COMPLIANCE - A CATALYST FOR GROWTH IN THE INDIAN PHARMA SECTOR - Building a Compliance Management Programme
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

COMPLIANCE - A
CATALYST FOR
GROWTH IN THE
INDIAN PHARMA SECTOR
Building a Compliance
Management Programme
©Legasis - Thought Paper
Pharma: A Multi-Billion Dollar Industry
The Indian pharmaceutical industry has been consistent in its growth trajectory, and is
expected to further grow into a 100-billion-dollar market by the end of 2025. The
domestic significance of the pharmaceutical industry can be estimated by the large
population that it caters to and the economy of scale that it achieves in doing so. On the
international front, pharmaceutical exports from India stand at a staggering 15 billion
dollars as per 2019-2020. The Indian biotechnology industry too has been valued at 65
billion dollars as of 2019 and is expected to grow into 150-billion-dollar market by 2025.
Figure 1Source: IBEF, 2021
©Legasis - Thought Paper 02
The Indian pharmaceutical industry has been consistent in its growth trajectory, and is
expected to further grow into a 100-billion-dollar market by the end of 2025. The
domestic significance of the pharmaceutical industry can be estimated by the large
population that it caters to and the economy of scale that it achieves in doing so. On the
international front, pharmaceutical exports from India stand at a staggering 15 billion
dollars as per 2019-2020. The Indian biotechnology industry too has been valued at 65
billion dollars as of 2019 and is expected to grow into 150-billion-dollar market by 2025.
Regulatory Overview
The pharmaceutical industry in particular CDSCO's licencing and controlling
is heavily regulated given the sensitive functions.
nature of its business operations. Form
manufacturing to advertisements, each 3. The National Pharmaceuticals
step is full of compliances. The patent Pricing Authority (NPPA)
framework in India in particular provides The NPPA was formed as a branch of the
for certain specific provisions to regulate Department of Pharmaceuticals of
the pharma patents, which play a key role Ministry of Chemicals and Fertilizers
in anti-trust issues as well. under the Drugs Price Control Order of
1995, with the objective of updating the
1. The Central Drug Standard pricing of regulated bulk medications and
Control Organisation (CDSCO) The formulation as well as to enforce prices
CDSCO is a department under the and guarantee medicine availability. The
Ministry of Health and Family Welfare NPPA helps maintain the prices of
and is in charge of the final regulatory decontrolled drugs to keep them at easy
delegation under the DGHS.i According availability.
to the provisions under Drugs and
Cosmetics Act, CDSCO is responsible for 4. The Directorate General of
approvals of drugs and conduct of clinical Health Services (DGHS)
trials. It is responsible for laying down the The Director-General of Health Services
