amino acid substitution pattern
that is otherwise difficult to access with classical Mannich type
approaches.
Scheme 15. Decarboxylative allylation for the enantioselective synthesis of
α-F-β-AA.
3.4. α-OR- and α-SR-β-AA Derivatives via Asymmetric
Electrophilic or Nucleophilic Heterofunctionalizations
The asymmetric synthesis of α-chalcogenated β-amino acids
can be achieved by a variety of different strategies,[43–64] as
outlined in the following chapters.
With respect to the introduction of various SR-groups the
electrophilic α-chalcogenation of suited β-AA precursors is a
very powerful approach for this task.[43–45] In 2006, Davies and
co-workers introduced a highly selective aza-Michael initiated
Scheme 13. Organocatalytic Mannich-type approaches for the synthesis of α- protocol for the synthesis of the α-sulfanylated β-AA 82
halogenated-β-AA. (Scheme 16A).[43] Hereby they made use of the addition of the
chiral Li-amide 43 to Michael acceptor 42 to form the chiral
An alternative strategy to construct the α-F-β-AA skeleton enolate 80 in situ, which can then be trapped with a suited
via an asymmetric C–C bond formation relies on the direct use electrophilic S-transfer reagent giving product 81. The chiral
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 208 © 2020 The Authors. Published by Wiley-VCH GmbH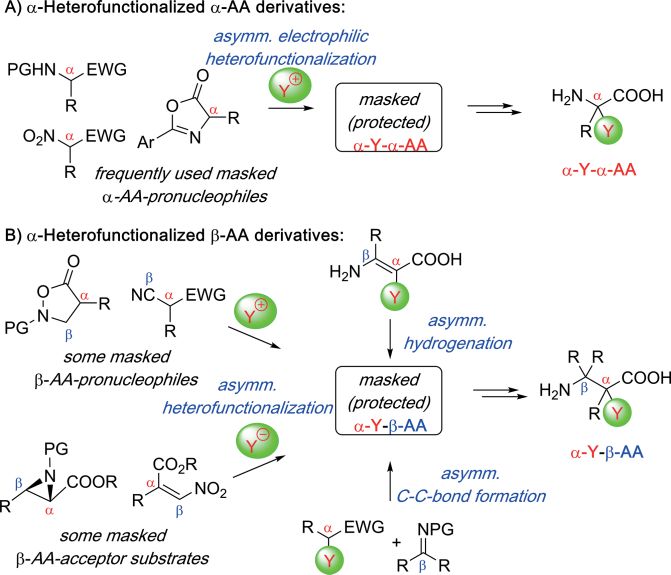
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
auxiliary can then be removed in a further step to access the containing products 91 could then be converted into the free
amino acid 82 with high enantiopurity. A versatile strategy to α-SR-β2,2-AA 92 by established methods (Scheme 17).
access β2,2-AA (β-AA with no additional substituents in the β-
position) was recently introduced by Brière's group, who re-
ported the use of isoxazolidin-5-ones 83 as easily available
masked β2,2-AA precursors that undergo enantioselective
α-sulfanylation reactions (Scheme 16B).[44] Key to success for
achieving high levels of enantioselectivities was the use of the
commercially available Maruoka phase-transfer catalyst C17
Scheme 17. Synthesis of α-SR-β2,2-AA via a nucleophilic stereospecific substi-
and the hereby accessed products 84 could easily be converted tution reaction.
into the free amino acids 85 upon reductive N–O-cleavage.
Building on this seminal contribution, the groups of Della Sala A conceptually different nucleophilic thiolation approach
and Aleman[45a] and Cahard and Waser[45b] simultaneously re- was developed by Xiao's group in 2009. Relying on chiral thio-
ported the α-trifluoromethylthiolation of 83 under rather simi- urea C19 organocatalysis, they succeeded in carrying out the
lar phase-transfer catalytic conditions (Scheme 16C and highly enantioselective thia-Michael addition of thiols 93 to the
Scheme 16D). Again the heterocyclic products 86 could be con- β-nitro acrylates 94 with as little as 0.3 mol-% of the catalyst
verted in the free α-SCF3-β-AA 87 or also incorporated in the (Scheme 18).[47] The resulting products 95 could then be re-
α-AA-β-AA dipeptide 89 directly,[45b] highlighting the general duced and deprotected giving the α-sulfanylated β-AA 96 in an
potential of this isoxazolidin-5-one heterocyclic platform. efficient manner.
Scheme 18. Asymmetric thia-Michael strategy to access α-SR-β2,2-AA.
