The Initial Evaluation of Patients After Positive Newborn Screening: Recommended Algorithms Leading to a Confirmed Diagnosis of Pompe Disease
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
The Initial Evaluation of Patients
After Positive Newborn Screening:
Recommended Algorithms Leading to a
Confirmed Diagnosis of Pompe Disease
Barbara K. Burton, MD,a David F. Kronn, MD,b Wuh-Liang Hwu, MD, PhD,c Priya S. Kishnani, MD,d
on behalf of the Pompe Disease Newborn Screening Working Group
abstract Newborn screening (NBS) for Pompe disease is done through analysis of acid α-glucosidase
(GAA) activity in dried blood spots. When GAA levels are below established cutoff values,
then second-tier testing is required to confirm or refute a diagnosis of Pompe disease. This
article in the “Newborn Screening, Diagnosis, and Treatment for Pompe Disease” guidance
supplement provides recommendations for confirmatory testing after a positive NBS result
indicative of Pompe disease is obtained. Two algorithms were developed by the Pompe
Disease Newborn Screening Working Group, a group of international experts on both NBS
and Pompe disease, based on whether DNA sequencing is performed as part of the screening
method. Using the recommendations in either algorithm will lead to 1 of 3 diagnoses: classic
infantile-onset Pompe disease, late-onset Pompe disease, or no disease/not affected/carrier.
Mutation analysis of the GAA gene is essential for confirming the biochemical diagnosis
of Pompe disease. For NBS laboratories that do not have DNA sequencing capabilities, the
responsibility of obtaining sequencing of the GAA gene will fall on the referral center. The
recommendations for confirmatory testing and the initial evaluation are intended for a
broad global audience. However, the Working Group recognizes that clinical practices,
standards of care, and resource capabilities vary not only regionally, but also by testing
centers. Individual patient needs and health status as well as local/regional insurance
reimbursement programs and regulations also must be considered.
aDepartment of Pediatrics, Northwestern University Feinberg School of Medicine, and the Division of Genetics, Birth Defects, and Metabolism, Ann & Robert H. Lurie Children’s Hospital,
Chicago, Illinois; bDepartment of Pathology and Pediatrics, New York Medical College, Valhalla, New York; cDepartment of Pediatrics and Medical Genetics, National Taiwan University
Hospital, and National Taiwan College of Medicine, Taipei, Taiwan; and dDivision of Medical Genetics, Department of Pediatrics, Duke University Medical Center, Durham, North Carolina
All authors analyzed and interpreted the data, critically reviewed and revised the manuscript, and approved the final manuscript as submitted; all authors are
members of the Pompe Disease Newborn Screening Working Group and have experience in newborn screening and in treating and caring for patients with Pompe
disease; and all authors provided input and reviewed and approved the content for all articles of the supplement.
DOI: https://doi.org/10.1542/peds.2016-0280D
Accepted for publication Mar 8, 2017
Address correspondence to Priya S. Kishnani, MD, Division of Medical Genetics, Department of Pediatrics, Duke University Medical Center, 595 LaSalle St, Durham, NC
27710. E-mail: priya.kishnani@duke.edu
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2017 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE: The authors have indicated they have no financial relationships relevant to this article to disclose.
