Sequencing, assembly and annotation of the whole-insect genome of Lymantria dispar dispar, the European gypsy moth - Oxford Academic Journals
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
2
G3, 2021, 11(8), jkab150
DOI: 10.1093/g3journal/jkab150
Advance Access Publication Date: 30 April 2021
Genome Report
Sequencing, assembly and annotation of the whole-insect
genome of Lymantria dispar dispar, the European gypsy
moth
Michael E. Sparks ,1,* Francois Olivier Hebert,2 J. Spencer Johnston,3 Richard C. Hamelin,2,4 Michel Cusson,2,5
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
Roger C. Levesque and Dawn E. Gundersen-Rindal1
2
1
USDA-ARS Invasive Insect Biocontrol and Behavior Laboratory, Beltsville, MD 20705, USA,
2
Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Quebec City, QC, G1V 0A6, Canada,
3
Department of Entomology, Texas A&M University, College Station, TX 77843, USA,
4
Department of Forest and Conservation Sciences, Faculty of Forestry, University of British Columbia, Vancouver, BC V6T 1Z4, Canada, and
5
Laurentian Forestry Centre, Canadian Forest Service, Natural Resources Canada, Quebec City, QC G1V 4C7, Canada
*Corresponding author: michael.sparks2@usda.gov
Abstract
The European gypsy moth, Lymantria dispar dispar (LDD), is an invasive insect and a threat to urban trees, forests and forest-related indus-
tries in North America. For use as a comparator with a previously published genome based on the LD652 pupal ovary-derived cell line, as
well as whole-insect genome sequences obtained from the Asian gypsy moth subspecies L. dispar asiatica and L. dispar japonica, the
whole-insect LDD genome was sequenced, assembled and annotated. The resulting assembly was 998 Mb in size, with a contig N50 of
662 Kb and a GC content of 38.8%. Long interspersed nuclear elements constitute 25.4% of the whole-insect genome, and a total of
11,901 genes predicted by automated gene finding encoded proteins exhibiting homology with reference sequences in the NCBI NR
and/or UniProtKB databases at the most stringent similarity cutoff level (i.e., the gold tier). These results will be especially useful in develop-
ing a better understanding of the biology and population genetics of L. dispar and the genetic features underlying Lepidoptera in general.
Keywords: whole-genome sequencing; PacBio long-read assembly; Illumina short-read polishing; automated gene finding; Lepidoptera;
European gypsy moth; gypsy moth genomics
Introduction earlier mitogenome-based research findings (Djoumad et al. 2017).
These resources will contribute to an improved understanding of
Feeding on over 300 species of woody plants, the European gypsy
lepidopteran genetics in general (Triant et al. 2018).
moth, Lymantria dispar dispar (LDD), poses significant economic
and ecological threats to urban trees and forests of North
America, particularly with respect to forest-related industries Materials and methods
(Leonard 1981). Having a representative genome sequence for this
The European gypsy moth genome size was estimated using previ-
forest pest is important to facilitate various aspects of biosurveil-
ously described methods (Johnston et al. 2019) applied to LDD New
lance, including discovery of traits associated with invasiveness
Jersey Standard Strain (NJSS) moth heads obtained from the USDA-
and mapping of population resequencing data to determine sour-
ces and pathways of spread (Roe et al. 2019; Hamelin and Roe APHIS Otis Laboratory (Buzzards Bay, MA, USA): four individuals
2020). An Illumina-based genome assembly of biomaterials pre- were processed apiece for each of the male and female sexes.
pared from the LDD-derived in vitro cell line, LD652 (Goodwin et al. Genomic DNA was prepared from a single female pupa, which had
1978), was recently published (Zhang et al. 2019); however, this cell been reared from an NJSS egg mass—this biomaterial was used for
culture may not accurately portray the comprehensive genomic sequencing a total of 96 SMRT-Cells by the University of
and/or genic complements of the intact LDD insect (Sparks et al. Washington PacBio Sequencing Services group (Seattle, WA, USA).