An efficient strategy for the synthesis of α-hydroxylated carb-
onyl derivatives is the nucleophilic ring-opening of glycidic es-
ters 97. By using amine nucleophiles, direct access to α-
hydroxy-β-AA derivatives is possible.[49,50] Most commonly, this
approach has been carried out in a diastereoselective manner,
by using enantioenriched epoxides 97.[49] In addition, Kureshy
et al. recently also showed that racemic epoxides 97 can be
resolved very efficiently by the addition of simple aniline deriva-
tives 98 in the presence of the chiral Cr-catalyst C20
(Scheme 19).[50]
Scheme 19. Kinetic resolution of epoxides 97 to access α-OH-β-AA deriva-
Scheme 16. Approaches for the asymmetric synthesis of α-SR-β-AA via elec- tives.
trophilic strategies.
Besides electrophilic α-sulfanylation approaches the addition
3.5. α-OR-β-AA Derivatives via Asymmetric C–C-Bond
of nucleophilic S-reagents to appropriately substituted β-AA
Formations
precursors has been successfully carried out to achieve the syn-
thesis of enantioenriched α-SR-β-AA derivatives.[46–48] As an Asymmetric Mannich-type approaches using α-oxygenated
early example, Avenoza, Peregrina, and co-workers showed that enolate precursors have been successfully used for the synthe-
thiols or thiolates can be added in a stereospecific manner to sis of various α-oxygenated β-amino acid derivatives over the
the chiral sulfamidates 90. The resulting Weinreb amide- last years.[51–54] Like it was discussed above for α-halogenated
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 209 © 2020 The Authors. Published by Wiley-VCH GmbH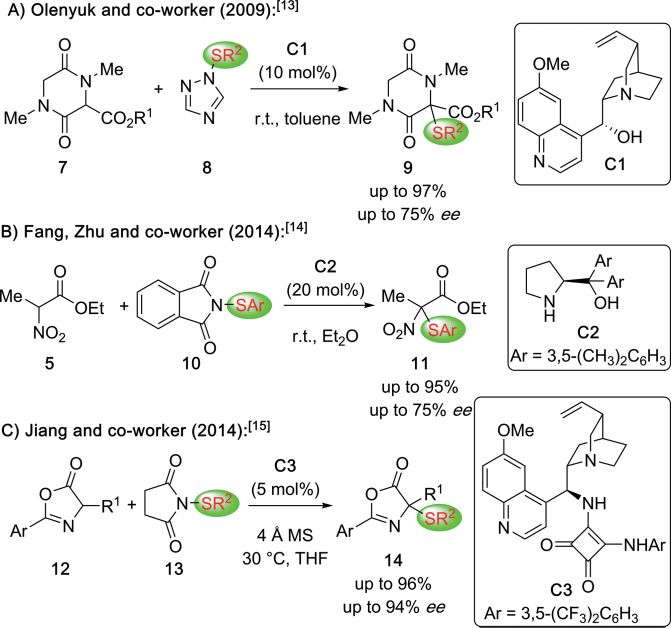
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
β-AA, organocatalyzed,[51,52] and (transition) metal-cata-
lyzed[53,54] strategies have been reported, all of them having
their specific advantages and application scopes.
In 2008, Córdova and co-workers reported the enamine-cata-
lyzed Mannich addition of aldehydes 100 to various imines 57
(Scheme 20A).[51] The resulting α-oxygenated-β-amino alde-
hydes 101 could then be oxidized to the amino acids 102
straightforwardly. Very recently, Takemoto's group developed a
very selective and highly divergent method for the addition
of glyoxylate cyanohydrin 103 to imines 57 (Scheme 20B).[52]
Depending on the modifications of the catalyst C22, either the
syn- or the anti-diastereomer of products 104 could be ac-
cessed with excellent levels of enantio- and diastereoselectivi-
ties, resulting in a powerful catalysis concept for the divergent
synthesis of highly functionalized small molecules.
Scheme 21. Metal-catalyzed Mannich approaches for the synthesis of α-oxy-
genated β-AA derivatives.
ester 113 using a chiral La-catalyst (Scheme 22B).[56] Again, the
nitro group could be reduced then and incorporation of the
product in the dipeptide Bestatin (117) was successfully dem-
onstrated. Recently, Shibasaki's group reported a broadly appli-
cable method for the addition of a variety of different nitro-
alkanes 111 to α-keto esters 112 giving the α-hydroxy-β-nitro-
esters 118 with excellent levels of enantio- and diastereoselec-
tivities (Scheme 22C).[57] Again, reduction of the nitro group
Scheme 20. Organocatalyzed Mannich approaches for the synthesis of was successfully demonstrated.
α-oxygenated β-AA derivatives.