standards for drugs and exercise control (DGHS) is a Central Health Services
over quality of drugs being marketed in officer. It is responsible for rendering
India and the ones being imported from technical advice on all medical and public
other countries. CSDSCO’s main job is to health matters to the Ministry of Health
bring uniformity in application of the Act and Family Welfare. The DGHS
over different regions of the country.ii co-ordinates with the Health Directorates
of all States/UTs for implementation of
2. The Drug Controller General various National Health Programmes
of India (DCGI) through its Regional Offices of Health and
It is a statutory authority for the purposes Family Welfare. The Directorate is also
of the Drugs and Cosmetics Act, 1940 and responsible to ensure compliance with
Rules, 1945 and is the head of CDSCO. International Health regulations.iii
The DCGI is responsible for carrying out
©Legasis - Thought Paper 03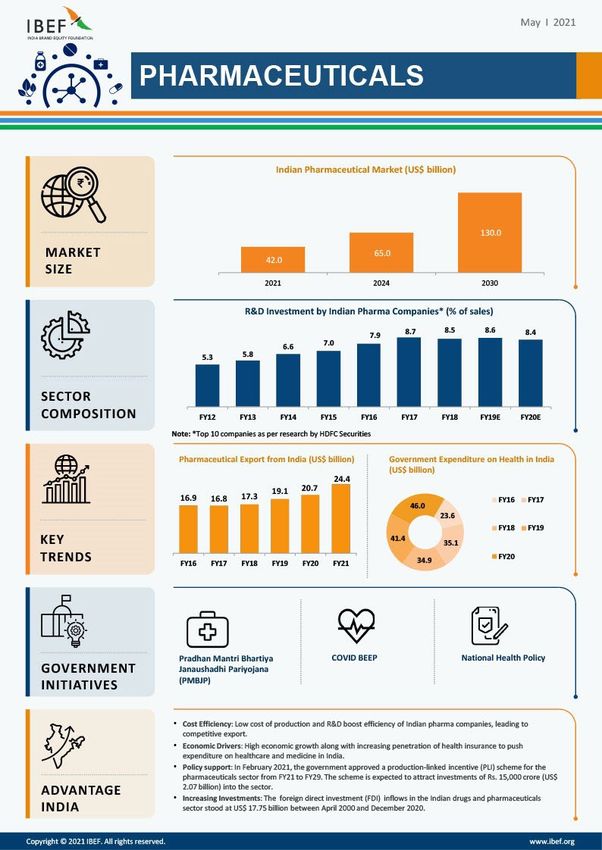
5. The Indian Council of Medical Research (ICMR)
ICMR is actively involved in developing ethical guidelines and policy that acts as
guidelines for biomedical researches being conducted in country. ICMR also helps in
developing tools for providing support in the conduct of training programs along with
capacity building exercises and aims at forming policies and guidelines by taking into
account the ideas of clinicians, researchers, ethics committee members along with legal
experts and society representatives.iv
Legislative Overview
The legal and regulatory environment The Drug (Price Control) Order, 2013 is
within which the pharmaceutical business an order issued to regulate the prices of
operates, comprises mainly of the drugs wherein it provides a list of
following laws: price-controlled drugs, procedures for
fixation of price of drugs and methods for
1. The Drugs and Cosmetics Act, implementation of prices fixed by the
1940 Government.
The Drugs and Cosmetics Act, 1940 is the
primary pharmaceutical statute and 4. The Drugs and Magic Reme-
further comprises of the Drugs and dies (Objectionable Advertise-
Cosmetics Rules, 1945 and the New Drugs ments) Act, 1954 and The Drugs and
and Clinical Trials Rules, 2019v framed Magic Remedies (Objectionable Ad-
thereunder. It seeks to: vertisements) Rules, 1955.
a. Regulate the clinical trial, The law prohibits advertisements of
import, manufacture, drugs/remedies that claim to have
distribution and sale of magical properties and secures the
drugs. consumers from being misled into
b. Ensure the availability of consuming medicines and drugs which
standard quality drugs and may harm their health under the pretext
cosmetics to the consumer. of treatment.
2. The Pharmacy Act, 1948 5. The Narcotic Drugs and
The pharmacy law of 1948, regulates the Psychotropic Substances Act, 1985
professional conduct of pharmacists and The legislation prohibits individuals from