2013). Whole-insect genomes of two subspecies of Asian gypsy Subread data were extracted in fastq format from bas.h5 archives
moths, L. dispar asiatica (LDA) and L. dispar japonica (LDJ), were also (without imposition of quality filters) using Pacific Biosciences’
recently sequenced using PacBio technology (Hebert et al. 2019). pbh5tools (https://github.com/PacificBiosciences/pbh5tools). Read
This study reports the sequencing of LDD at the whole-insect level correction, trimming and assembly were performed using version
using PacBio and Illumina technologies, done specifically to enable 1.9 of the CANU assembler (Koren et al. 2017). Subread sets were
nuclear genome-informed population genetics and comparative compiled into BAM-formatted archives using the bax2bam utility
genomics analyses among these subspecies, thereby augmenting from Pacific Biosciences’ SMRT Analysis software suite (https://www.
Received: February 05, 2021. Accepted: April 26, 2021
Published by Oxford University Press on behalf of Genetics Society of America 2021. This work is written by US Government employees and is in the public
domain in the US.2 | G3, 2021, Vol. 11, No. 8
pacb.com/support/software-downloads) with the following pulse with a reference protein (also of at least 150 aa in length) such that
feature options enabled: DeletionQV, DeletionTag, InsertionQV, 75% or more of aligned residues are positively similar, and the hit
IPD, MergeQV, SubstitutionQV, PulseWidth and SubstitutionTag. length-to-subject sequence length ratio is 90% or greater. Silver-tier
BAM-formatted subreads were aligned to assembled genomic entries are a minimum of 100 amino acid residues long with a hit
templates using the BLASR long read aligner via the pbalign wrapper spanning at least 75% of a reference protein sequence’s length (also
(https://github.com/PacificBiosciences/blasr; https://github.com/ at least 100 aa in length), and bronze-tier entries are similar, but
PacificBiosciences/pbalign), the results of which were subsequently with the hit covering 30% or more of the reference protein’s length.
used as inputs to the Arrow genome polishing algorithm as imple- The annotation pipeline was also used to independently analyze
mented in the Pacific Biosciences GenomicConsensus package the LDA and LDJ genomes (available from https://osf.io/unz2v/files/ as
(https://github.com/PacificBiosciences/GenomicConsensus). Three “lda.genome.v0.3.arrow-polished.fasta” and “ldj.genome.v0.3.arrow-
rounds of Arrow-based assembly polishing were performed. polished.fasta”, respectively), as well as the two LD652 cell line assem-
An additional library was prepared from a single non-sexed, blies described in Zhang et al. (2019): one of these LD652 assemblies
3rd-instar larval insect reared from an NJSS egg mass. A set of incorporates information from Novogene-prepared Hi-C libraries
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
359,593,922 Illumina PE100 short read pairs was sequenced from (“LD652_Lymantria_dispar.FINAL.fasta”, obtained from http://prodata.
this library by the Georgia Genomics Facility (Athens, GA, USA) swmed.edu/download/pub/LD652/) and the other (available from
and aligned to the Arrow-polished template with bowtie2 GenBank under assembly accession GCA_004115105.1), does not.
(Langmead and Salzberg 2012), the results of which were used as Predicted protein sequences from each of the gold, silver and bronze
input to the Pilon polishing program operating in its diploid mode tiers were combined in an assembly-specific manner, and these were
(Walker et al. 2014). The fully polished assembly was then proc- then globally pooled and clustered with CD-HIT (Fu et al. 2012) using a
essed with Purge Haplotigs (Roach et al. 2018) to minimize resid- sequence identity threshold of 0.975. Clusters having exactly one rep-
ual heterogeneity; empirically selected read depth cutoffs of low resentative sequence from each of the five assemblies were identified,
¼ 19, mid ¼ 58, and high ¼ 107 were used. The genome was then providing a set of putative single-copy gene models universally pre-
submitted to the NCBI WGS division, which filtered for potential sent in these L. dispar genomes.