Over the years, Shibasaki's group contributed like maybe no
other to the development of robust and highly selective metal-
catalyzed Mannich type approaches using diversely functional-
ized enolate precursors (see also Scheme 12 for the already
discussed synthesis of α-halogenated-β-AA via Mannich reac-
tions).[35,36,54] With respect to the synthesis of α-OH-β-AA deriv-
atives, they succeeded in controlling the addition of the free-
OH-containing amide 105 to various imines 57 by using a chiral
indium catalyst system (Scheme 21A).[54a] The resulting pyrrole-
amides 106 could then be transferred into the esters 107
straightforwardly. More recently, they also expanded their chiral
Cu-catalyzed Mannich protocol (compare with Scheme 12B) to-
wards amides 108 (Scheme 21B),[54b] giving the masked α-oxy-
genated β-AA derivatives 109 with very high levels of enantio-
selectivity and almost perfect control of diastereoselectivity.
Besides Mannich-type reactions, also asymmetric nitro-aldol
additions (Henry reactions) of various nitroalkanes 110 or 111
to α-oxo-esters 112 or 113 have been successfully used to ob-
tain chiral α-hydroxylated β-AA derivatives (Scheme 22).[55–57]
In 2002, Jørgensen's group developed the Cu-catalyzed Henry
Scheme 22. Asymmetric Henry-type reactions for the syntheses of α-OH-β-
reaction of nitromethane (110) with different α-keto esters 112 AA derivatives.
(Scheme 22A).[55] The resulting nitro esters 114 were obtained
with high selectivities and could easily be reduced to the free Next to the classical Henry-type addition reactions of nitro-
β-amino acid esters 115 then. Some years later, Barua and co- alkanes to α-oxo-esters shown in Scheme 22, also the enantio-
workers reported the addition of nitroalkane 111 to the α-oxo- selective addition of hydrazone derivatives like compound 119
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 210 © 2020 The Authors. Published by Wiley-VCH GmbH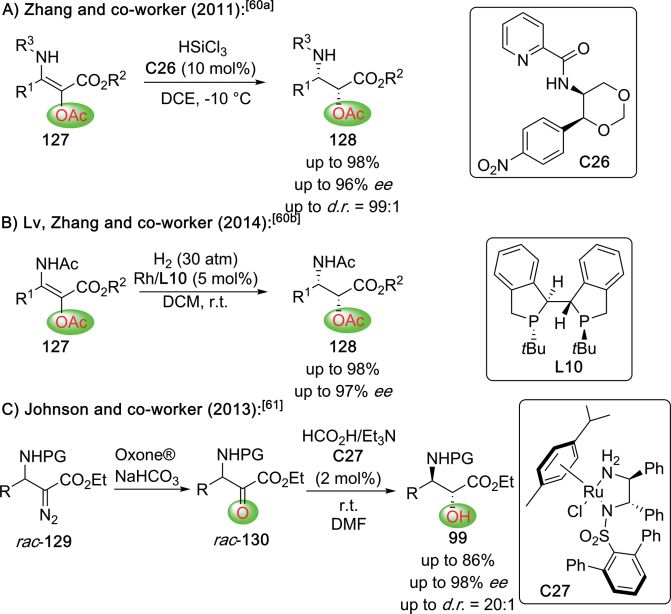
EurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
to α-keto esters 112 was reported for the successful enantio- 3.6. α-OR-β-AA Derivatives via Reductive Approaches
selective synthesis of α-OH-β-AA derivatives (Scheme 23). By
utilizing the chiral H-bonding catalysts C23 or C24, the groups Besides the asymmetric C-X or C–C bond forming reactions dis-
of Fernandez and Lassaletta succeeded in carrying out the addi- cussed so far, asymmetric reductive approaches of already ap-
tion of 119 to 112 with excellent enantioselectivities and sub- propriately constituted sp2-hybridized β-AA precursors repre-
sequent transformations then gave access to the corresponding sent another powerful way to access chiral α-oxygenated-β-AA
α-OH-β-AA derivatives 115 in an elegant manner.[58] derivatives (Scheme 25).[60,61] Around 10 years ago, Zhang's
group introduced a highly selective protocol for the asymmetric
hydrosilylation of the α-O-acylated β-aminoesters 127 by using
the chiral Lewis base catalyst C26 (Scheme 25A).[60a] A few years
later, Zhang, Lv and their co-workers then showed that the
same substrates can also be hydrogenated with excellent
enantio- and diastereoselectivities employing asymmetric
Rh-catalysis (ligand L10; Scheme 25B).[60b] An alternative reduc-
tive approach which directly yielded the free-OH-containing
β-amino ester 99 was reported by Johnson's group in 2013
(Scheme 25C).[61] Hereby, they started from the racemic
α-diazoesters 129, which were first converted in the α-oxo-
esters 130, followed by a Ru complex C27-catalyzed transfer
hydrogenation.