quality of service rendered by engaging in an activity consisting of
pharmacists. The enactment made it production, cultivation, sale, purchase
mandatory for pharmacists to achieve a and transport of any narcotic drug or
minimum standard of qualification to psychotropic substance. It provides for
make them eligible for practice. forfeiture of property derived from or
used in, illicit traffic in narcotic drugs and
3. The Essential Commodities psychotropic substances.
Act, 1955 and Drugs (Price Control)
Order, 2013 (DPCO) 6. The Patent Act, 1970
©Legasis - Thought Paper 04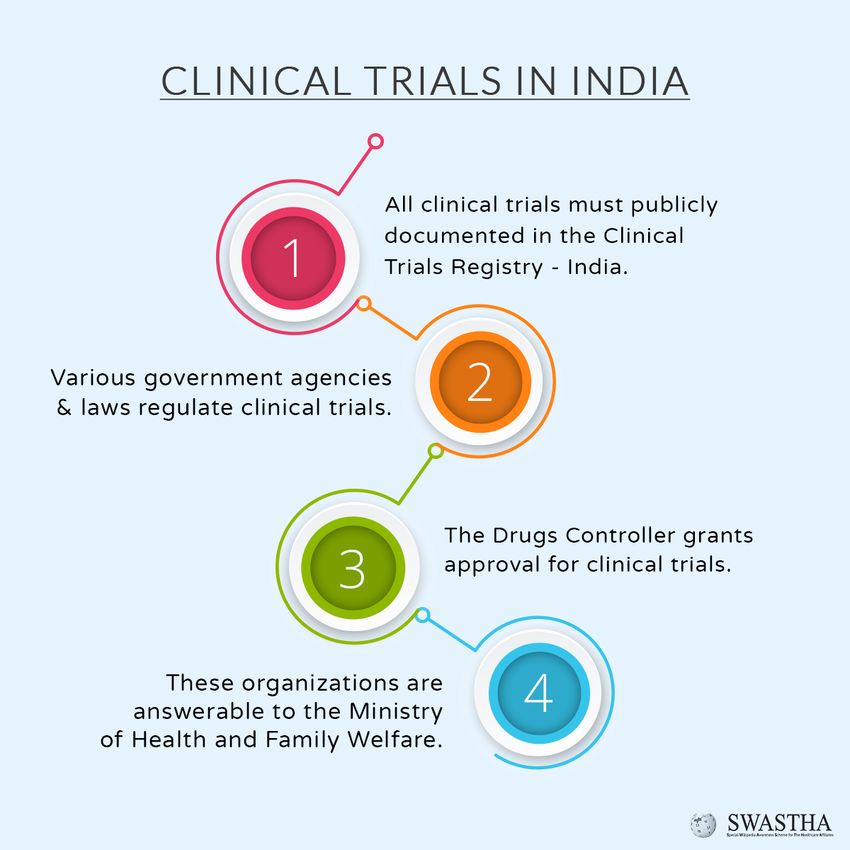
Section 92A of the Patent Act, 1970 calls for issue of compulsory license for manufacture
and export of patented pharmaceutical products. Patents give the pharmaceutical
company exclusive rights to exclude other from unauthorised production, manufacturing
and importing the invention.
Major Compliance Requirements
In The Pharmaceutical Sector
I. Good Manufacturing Practices (read with Drugs and Cosmetics Act
Good Manufacturing Practices (GMP) is a 1940), and became mandatory on July 1,
method for ensuring that goods are 2005.
consistently produced and regulated in
Schedule M categorises the
accordance with quality standards. It is
different legal criteria that must be met by
intended to reduce any hazards associated
medicines, medical devices, and other
with pharmaceutical manufacture that
types of products. These include
cannot be eliminated by testing the
infrastructure, premises, ESH measures,
finished product. GMP encompasses
manufacturing and operation controls,
every element of manufacturing, from raw
quality control and assurance, and
materials, facilities, and equipment to
stability and validation studies.x
employee training and personal hygiene.vi
After a joint examination of the
GMP guidelines were established by the premises by the CDSCO and state
World Health Organization in 1975 to help regulatory agencies, the manufacturer will
regulatory agencies in various countries in get a certificate of current good
ensuring consistency in quality, safety, manufacturing practise (cGMP).
and effectiveness requirements when
A No-Objection Certificate (NOC)
importing and exporting medicines and
and a Certificate of Origin (CO) will be
associated goods.vii The Indian GMP
required to export any produced items.
criteria, as specified in Schedule M of the
Form 28 and Schedule M compliance
Drugs and Cosmetics Act, 1940, were
must be presented to the DCGI in order to
based on the WHO GMP
get an NOC.