vector contaminants and mitochondrial sequences. The NCBI-
vetted assembly was compared with the endopterygota_odb9 Data availability
database using BUSCO (Sima ~ o et al. 2015) to assess genome qual- All data used in the preparation of the final LDD assembly are
ity in terms of known single-copy orthologs. Repetitive content available at the NCBI under BioProject accession number
was identified using RepeatModeler (Smit and Hubley 2020)— PRJNA694036: PacBio subreads and Illumina short-read data are
which wraps the LTRharvest (Ellinghaus et al. 2008), RepeatScout available at the SRA database (accession numbers SRR13505170-
(Price et al. 2005) and RECON (Bao and Eddy 2002) pipelines—and SRR13505265, and SRR13518687-SRR13518688, respectively). The
masked using RepeatMasker (Smit et al. 2019). Whole Genome Shotgun project has been deposited at DDBJ/
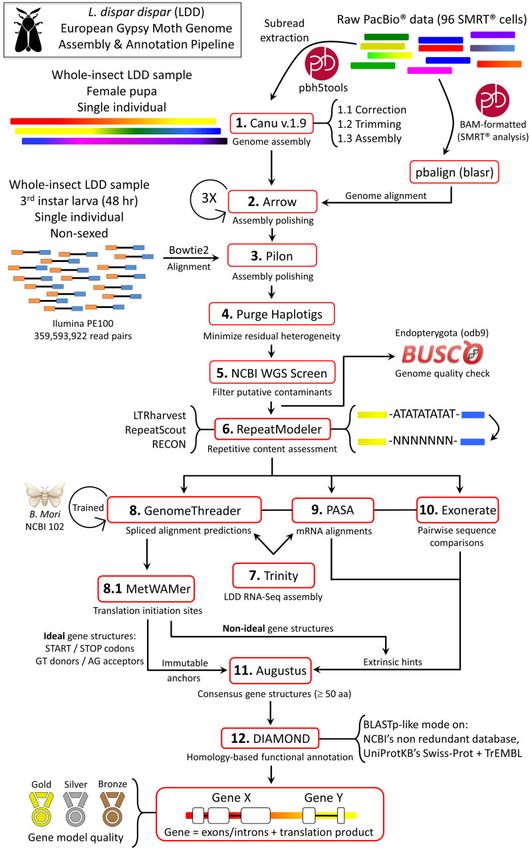
Gene annotation was conducted using a modified version of ENA/GenBank under the accession JAEVLZ000000000. The ver-
the insect-specific pipeline described in Hebert et al. (2019) (see sion described in this paper is JAEVLZ010000000. In addition, the
Figure 1). In addition to extrinsic hints provided to Augustus whole-genome assembly and automated protein-coding gene
(Stanke et al. 2006) by Exonerate (Slater and Birney 2005) and finding results (see below) are available at the Open Science
PASA (Haas et al. 2003), proteins predicted by GenomeThreader Framework (OSF) repository: https://doi.org/10.17605/OSF.IO/
(Gremme et al. 2005), used to align Trinity-based RNA-Seq assem- JX2VS. Lastly, interested parties are welcome to contact the cor-
blies (Haas et al. 2013) to genomic templates and compute con- responding author to request relevant data and software.
sensus gene structures from the results, were also used. In
particular, GenomeThreader’s splice site models were retrained
using BSSM4GSQ (Sparks and Brendel 2005) with Bombyx mori data
Results and discussion
(obtained from NCBI’s Annotation Release 102; https://www.ncbi. The fully processed, NCBI-vetted genome assembly of the whole-
nlm.nih.gov/genome/annotation_euk/Bombyx_mori/102/) and its insect European gypsy moth (i.e., LDD) exhibited a size of 998 Mb,
predictions were post-processed with MetWAMer (https://github. a contig N50 of 662 Kb and a GC content of 38.8% (see Table 1).
com/scentiant/MetWAMer; Sparks and Brendel 2008) to identify The assembly size is 1.015 times larger than the 983 Mb mean es-
translation initiation sites. If a GenomeThreader-derived con- timate obtained from a sample of four females using flow cytom-
sensus gene structure was ideal (i.e., canonical translation start etry [female: 983.2 6 6.7 Mb (n ¼ 4) and male: 993.3 6 6.2 Mb
and stop codons were present and, for multi-exon models, (n ¼ 4)]. Of a total of 2,442 known BUSCOs in the Endopterygota,
introns uniformly exhibited GT donors and AG acceptors), it was 95.5% were complete (93.7% single-copy and 1.8% duplicated),
provided to Augustus as an immutable anchor; otherwise, it was 2.7% were fragmented and 1.8% were missing. Long interspersed
supplied as a hint. Both of the alternatives-from-evidence and nuclear elements (LINEs) constitute 25.40% of the LDD genome
alternatives-from-sampling Augustus flags were set to false. (see Table 2). This amount is only slightly less than the 25.74% and
RNA-Seq data used to prepare Trinity assemblies were obtained 26.86% fractions of LINEs observed by the revised annotation pipe-
from Sparks et al. (2013) and other unpublished studies con- line as applied to LDA and LDJ, respectively, although it is notably
ducted by MES and DEGR. greater than the 18.96% and 17.34% amounts observed for the
Protein sequences predicted by the overall pipeline were con- LD652 assemblies with and without Hi-C library integration, re-
strained to 50 or more amino acids (aa) in length. These were then spectively (see Table 3). Similarly, although LDD, LDA and LDJ ex-
compared with the NCBI NR and UniProtKB’s Swiss-Prot and TrEMBL hibit commensurate levels of total repetitive DNA (68.74%, 67.45%
protein databases (UniProt Consortium 2019) using the DIAMOND and 67.09%, respectively), the LD652 assemblies comprised lesser
aligner in its BLASTp-like mode (Buchfink et al. 2015). Gene models amounts of such content (with Hi-C ¼ 59.76%, sans Hi-C ¼ 59.66%).