Scheme 23. Asymmetric addition of hydrazone derivatives 119 to α-keto
esters 112.
An alternative, conceptually remarkable, approach to access
the α-oxygenated-β-AA skeleton in an asymmetric fashion was
developed by Johnson and co-workers in 2004.[59] Starting from
acylsilanes 121 and cyanoformates 122, they succeeded in ac-
cessing the silyl ethers 123 via an Al(salen) complex C25-cata-
lyzed cyanation – 1,2-Brook rearrangement – C-acylation se-
quence as outlined in Scheme 24. Hereby, the catalyst first
forms a nucleophilic CN-species with 122, which gives 125
upon addition to starting material 121. This intermediate then
undergoes a 1,2-Brook rearrangement towards the silyloxy
nitrile anion 126, which finally performs the asymmetric C-
acylation giving product 123. A final reduction of the nitrile
group was also successfully reported, thus resulting in an effi-
cient and, compared to the other approaches described before,
complementary synthesis strategy for the asymmetric forma-
tion of
α-oxygenated-β-AA 124 from simple starting materials. Scheme 25. Reductive approaches for the synthesis of α-oxygenated-β-AA
derivatives.
3.7. α-OR- and α-SR-β-AA Derivatives by Miscellaneous
Approaches
The asymmetric aminohydroxylation of α,β-unsaturated esters
provides a direct entry to α-hydroxy-β-AA derivatives, as dem-
onstrated by McLeod and co-workers in 2008 for esters 131
already (Scheme 26).[62] Noteworthy, in that specific study the
major target was on the utilization of products 134 to access
various 3- and 4-amino sugars, rather than on β-amino acid
chemistry. Nevertheless, this example clearly underscores the
Scheme 24. Asymmetric synthesis of α-oxygenated-β-AA derivatives starting potential of asymmetric aminohydroxylation reactions for
from acylsilanes 121 and cyanoformates 122. α-OH-β-AA syntheses.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 211 © 2020 The Authors. Published by Wiley-VCH GmbHEurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
covering this important compound class have been reported.[7]
Given these detailed previous overviews, we will only try to
give some general outline of the most commonly applied meth-
ods to synthesize these compounds in the following chapters,
with a special focus on those (more recent) reports that have
not been discussed in the existing reviews.[7]
The asymmetric synthesis of α,β-diamino acids via asymmet-
ric α-amination reactions of suited β-AA-based building blocks
has received relatively little attention so far,[7,65–67] with interest-
ing organocatalytic approaches by the groups of Greck[65] and
Scheme 26. Asymmetric aminohydroxylation for the synthesis of α-OH-β-AA
Brière[66] standing out (Scheme 29). In 2011, Greck′s research
derivatives.
group reported a stereoselective one pot Mannich-reaction/
α-amination protocol to assemble the parent α,β-diamino carb-
Another interesting methodology for the construction onyl skeleton (Scheme 29A).[65] Carrying out the Mannich reac-
of β-AA derivatives was recently reported by Shao, He, and co- tion of acetaldehyde (141) with imine 57 under enamine cataly-
workers (Scheme 27).[63] Starting from the α-SR-acrylate 135 sis (using catalyst C29) first, followed by direct α-amination with
and the isocyanide 136, they carried out an asymmetric reagent 19 gave product 142 with excellent stereoselectivity.
Ag-catalyzed (3+2) cyclization reaction, which, after reductive Carefully optimized downstream chemistry then gave access to
workup, gave the α-SR-β-proline derivatives 137 with excellent the syn-configured α,β-diamino acid 143.[65] Very recently,
enantio- and good diastereoselectivities.
Brière and co-workers developed an asymmetric α-amination
reaction of α-substituted isoxazolidin-5-ones 83 with diazo-
carboxylate 19 (Scheme 29B).[66] The reaction proceeded
smoothly by using the Maruoka catalyst C17 and reductive
N–O cleavage of the primary reaction product 144 then yielded
the β2,2-amino acid derivative 145 with satisfying enantioselec-
tivity.
Scheme 27. Asymmetric [3+2]-cyclization reaction for the synthesis of α-SR-
β-proline derivatives.