recommendations of 1982, and were later
updated in 2001 with the goal of To get a CO, an application must be
establishing qualitative standards for drug submitted to a recognised agency, such as
manufacture.viii the Chamber of Indian Industry's offices
(CII). CO is a document that certifies that
Whether GMP is mandatory for the the items being exported were entirely
Indian Pharmaceutical Sector? sourced, produced, or manufactured in
India. This certificate is necessary to
GMPs are included in Schedule ‘M’
comply with India's WTO requirements.xi
of the Drugs and Cosmetics Rules, 1945ix
©Legasis - Thought Paper 05Key Clauses in Good Manufacturing areas should not be used for storage of
Practices materials.
The factory building(s) for The records shall be made or
manufacture of drugs shall be so situated completed at the time of each operation in
and shall have such measures as to avoid such a way that all significant activities
risk of contamination from external concerning the manufacture of
environment. pharmaceutical products are traceable.
Records and associated Standard
The building(s) used for the factory
Operating Procedures (SoP) shall be
shall be designed, constructed, adapted
retained for at least one year after the
and maintained to suit the manufacturing
expiry date of the finished product.
operations so as to permit production of
drugs under hygienic conditions.
II. Drug Approval
The disposal of sewage and
A company desirous of
effluents (solid, liquid and gas) from the
manufacturing or importing a new drug
manufactory shall be in conformity with
has to file an application for issue of
the requirements of Environment
license from DCGI and file a Form 44
Pollution Control Board.
along with submission of data that is
Rest and refreshment rooms shall required to be submitted according to
be separate from other areas and must not guidelines provided under Schedule Y of
lead directly to the manufacturing and Drugs and Cosmetics Act 1940 and Rules
storage areas. 1945.
Manufacturing is to be conducted Clinical trials have to be conducted
under direct supervision of competent according to specified guidelines so that
technical staff with prescribed the safety of drug among Indian
qualifications and practical experience. population can be proved. According to
The head of the Quality Control section 2.4 (a) of Schedule Y of Drugs and
Laboratory shall be independent of the Cosmetics Act 1940 and Rules 1945 “those
manufacturing unit. drug substances which are discovered in
India all phases of clinical trials are
The contents of all vessels and
required.”xii
containers used in manufacture and
storage during the various manufacturing The regulations under Drugs and
stages shall be conspicuously labelled with Cosmetics Act 1940 and its Rules 1945,
the name of the product, batch no., batch 122A, 122B and 122D and further
size and stage of manufacture. Appendix I, IA and VI of Schedule Y lay
down the parameters for information that
Manufacturing environment shall
must be submitted for taking approval of
be maintained at the required levels of
manufacturing or sale of a new drug.
temperature, humidity and cleanliness.
After completing the clinical trials,
Premises shall be cleaned and
Form 44 along with clinical testing data
maintained in an orderly manner, and free
has to be filed. The information regarding
from accumulated waste, dust, debris and
marketing status of drugs in countries
other similar material. Manufacturing
©Legasis - Thought Paper 06across world have to be submitted along drug is tested on 100 patients in about 3 to
with information for prescribing samples, 4 centres. However, if the drug is new and
testing of protocols along with product not marketed in any other country, at least
monograph, labels, and cartons. The 500 patients across 10-15 centres have to
review of application usually takes in be used for conducting trial.
12-18 months.xiii
The stages of drug approval may be
briefly summed up as follows:
1. Submitting clinical trial application
for the evaluation of safety and efficacy.
2. The permission of New Drug
Approval can be obtained after fulfilling
requirements.
3. The changes taking place in product
quality and safety after approval have to
be analysed.