were sorted into gold, silver and bronze tiers based on their encoded The protein-coding gene annotation pipeline identified 75,018
products’ similarity to exemplar proteins: gold-tier entries are at unique LDD genes, of which 11,901 sorted to the gold tier, 17,598 to
least 150 aa in length and exhibit a single high-scoring segment pair the silver, and 26,773 to bronze (the remaining 18,746 “non-podium”M.E. Sparks et al. | 3
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
Figure 1 L. dispar dispar genome assembly and annotation pipeline. The automated pipeline used to assemble the European gypsy moth genome, as well
as annotate repetitive content and nuclear protein coding genes, is a modified version of the methods used for annotating the Asian gypsy moth
genomes, L. dispar asiatica and L. dispar japonica (Hebert et al. 2019). (The Bombyx mori image is copyright Freepik 2018 and reproduced here with
permission.)4 | G3, 2021, Vol. 11, No. 8
Table 1 Genome assembly statistics following initial Canu assembly, each of three rounds of Arrow polishing, a round of Pilon short-
read polishing, primary contig identification using the Purge Haplotigs pipeline, and purging of mitochondrial sequences and putative
vector contaminants by software filters at the NCBI WGS division
Unpolished Arrow polished Arrow polished Arrow polished Pilon Purge Haplotigs NCBI filters (final
assembly (Canu) (round 1) (round 2) (round 3) polished processed version)
N50 (a) 355,683 355,730 355,728 355,719 354,801 661,876 661,876
N90 (a) 26,157 26,161 26,163 26,167 26,130 91,592 91,592
GC (%) 38.88 38.89 38.89 38.89 38.89 38.83 38.83
Total size 1,280,392,172 1,281,096,304 1,281,192,033 1,281,236,029 1,279,160,664 998,776,906 998,430,749
Min contig 1,028 1,028 1,028 1,029 1,029 1,039 1,039
Max contig 8,311,499 8,312,296 8,312,357 8,312,335 8,267,660 8,267,660 8,267,660
Contig count 16,115 16,115 16,115 16,115 16,115 4,625 4,622
SMRT cells 96
Subread count (b) 20,008,385
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
Bases in subreads (b) 113,177,874,503
Genome size estimate 983,200,000
Sequencing coverage 115.11
a
N50 was calculated using the definition provided in Miller et al. (2010), with N90 being similarly defined.
b
Subreads determined using bax2bam (from smrtlink_7.0.0.63985) w/options “--subread” and
“--pulsefeatures¼DeletionQV,DeletionTag,InsertionQV,IPD,MergeQV,SubstitutionQV,PulseWidth,SubstitutionTag”.