In addition to using α-keto esters as acceptors as outlined
above (see Scheme 22 and Scheme 23), enolizable derivatives
like compounds 138 can also be utilized to access α-OH-β-AA
derivatives via asymmetric α-amination approaches, as reported
by Jørgensen's group in 2002 already.[64] By controlling the α-
amination of 138 with diazocarboxylate 19 by using the chiral
Cu-catalyst C28, they achieved high levels of enantioselectivi-
ties for the formation of products 139 (Scheme 28). The keto-
functionality could then be reduced with high diastereoselecti-
vity, providing a direct route to the α-OH-β-AA derivative 140.
Scheme 29. Asymmetric α-amination methods of β-AA precursors.
Besides these asymmetric electrophilic α-amination reac-
tions of suited β-AA precursors, also the asymmetric α-phos-
Scheme 28. Asymmetric amination of α-keto esters 138. phorylation of cyanoacetates 30 with diarylphosphine chlorides
146 was successfully carried out en route to α-P-β-AA deriva-
tives, as reported by Jørgensen and co-workers (Scheme 30).[68]
By using the dimeric Cinchona alkaloid catalyst C30, they intro-
3.8. α-NR2- and α-PR2-β-AA Derivatives via Asymmetric
duced a protocol for the direct formation of products 147,
Electrophilic α-Heterofunctionalizations
which could then be transferred into the protected α-phos-
The synthesis and chemistry of α,β-diamino acids have at- phorylated-β-AA 148 directly by means of established func-
tracted much attention in the past and comprehensive reviews tional group interconversion reactions.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 212 © 2020 The Authors. Published by Wiley-VCH GmbHEurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
acidic hydrolysis, the obtained anti-configured β-amino-α-azido
products were then converted straightforwardly to the corre-
sponding diamino acids 151 by reduction of the azido group
under Pd-catalysis. On the other hand, syn-diamino esters 153
were synthesized successfully by He's and Zhang′s research
groups very recently (Scheme 31B).[71] By the reaction of N-
phosphinoylimines 57 with glycine aldimines 152 under bifunc-
tional Cu-urea catalysis the desired syn-products 153 were ob-
tained with very high levels of diastereo- and enantioselectivity,
thus providing a powerful alternative to Shibasaki's anti-selec-
tive protocol.
Scheme 30. Asymmetric synthesis of α-P-β-AA derivatives 148. With respect to the utilization of ketimines, Xie and co-work-
ers developed a highly diastereo- and enantioselective syn-
thetic route to access Mannich products 156 by reacting imine
3.9. α,β-Diamino Acid Derivatives via Asymmetric
154 with glycine Schiff base 155 using Cu(OAc)2 in combination
C–C-Bond Formations
with ligand L15 (Scheme 32A).[72] This methodology gave ac-
In contrast to the rarely investigated above mentioned α-amin- cess to the highly functionalized α,β-diamino acid derivatives
ation of β-AA precursors, the asymmetric construction of the 156 with excellent selectivities, and further protecting group
α,β-diamino acid skeleton via Mannich type reactions has been hydrolysis was demonstrated as well. Very recently, the groups
explored in much detail. Given the variety of different catalysis of Yang and Deng[73] and Wu[74] independently reported the
protocols to achieve asymmetric Mannich type reactions,[69] this addition of glycine Schiff bases 155 to isatin-derived ketimine
methodology is without doubt one of the most versatile strate- derivatives 157 under Cu(I)-catalysis (Scheme 31B). Noteworthy,
gies to access the α,β-diamino acid scaffold from simple start- the diastereoselectivity of this transformation could be effi-
ing materials.[7,70–85] ciently controlled by nature of the employed ligand (and condi-
Cu-catalyzed Mannich reactions between α-amino-function- tions), thus resulting in a highly divergent catalysis concept to
alized carbonyl derivatives and either aldimines or ketimines access these densely functionalized target molecules.
have been extensively explored by several groups over the
course of the last decades (Scheme 30 and Scheme 31).[7,70–74]
In analogy to their elegant protocols to access α-halogenated-
and α-oxygenated-β-AA derivatives via Mannich approaches
(compare with Scheme 12 and Scheme 21), Shibasaki′s group
developed a highly stereoselective anti-Mannich strategy to ac-
cess α,β-diamino acid derivatives as well. Using simple Cu-sour-
ces in combination with ligand L13 they were able to access
amides 150 by reacting aldimines 57 with α-azido-azaind-
oline 149 with excellent selectivities (Scheme 31A).[70] After
Scheme 32. Cu-catalyzed asymmetric Mannich reactions with ketimines.