4. Quality information for submitting
drug for New Drug Approval has to be
Figure 2 Source: AbhiSuryawanshi, CC BY-SA 4.0
obtained.xiv , via Wikimedia
Commons
III. Clinical Trials
A clinical trialxv is the systematic study of a Compliance requirements in
new drug in human subjects to generate conducting clinical trials are as
data for discovering and/or verifying follows:
pharmacological effects with the objective DCGI Approval
of determining safety and efficacy of the
new drug. Clinical trials in India take Approval from Institutional
place in accordance with New Drugs and Ethics Committee
Clinical Trial Rules, 2018xvi and ICMR’s All clinical trials need to have
Ethical Guidelines for Research Involving approval from the IEC.
Human Participants.xvii
Investigators and Administrators of
Trial in Phases: Phase I clinical trials Academic Institutes should ensure that
have to be conducted with an aim of their Institutional Ethics Committees
determining maximum dose tolerable in (IECs) are registered with the central
human and the adverse reactions that can licensing authority and the registration
occur due to the drug on healthy human renewed at the end of 3 years.xviii
volunteers. Phase II trial is to determine IECs function according to
the therapeutic use and effective ranges of standard operating procedures that are
drug doses in about 10-12 patients at every usually available on their websites.
level of dose. The safety and efficacy of Projects submitted essentially undergo
drug is tested in Phase III trials as the
©Legasis - Thought Paper 07two broad types of review – Full board or occurrence.
full committee review or expedited review.
Site preparedness
Registration of the clinical
The regulator can inspect the site at
trial with the Clinical Trials Registry
any time and can cancel the trial
of India
permission and discontinue the study.
The CTRI is a free, online portal Therefore, preparedness of the study site
that allows both investigator-initiated and at all times must be ensured.
regulatory studies to be registered.
IV. Protection of Intellectual
For Regulatory Clinical Trials,
Property
registration in CTRI is mandatory from
Drug discovery forms a critical element
June 2009.
for success amidst pharmaceutical
Registration must be done before companies as they invest heavily in R&D
the first participant is enrolled. of potential drugs which can cure certain
ailments that lack precise treatment. Such
Obtain informed consent from
research undertakings on the part of
participants
pharmaceutical companies benefits not
Investigators must ensure that only the end consumer but is also a key
written, informed consent is obtained element to gain competitive edge in the
from all participants in a clinical trial. market. These innovations lead to
For trials that involve vulnerable discovery of new life-saving drugs and
participants (e.g. children or patients) and thus have to be protected through
involve a new chemical entity or a new intellectual property rights (IPR). Such
molecular entity, the investigators in patents on new molecular combinations,
addition have to ensure audio visual provide the pharmaceutical companies
recording of the informed consent exclusive rights to market their discovered
process. drugs and prevent other to manufacture,
sell or make these drugs for a period of 20
Report serious adverse events years.
that occur during a clinical trial
An SAE is defined as an untoward IPR protections work in the
medical occurrence during a clinical trial following ways –
that is associated with death, in patient a. Promotes fair and effective
hospitalisation (if the study was done on incentive for innovation.
outpatient basis), prolongation of b. Protects pharma companies from
hospitalisation (if the study was potential infringements.
conducted on in-patient basis), persistent
or significant disability or incapacity, a c. Provides strong enforcement
congenital anomaly or birth defect or is arsenal to defend infringed patents.
otherwise life-threatening.
Trademark Protection for
Investigator should report all SAEs pharmaceutical products
to the DCGI (for regulatory studies), the
sponsor and the IEC, within 24 h of their Section 9(a) of the Trademark Act
©Legasis - Thought Paper 08of 1999 prohibits the registration of apparatus, on the other hand, may be
trademarks that are descriptive or lack granted a patent.xxiv
distinctiveness, that is, are unable to
differentiate one source's products from There are two types of licencing
another.xix arrangements under the Patents Act:
In the case of pharmaceutical 1. Compulsory licencing – A
trademarks, the brand name or drug name compulsory license is requested by a
is typically sourced from the drug's person who wishes to work on a patented
treatment, salt composition, or any other innovation, after at least 3 years have
related medical term, and thus does not passed since the patent seal.