Table 2 Repetitive content of the L. dispar dispar genome assembly as identified using RepeatModeler/RepeatMasker
Number of elements Length occupied (bp) Percentage of sequence (%)
Bases masked 686,312,151 68.74
Retroelements 1,688,175 336,176,424 33.67
SINEs 150,440 12,344,035 1.24
Penelope 214,022 28,041,873 2.81
LINEs: 1,210,601 253,631,498 25.40
CRE/SLACS 1,123 414,475 0.04
L2/CR1/Rex 120,505 29,326,164 2.94
R1/LOA/Jockey 289,120 100,744,241 10.09
R2/R4/NeSL 3,328 1,806,751 0.18
RTE/Bov-B 392,519 58,464,284 5.86
L1/CIN4 0 0 0.00
LTR elements: 327,134 70,200,891 7.03
BEL/Pao 97,919 24,858,437 2.49
Ty1/Copia 20,415 6,797,796 0.68
Gypsy/DIRS1 114,861 23,908,124 2.39
Retroviral 455 56,012 0.01
DNA transposons 410,996 75,109,994 7.52
hobo-Activator 39,043 10,065,906 1.01
Tc1-IS630-Pogo 324,022 56,591,826 5.67
En-Spm 0 0 0.00
MuDR-IS905 0 0 0.00
PiggyBac 1,258 410,331 0.04
Tourist/Harbinger 6,160 810,421 0.08
Other (Mirage, P-element, Transib) 4,091 523,746 0.05
Rolling-circles 746,035 123,018,327 12.32
Unclassified 1,043,151 151,324,752 15.16
Total interspersed repeats 562,611,170 56.35
Small RNA 4,846 382,666 0.04
Satellites 192 130,399 0.01
Simple repeats 3,585 169,589 0.02
genes failed to satisfy the criteria for any of these levels, thus repre- Given that the LD652 cell line was derived from ovarian tissue
senting either false positive gene calls or novel sequences not yet pre- over four decades ago (Goodwin et al. 1978) and has undergone
sent in reference databases). Reannotation of the LDA and LDJ long-term storage, multiple serial passages and exposure to un-
genomes with this updated annotation system yielded commensu- known selective pressures in the interim (Lynn 2000, 2006), the
rate counts of genes encoding proteins corroborated by records in the possibility exists that loss of chromosomal-level genetic material
NCBI and UniProtKB databases (i.e., gold, silver and bronze tier mod- may partially explain the discrepancies in genome size, repetitive
els): LDA ¼ 9,581 þ 16,081 þ 27,583 ¼ 53,245 “podium-level” genes, content and gene content observed between LD652 and LDD
LDJ ¼ 57,331 and LDD ¼ 56,272 (see Table 3). These amounts are (Table 3). More likely, however, is that the short-read technology
roughly double what was observed for the LD652 assemblies: 24,345 used to sequence LD652 (Zhang et al. 2019) was comparably less
unique protein-coding gene loci were identified in the Hi-C library- effective at characterizing such a highly repetitive genome as
based assembly and 26,504 in the other version. Clustering of pooled, that of L. dispar than were the longer PacBio reads used in this
podium-level sequences identified a total of 3,265 putative single- study (and in Hebert et al. 2019). What is more, the lower levels of
copy proteins shared across all five of these L. dispar genomes. BUSCOs detected and fewer protein-coding genes predicted in theM.E. Sparks et al. | 5
Table 3 A comparison of descriptive statistics assessing assembly quality, and repetitive element annotation and gene finding results
LDD LDA LDJ LD652 (Hi-C) LD652
Size (bp) 998,430,749 921,904,509 1,000,530,546 788,637,865 864,963,299
Contig count 4,622 8,189 11,303 134,187 135,446
N50 661,876 212,365 137,117 5,068,179 249,594
N90 91,592 40,935 37,304 2,453 3,108
GC (%) 38.83 38.60 38.51 35.18 35.25
LINE elements 1,210,601 1,073,465 1,439,736 902,373 879,467
LINEs (%) 25.40 25.74 26.86 18.96 17.34
bp masked 686,312,151 621,829,597 671,280,650 471,271,105 516,072,603
bp masked (%) 68.74 67.45 67.09 59.76 59.66
Gold tier 11,901 9,581 8,834 5,341 5,896
Silver tier 17,598 16,081 16,445 5,289 5,819
Bronze tier 26,773 27,583 32,052 13,715 14,789
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
Non-podium 18,746 21,614 27,695 22,491 24,131
BUSCO (%) 98.2 96.4 97.7 87.7 93.9
Assembly sources and methods used are as described in the Materials and Methods section. Reported gold-, silver- and bronze-tier, as well as non-podium level,
values correspond to the number of unique protein-coding loci identified at the respective quality level by the gene annotation pipeline. Reported BUSCO values
correspond to the fraction of complete (i.e., single-copy and duplicated) and fragmented BUSCOs detected.