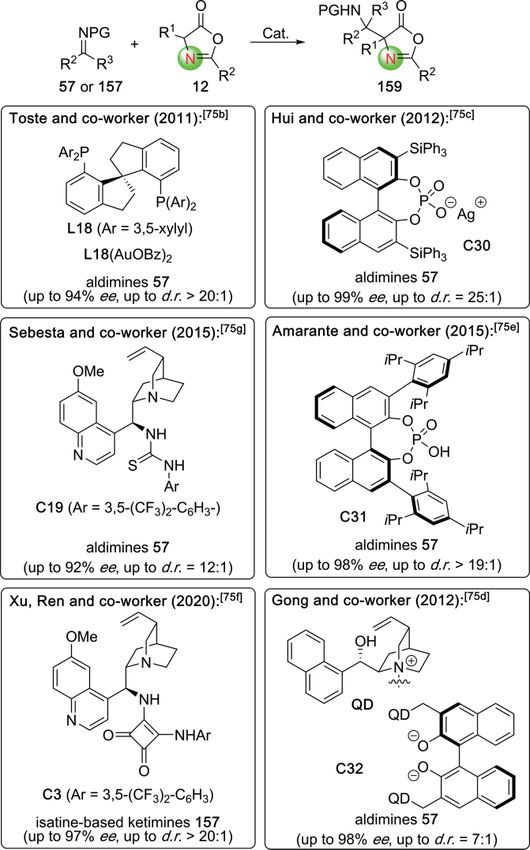
Azlactones 12 are amongst the most frequently employed
masked α-amino acid starting materials for asymmetric synthe-
ses (see Scheme 3 and Scheme 4 for already discussed exam-
ples). Accordingly, it comes as no surprise that they have also
been heavily utilized for asymmetric Mannich-type approaches
to access chiral α,β-diamino acid derivatives (Scheme 33).[7,75–77]
A powerful asymmetric procedure was published in 2011 by
Toste′s group, who applied the L18(AuOBz)2 complex to control
the asymmetric reaction between azlactones 12 and aldimines
Scheme 31. Cu-catalyzed asymmetric Mannich reactions with aldimines. 57.[75b] Shortly after, Hui and co-workers utilized the chiral
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 213 © 2020 The Authors. Published by Wiley-VCH GmbHEurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
phosphate-based Ag-catalyst C30 to carry out these Mannich nucleophilic ring-opening reactions, were reported as well, illus-
reactions with very high enantioselectivities as well.[75c] Besides trating the power of these methods to access a multitude of
such transition metal-catalyzed strategies, the use of chiral or- chiral α,β-diamino acid derivatives from simple starting materi-
ganocatalysts was found to be rather promising for these target als with high levels of stereocontrol.
transformations either. As an early example, therefore, Gong In 2016, Terada and co-workers investigated the asymmetric
and co-workers reported the successful use of the chiral bis- addition of azlactones 12 and thiazolones 161 to the acetalde-
betaine catalyst C32 in 2012 already.[75d] Switching to an alter- hyde-based enamide 160 under chiral phosphoric acid catalysis
native activation mode, Amarante's group utilized the chiral (enamide 160 tautomerizes to the corresponding imine under
phosphoric acid C31 as a versatile catalyst for these reactions acidic conditions).[77] Interestingly, while the classical azlactones
in 2015 then.[75e] At the same time, Sebesta's group also found 12 performed with very low selectivities only, their sulfur-
the Cinchona alkaloid-based thiourea C19 being well-suited for containing analogs 161 could be employed with very high
Mannich additions between 12 and aldimines 57,[75g] thus enantio- and diastereoselectivities when using the chiral
demonstrating the rather general potential of different organo- phosphoric acid catalyst C33, giving access to the masked α,β-
catalytic strategies to control these frequently investigated tar- diamino acids 162 with excellent selectivities (Scheme 34).[77]
get reactions. Finally, very recently this assembly strategy was
also successfully expanded to ketimines as acceptors. By using
the chiral bifunctional squaramide C3 as an organocatalyst Xu,
Ren, and their research groups succeeded in utilizing isatine-
based ketimines 157 in a highly selective Mannich reaction
with azlactones 12.[75f ] In most of these reports summarized in
Scheme 33 further manipulations of the products 159 like, e.g.
Scheme 34. Asymmetric phosphoric acid-catalyzed addition of thiazolones
161 to enamides 160.