have an innate unique character; however,
2. Licences of right - In some cases,
"distinctiveness" is a requirement for a
the central government can order that the
mark to be eligible for a trademark.xx
patent be endorsed with the expression
Further, a trademark cannot be any "licences of right" after 3 years from the
name of chemical elements, compounds, date of the patent's sealing. This has the
or International Non-proprietary Names effect of allowing anybody interested in
(INNs)xxi issued by the WHO and notified working on the patented innovation in
by the Registrar of Trademarks in 2012, or India to obtain a licence from the
which are deceptively similar to the INNs . patentee.xxv
Since, the INNs mentioned are generic
names for active medicinal components,
no pharmaceutical corporation may claim
exclusive rights to them, and they can thus
be used by anybody .
Patent protection for
pharmaceutical products
Pharmaceutical and related
discoveries must pass the benchmark
outlined in Section 3 of the Patents Act,
1970, specifically Sections 3 (d), (e), and
(i). Even if such innovations meet the
patentability standards, Section 3 of the
Patents Act of 1970 defines which
inventions are not patentable subject
matter.xxiii
In the domain of pharmaceutical,
method of treatment claims is frequently
made under the pretence of composition
claims. It is worth noting, however, that in
India, any claim relating to treatment is
not patentable subject matter. Surgical,
therapeutic, or diagnostic equipment or
©Legasis - Thought Paper 09Emerging Compliance Issues in
the Pharmaceutical Sector
1
The introduction of National List of Essential Medicines, 2015 carries with
itself a compliance burden to be discharged by the companies with respect to
price control. In addition to this, mandatory barcoding of the drugs for export
too has gained momentum which adds on to the compliance obligations.
2
To further check improper promotions of drugs to medical practitioners (via
medical representatives), a Uniform Code for Marketing Practices of
Pharmaceutical Companies has been introduced, which creates a new set of
compliance obligations in itself.
The Central Government recently released National Guidelines for Data
3 Quality in Surveysxxvi with an aim of providing principles and guidelines to
ensure that data is of quality in surveys and in areas of demographic, health and
nutrition surveys especially. These guidelines provide steps that must be
followed in three phases of field-based study that is very critical including
preparatory, during and post data collection.
Managing Compliance Effectively
The present pandemic of Covid-19, made a significant impact on the Indian
pharmaceutical industry, and bolstered the growth of industry in different avenues.
Significant manufacturing of vaccines and drugs demand, created by individuals and
government alike has increased the supply chain efficiency of the sector. On the other
hand, prices of non-Covid related material have been increased which has interrupted
production cycles and increased shipping costs of such medicines.
The compliance burden on the pharma industry is more than in other sectors. Apart from
the day-to-day compliances of Labour, Land, Finance and corporate, they have additional
obligation of complying with special regulations owing to the nature of the work they
undertake. Further, the industry is open to strict scrutiny and large risks in case things go
awry.
Managing risk, therefore, becomes a large part of the compliance process. In order to
manage risk, usually three techniques are resorted to: Mitigating, Avoiding and
Eliminating. A healthy compliance management program ensures that these three
techniques are applied in some way or the other. Further, outsourcing Compliance
management to a specialized agency has been prevalent in most sectors as it contributes
to transferring the additional burden, hence, reducing the possibility of running into a
regulatory issue.