LD652 genomes suggest that the longer PacBio reads were also Conflicts of interest
more effective at recovering coding (and hence, typically less re-
None declared.
petitive) regions of the LDD, LDA and LDJ genomes in a highly
contiguous manner. Resequencing of LD652 using longer-read
technologies will help with resolving this ambiguity. Literature cited
In L. dispar, females constitute the heterogametic sex (Sahara
et al. 2012), which in part motivated the decision to sequence a Bao Z, Eddy SR. 2002. Automated de novo identification of repeat se-
single female LDD pupa using long reads—representative se- quence families in sequenced genomes. Genome Res. 12:
quence from both sex chromosomes would thereby be captured. 1269–1276.
Among Asian gypsy moths, females are also flight capable Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein align-
(Keena et al. 2008; Srivastava et al. 2021), whereas European gypsy ment using DIAMOND. Nat Methods. 12:59–60.
moth females lack this capacity for flight. (Gypsy moth males are Djoumad A, Nisole A, Zahiri R, Freschi L, Picq S, et al. 2017.
flight capable.) This results in a comparatively slower rate of host Comparative analysis of mitochondrial genomes of geographic
range expansion in North America for European gypsy moth pop- variants of the gypsy moth, Lymantria dispar, reveals a previously
ulations, and a corresponding increase in concern for preventing undescribed genotypic entity. Sci Rep. 7:14245.
the establishment of Asian gypsy moths. The genomic sequence Ellinghaus D, Kurtz S, Willhoeft U. 2008. LTRharvest, an efficient and
resources contributed by this study should, upon detailed func- flexible software for de novo detection of LTR retrotransposons.
tional comparisons with the genomes of LDA and LDJ in particu- BMC Bioinformatics. 9:18.
lar, help identify genetic determinants controlling flight in this Fu L, Niu B, Zhu Z, Wu S, Li W. 2012. CD-HIT: accelerated for cluster-
important nuisance insect. Such results could find potential use ing the next-generation sequencing data. Bioinformatics. 28:
as molecular diagnostic markers to assist with the identification 3150–3152.
and interception of Asian gypsy moths in cargo containers, for in- Goodwin RH, Tompkins GJ, McCawley P. 1978. Gypsy moth cell lines
stance. divergent in viral susceptibility. I. Culture and identification. In
Vitro. 14:485–494.
Gremme G, Brendel V, Sparks ME, Kurtz S. 2005. Engineering a soft-
Acknowledgments ware tool for gene structure prediction in higher organisms. Inf
The authors thank Daniel Kuhar for technical assistance with in- Softw Technol. 47:965–978.
sect rearing and DNA extraction. The constructive suggestions Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, et al. 2003.
provided by two anonymous reviewers substantially improved Improving the Arabidopsis genome annotation using maximal
this report. Mention of trade names or commercial products in transcript alignment assemblies. Nucleic Acids Res. 31:
this publication is solely for the purpose of providing specific in- 5654–5666.
formation and does not imply recommendation or endorsement Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, et al.
by the U.S. Department of Agriculture. The USDA is an equal op- 2013. De novo transcript sequence reconstruction from RNA-Seq:
portunity provider and employer. reference generation and analysis with Trinity. Nat Protoc. 8:
1494–1512.
Hamelin RC, Roe AD. 2020. Genomic biosurveillance of forest inva-
Funding sive alien enemies: a story written in code. Evol Appl. 13:95–115.
Primary funding for this work came from USDA-ARS base project Hebert FO, Freschi L, Blackburn G, Béliveau C, Dewar K, et al. 2019.
8042-22000-315-00-D. Additional sources of funding include Expansion of LINEs and species-specific DNA repeats drives ge-
Genome Canada’s Large-Scale Applied Research Project (LSARP nome expansion in Asian gypsy moths. Sci Rep. 9:16413.
project # 10106), Biosurveillance of Alien Forest Enemies Johnston JS, Bernardini A, Hjelmen CE. 2019. Genome size estimation
(bioSAFE; https://www.biosafegenomics.com/), and a Genomics and quantitative cytogenetics in insects. In: S Brown, M Pfrender,
Research and Development Initiative (GRDI; Government of editors. Insect Genomics, Methods in Molecular Biology. New
Canada) grant awarded to M.C. York, NY: Humana Press, p. 15–26.6 | G3, 2021, Vol. 11, No. 8
Keena MA, Côté M-J, Grinberg PS, Wallner WE. 2008. World distribu- completeness with single-copy orthologs. Bioinformatics. 31:
tion of female flight and genetic variation in Lymantria dispar 3210–3212.