Besides the use of azlactones 12 as masked α-amino acid
starting materials for α,β-diamino acid syntheses, the glycine
Schiff bases 155 have been used very extensively for asymmet-
ric Mannich reactions, i.e. in combination with asymmetric ion-
pairing catalysis.[78,79] Most of the seminal reports in this field
have been summarized in previous reviews on α,β-diamino
acids[7] and will therefore not be covered herein anymore. How-
ever, one more recent report that really underscores the poten-
tial of this non-covalent organocatalytic activation strategy was
reported by Maruoka's group in 2015 (Scheme 35).[79] By start-
ing from the stable aminals 163, they developed a highly effi-
cient protocol to generate the corresponding N-Boc aldimines
in situ under basic conditions, thus avoiding the preformation
of these usually sensitive imines. The in situ formed imines then
reacted with Schiff bases 155 with very high diastereo- and
enantioselectivities in the presence of the chiral ammonium salt
ion-pairing catalyst C34, giving the α,β-diamino ester 164 upon
hydrolysis of the imine group then.
Scheme 35. Asymmetric ammonium salt-catalyzed Mannich reaction using
aminals 163 as starting materials.
An interesting direct Mannich approach towards orthogo-
nally N-protected α,β-diamino esters was developed by Baud-
oux, Rouden, and co-workers in 2017 (Scheme 36).[80] By using
Scheme 33. Asymmetric azlactone-based Mannich reactions with aldimines the α-amido malonic acid half ester 165 in a decarboxylative
and ketimines. Mannich reaction with aldimine 57, they were able to directly
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 214 © 2020 The Authors. Published by Wiley-VCH GmbHEurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
access the diamino ester 166 in a racemic manner first.[80a] In
addition, the newly designed Cinchona alkaloid-based thio-
amide catalyst C35 gave some promising levels of enantioselec-
tivity for both diastereomers, demonstrating the general poten-
tial of this interesting concept.
Scheme 36. Decarboxylative organocatalytic Mannich approach for the syn-
theses of α,β-diamino esters 166.
Scheme 38. Diazoester 170-based asymmetric Mannich approaches.
A very inspiring biomimetic concept demonstrating the di-
rect use of free amine-containing α-aminoesters 167 for Man- Besides Mannich approaches of appropriately decorated α-
nich reactions was recently reported by the groups of Yuan and amino acid derivatives as outlined so far, asymmetric aza-Henry
Zhao (Scheme 37).[81] type reactions utilizing different nitroalkanes have been suc-
cessfully applied to access the α,β-diamino acid scaffold as
well.[7,86–88] Retrosynthetically, it can be differentiated between
two complementary strategies. First, α-nitroesters like com-
pound 5 can be added to aldimines 57 in the presence of a
chiral (organo)-catalyst, as reported by the groups of Huang
and Dong[88a] and He, Liu, and co-workers (Scheme 39A).[88b] In
both cases, chiral ion-pairing catalysts were successfully em-
ployed, either the bis-thiourea-containing guanidinium salt
C39, or the Cinchona alkaloid-based quaternary ammonium
salt C40. On the other hand, a simple nitroalkane, i.e. nitro-
methane (110), can be added to an α-imino ester like com-
pound 176 to access the target α,β-diamino ester skeleton as
well (Scheme 39B).[87] As demonstrated by Lin and Duan re-
Scheme 37. Direct Mannich addition of free amine-containing esters 167.
By utilizing the chiral pyridiniumcarbaldehyde C36 as a cata-
lyst, they succeeded in developing a protocol for the direct
highly stereoselective addition of 167 to 57 with very low cata-
lyst loadings. The chiral catalyst hereby forms a Schiff base with
167 under the reaction conditions, mimicking the behavior of
Nature's transamination and amino acid decarboxylation co-
factor pyridoxal phosphate.[81] This chiral enolate species then
adds to the imine 57 with excellent face-differentiation, fol-
lowed by hydrolysis of the catalyst 36 again.
A versatile strategy for the synthesis of α,β-diamino esters
utilizing α-diazoesters 170 as starting materials were intro-
duced by Hu's group around 10 years ago (Scheme 38).[82,83]
By using a catalyst system consisting of Rh2(OAc)2 and a chiral
phosphoric acid (C37 or C38) together with tartaric acid as a
co-catalyst, the products 172 could be accessed directly from
diazoester 170, carbamate 171, and imines 57. Hereby the Rh-
carbenoid species 173 is formed first, which is trapped by the
addition of 171 resulting in the ylide-type intermediate 174.
The latter then undergoes the chiral phosphoric acid controlled
Mannich reaction with 57. Noteworthy, the diastereoselectivity
of the reaction can be controlled very efficiently by nature of
the catalyst. While C37 favors the syn-product, C38 allows for Scheme 39. Asymmetric aza-Henry type reactions to access the α,β-diamino
the opposite anti-diastereoselectivity (Scheme 38).[82,83] acid motive.
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 215 © 2020 The Authors. Published by Wiley-VCH GmbHEurJOC Minireview
European Journal of Organic Chemistry doi.org/10.1002/ejoc.202001077
cently, this reaction can be controlled efficiently by using the
bifunctional ammonium salt C41 as a catalyst, giving products
177 with excellent enantioselectivities.