©Legasis - Thought Paper 10Legasis Celebrates and Hosts
COMPLIANCE - A CATALYST FOR
GROWTH OF PHARMA SECTOR
IN INDIA
6 AUGUST
2021
COMPLIANCE 10/10
SYMPOSIUM AND AWARDS
8TH EDITION
10.10.2021
References
i Parvathi K. Iyer, Regulatory Issues in the Indian Pharmaceutical Industry, India, Science and
Technology, https://www.nistads.res.in/all-html/Regulatory%20Is
sues%20in%20the%20Indian%20Pharmaceutical%20Industry.html
ii https://cdsco.gov.in/opencms/opencms/en/Home/
iii https://dghs.gov.in/content/249_1_Objective.aspx
iv http://www.vmmc-sjh.nic.in/writereaddata/Role%20of%20ICMR.pdf
v Nupur Chowdhury, Pallavi Joshi, Arpita Patnaik, Beena Saraswathy, Administrative Structure
and Functions of Drug Regulatory Authorities in India, Working Paper 309, Indian Council for
Research on International Economic Relations, 4-16 (Sept., 2015) https://icrier.org/pdf/Work
ing_Paper_309.pdf
vi WHO good manufacturing practices. In: Quality assurance of pharmaceuticals. A compendium of
guidelines and related materials. Good manufacturing practices and inspection, Vol. 2, 2nd
updated ed. Geneva, World Health Organization, 2007. https://apps.who.int/iris/han
dle/10665/43532
vii Supra note i
viii Good Manufacturing Practices for Pharmaceutical sector, Journals of India, (Mar. 3, 2021)
©Legasis - Thought Paper 11https://journalsofindia.com/good-manufacturing-practices-for-pharmaceutical-sector/
ix Schedule M, Drugs and Cosmetics Rules, 1945 https://cdsco.gov.in/opencms/export/sites/CD
SCO_WEB/Pdf-documents/acts_rules/2016DrugsandCosmeticsAct1940Rules1945.pdf
x Probir Roy Chowdhury, Outsourcing Biopharma R&D to India 92-95, (2011) Oxford Publications
Ltd. https://www.sciencedirect.com/science/article/pii/B9781907568084500081
xi Ibid
xii https://www.pharmatutor.org/articles/new-drug-approval-procedure-india
xiii https://www.globalresearchonline.net/journalcontents/v13-2/004.pdf
xiv https://www.jpsr.pharmainfo.in/Documents/Volumes/vol9Issue10/jpsr09101759.pdf
xv https://www.rgcb.res.in/uploads/2014/07/Schedule-Y.pdf
xvi https://cdsco.gov.in/opencms/export/sites/CDSCO_WEB/Pdf-docu
ments/NewDrugs_CTRules_2019.pdf
xvii https://main.icmr.nic.in/sites/default/files/guidelines/ICMR_Ethical_Guidelines_2017.pdf
xviii https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7034142/
xix Chandra Nath Saha, Sanjib Bhattacharya, Intellectual property rights: An overview and
implications in pharmaceutical industry, J Adv Pharm Technol Res. (Apr-Jun, 2011)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3217699/
xx Pharmaceutical Trademarks, VideAim IP,
https://videaimip.com/blog/trademark-and-pharmaceuticals/
xxi Section 13, Prohibition of registration of names of chemical elements or international
non-proprietary Names, Trademark Act, 1999
xxii Supra Pt. 3
xxiii Section 3, What are not inventions, Patent Act, 1970
https://ipindia.gov.in/writereaddata/Portal/IPOAct/1_31_1_patent-act-1970-11march2015.pdf
xxiv Nilesh Zacharias, Sandeep Farias, Patents And The Indian Pharmaceutical Industry, Mondaq (No.
20, 2019) https://www.mondaq.com/india/patent/865888/patents-and-the-indian-
pharmaceutical-industry?type=mondaqai&score=66
xxv Poonam Chetry, Interpretation Of Section 3 (e) And Significance Of Synergistic Data, Mondaq,
(July 8, 2021) https://www.mondaq.com/india/patent/1089098/interpretation-of-section-
3-e-and-significance-of-synergistic-data?type=mondaqai&score=53
xxiv https://ndqf.in/wp-content/uploads/2021/07/National-Guidelines-for-DATA-QUALITY-
in-Surveys.pdf
©Legasis - Thought Paper 12You can also read

















