(Lepidoptera: Lymantriidae). Environ Entomol. 37:636–649. Slater GSC, Birney E. 2005. Automated generation of heuristics for bi-
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, et al. 2017. ological sequence comparison. BMC Bioinformatics. 6:31.
Canu: scalable and accurate long-read assembly via adaptive Smit AFA, Hubley R. 2020. RepeatModeler. Version 2.0.1, obtained
k-mer weighting and repeat separation. Genome Res. 27: from https://www.repeatmasker.org/RepeatModeler/
722–736., Smit AFA, Hubley R, Green P. 2019. RepeatMasker. Version 4.1.0,
Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with obtained from http://www.repeatmasker.org/RepeatMasker/.
Bowtie 2. Nat Methods. 9:357–359. Sparks ME, Blackburn MB, Kuhar D, Gundersen-Rindal DE. 2013.
Leonard DE. 1981. Bioecology of the gypsy moth. In: Doane CC, Transcriptome of the Lymantria dispar (gypsy moth) larval midgut
McManus ML, editors. The Gypsy Moth: Research Toward in response to infection by Bacillus thuringiensis. PLoS One. 8:
Integrated Pest Management. Technical Bulletin 1584—U.S. Dept. e61190.
Sparks ME, Brendel V. 2005. Incorporation of splice site probability
Downloaded from https://academic.oup.com/g3journal/article/11/8/jkab150/6261075 by guest on 15 October 2021
of Agriculture (USA). Washington, DC: U.S. Department of
models for non-canonical introns improves gene structure pre-
Agriculture, Forest Service, Science and Education Agency,
diction in plants. Bioinformatics. 21:iii20–30.
Animal and Plant Health Inspection Service, p. 9–29.
Sparks ME, Brendel V. 2008. MetWAMer: eukaryotic translation initi-
Lynn DE. 2000. Effects of long- and short-term passage of insect cells
ation site prediction. BMC Bioinformatics. 9:381.
in different culture media on baculovirus replication. J Invertebr
Srivastava V, Keena MA, Maennicke GE, Hamelin RC, Griess VC.
Pathol. 76:164–168.
2021. Potential differences and methods of determining gypsy
Lynn DE. 2006. Lepidopteran cell lines after long-term culture in al-
moth female flight capabilities: implications for the establish-
ternative media: comparison of growth rates and baculovirus
ment and spread in novel habitats. Forests. 12:103.
replication. In Vitro Cell Dev Biol Anim. 42:149–152.
Stanke M, Schöffmann O, Morgenstern B, Waack S. 2006. Gene pre-
Miller JR, Koren S, Sutton G. 2010. Assembly algorithms for next-gen-
diction in eukaryotes with a generalized hidden Markov model
eration sequencing data. Genomics. 95:315–327. that uses hints from external sources. BMC Bioinformatics. 7:62.
Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat Triant DA, Cinel SD, Kawahara AY. 2018. Lepidoptera genomes: cur-
families in large genomes. Bioinformatics. 21:i351–358. rent knowledge, gaps and future directions. Curr Opin Insect Sci.
Roach MJ, Schmidt SA, Borneman AR. 2018. Purge Haplotigs: allelic 25:99–105.
contig reassignment for third-gen diploid genome assemblies. UniProt Consortium. 2019. UniProt: a worldwide hub of protein
BMC Bioinformatics. 19:460. knowledge. Nucleic Acids Res. 47:D506–D515.
Roe AD, Torson AS, Bilodeau G, Bilodeau P, Blackburn GS, et al. 2019. Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, et al. 2014. Pilon: an
Biosurveillance of forest insects: part I—integration and applica- integrated tool for comprehensive microbial variant detection
tion of genomic tools to the surveillance of non-native forest and genome assembly improvement. PLoS One. 9:e112963.
insects. J Pest Sci. 92:51–70., Zhang J, Cong Q, Rex EA, Hallwachs W, Janzen DH, et al. 2019. Gypsy
Sahara K, Yoshido A, Traut W. 2012. Sex chromosome evolution in moth genome provides insights into flight capability and viru-
moths and butterflies. Chromosome Res. 20:83–94. s-host interactions. Proc Natl Acad Sci USA. 116:1669–1678.
~ o FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov
Sima
EM. 2015. BUSCO: assessing genome assembly and annotation Communicating editor: B. OliverYou can also read

















