3.10. α-NR2-β-AA Derivatives Starting from Alkenes
As outlined in Scheme 25 for the synthesis of α-oxygenated-β-
AA derivatives, the stereoselective reduction of appropriately
substituted α,β-unsaturated carbonyl compounds is a powerful
strategy to access chiral β-AA surrogates. In analogy to those
already illustrated approaches, also the stereoselective reduc-
tion of α,β-diamino-α,β-unsaturated carboxylic acid derivatives
can be successfully carried out to access enantioenriched α,β-
Scheme 41. Asymmetric aza-Michael additions to chiral auxiliary-containing
diamino acids.[89,90] For example, compounds 178 can be con- acceptors.
verted into the orthogonally N-protected esters 179 with very
high enantioselectivities and good diastereoselectivities by em-
ploying a C42-catalyzed hydrosilylation, as developed by
Zhang's group in 2013 (Scheme 40).[90]
Scheme 42. Asymmetric phosphoric acid-catalyzed formal (3+2)-cyclization.
Scheme 40. Asymmetric hydrosilyation of α,β-unsaturated esters 178. 4. Synopsis
Besides carrying out asymmetric reductions of α,β-diamino As outlined in this short review, a broad variety of different
alkenes as shown in Scheme 40, another synthetically usefully asymmetric synthesis and catalysis concepts to access poten-
alkene-based strategy towards α,β-diamino acids are aza- tially biologically active α-heterofunctionalized α- and β-amino
Michael-initiated approaches (Scheme 41 and Scheme 42).[91–94] acid derivatives have been introduced over the last decades.
Several complementary, either catalyst- or chiral auxiliary- While the most commonly employed approaches for α-amino
controlled asymmetric protocols have been reported to achieve acid derivatives obviously rely on electrophilic α-heterofunc-
this transformation. One of the most commonly employed strat- tionalization reactions of suited (masked) α-amino acids, a
egies is to carry out the addition of suited N-nucleophiles to much broader diversity of conceptually different strategies to
chiral Ni complexes 180, as demonstrated by numerous re- access β-amino acids are available. Here not only asymmetric
search groups over the last years (Scheme 41A).[92] These reac- electrophilic α-heterofunctionalizations, but also asymmetric
tions usually proceed with high diastereoselectivities and the C–C-bond forming reactions, i.e. Mannich type reactions have
resulting products 181 can easily be hydrolyzed to the free been used very successfully. In addition, it should be empha-
amino acids 182 then, thus resulting in a rather broadly applica- sized that not only asymmetric catalysis approaches, but some-
ble and general synthesis strategy. Very recently, Navo, Pere- times also chiral auxiliary-controlled methods have been em-
grina, and co-workers then reported another very useful auxil- ployed with excellent selectivities, thus providing a comple-
iary approach by adding secondary amines to the novel chiral mentary approach, i.e. where catalytic methods are still limited.
acceptor molecules 183, which resulted in the formation of sin- All these methods have their obvious benefits but often also
gle diastereomers of products 184, which again could easily be limitations and it is clear that with respect to potential larger
hydrolyzed to obtain the targets 182 with more or less perfect scale syntheses the single methods have to be evaluated with
enantiopurity (Scheme 41B).[93] respect to their potential and limitations, e.g. with respect to
In addition to these very powerful and often used auxiliary costs and atom efficiency, to mention two relevant points of
methods, also catalytic asymmetric approaches have been re- consideration only. Thus, by looking at the sometimes high
ported. In 2016, Takemoto et al. introduced a highly selective loadings of complicated and/or expensive catalysts or the costs
chiral phosphoric acid C33-catalyzed (3+2)-cyclization reaction and the difficulties in removing and recovering the chiral auxil-
between the vinylogous azlactones 185 and hydroxylamides iaries that are necessary it is clear that, despite all the progress
186 (Scheme 42).[94] The hereby obtained products 187 can that was made in this field, further developments are required
then easily undergo nucleophilic ring-opening, giving again ac- to really make the methods interesting for industry-related ap-
cess to the parent α,β-diamino acid scaffold, as exemplified for proaches as well. In addition, it becomes obvious that some
compounds 188 again. classes of α-heterofunctionalized amino acids have still been
Eur. J. Org. Chem. 2021, 202–219 www.eurjoc.org 216 © 2020 The Authors. Published by Wiley-VCH GmbHYou can also read

















