The guidelines/recommendations in this article are not American Academy of Pediatrics policy, and publication herein does not imply endorsement. 2017;
140(s1):e20160280D
Downloaded from www.aappublications.org/news by guest on November 4, 2021
SUPPLEMENT ARTICLE PEDIATRICS Volume 140, Number s1, July 2017:e20160280Delays in the clinical diagnosis of reports from the newborn screening with decreased GAA activity in the
Pompe disease are frequent due (NBS) pilot program in Taiwan, when blood (leukocytes, DBSs, isolated
to the rarity of the disorder, its compared with patients with classic lymphocytes) or another tissue with
heterogeneous clinical presentation, IOPD diagnosed clinically, where the presence of 2 known pathogenic
and overlap of signs and symptoms delays in diagnosis clearly lead to GAA variants in trans. A probable
with other neuromuscular disorders. a more guarded prognosis.2,3 With diagnosis of Pompe disease can be
These delays range from a few LOPD, patients diagnosed early in made if there is decreased enzyme
months in patients with classic the presymptomatic phase of the activity, but molecular studies are
infantile-onset Pompe disease (IOPD) disorder will benefit from avoiding ambiguous due to the presence of
to many years in patients with the many years of diagnostic odyssey molecular variants of unknown
late-onset Pompe disease (LOPD). they follow after they present with significance (VUS). In IOPD, the
A published analysis of data from signs and symptoms of LOPD. Patients presence of clinical findings of Pompe
1003 patients in the Pompe Registry identified through NBS can be started disease in the presence of decreased
reported diagnostic delays of 1.4 on ERT when there is early evidence of enzyme activity is a confirmation
months in patients with classic disease progression. of diagnosis. In cases of LOPD
IOPD, 12.6 years among non-classic NBS for Pompe disease is done where there is confirmed decreased
infantile and juvenile patients, and through analysis of GAA enzyme enzymatic activity, patients will
6 years for patients with LOPD.1 activity in dried blood spots (DBSs). If need to be managed closely for the
With the availability of enzyme levels of GAA are found to be normal, development of signs or symptoms
replacement therapy (ERT) with then the patient is classified as of disease, even when molecular
alglucosidase alfa (recombinant unaffected and no additional testing changes are known because there is
human acid α-glucosidase [rhGAA]) is required or performed. If the GAA considerable variation in how and
for Pompe disease and new levels are below established cutoff when patients will present.
treatments on the horizon, timely and values, molecular testing of the GAA Sometimes in patients suspected
accurate diagnosis can lead to early gene may be performed on second- to have LOPD, only 1 pathogenic
initiation of ERT before the onset tier screening. variant is found along with low
of irreversible pathologic changes.
Here, 2 algorithms for confirmatory GAA activity. In these cases, if
Classic IOPD is rapidly progressive,
testing that will confirm or refute the low GAA level is not due to a
and any delay can have a negative
a diagnosis of Pompe disease are pseudodeficiency allele, which can
impact on outcomes. Even in cases
provided, based on whether DNA cause low GAA activity measured in
of LOPD that are considered to be
sequencing is performed by the enzyme assays, but is not associated
in the early stages of the disease
NBS laboratory (Figs 1 and 2). The with Pompe disease, then measuring
clinically, there often is significant
confirmatory testing discussed GAA activity in another sample (eg,
damage to the muscles as noted by
includes recommended clinical blood, fibroblasts, or muscle biopsy
studies such as whole-body MRI
and laboratory procedures. The tissue) may be necessary to confirm a
and muscle ultrasound. Thus, any
recommendations for confirmatory diagnosis.4,5
delay in treatment initiation can be
significant, even for cases that are testing after a positive NBS result Nomenclature for classifications of
felt to be identified early or those in and the 2 algorithms were developed patients with Pompe disease has
which the patients appear to be in by the Pompe Disease Newborn been problematic, confusing, and
good clinical condition. Screening Working Group, a group used inconsistently and variably in
of international experts on both NBS the published literature and within
and Pompe disease. the medical community. Pompe
NEWBORN SCREENING AND POMPE These guidelines and disease must be considered as a
DISEASE recommendations do not necessarily continuum of disease that varies
In March 2015, the US Secretary of reflect the policy of the American by age of onset, symptoms, organ
Health and Human Services decided Academy of Pediatrics, and involvement, and degree of muscle
to adopt the Advisory Committee on publication herein does not imply involvement and pathology.6–8 The
Heritable Disorders in Newborn and endorsement. subtypes of Pompe disease are not
Children recommendation to add always clearly delineated based
Pompe disease to the Recommended on age at presentation because of
Uniform Screening Panel (RUSP). WHAT CONSTITUTES A DIAGNOSIS OF the heterogeneous nature of the
Early detection of patients with classic POMPE DISEASE? disease. Clinical presentation and
IOPD and initiation of ERT have clear Ideally, a diagnosis of Pompe manifestations, therefore, also
benefits as seen in these patients in disease is confirmed in a patient must be carefully considered and
Downloaded from www.aappublications.org/news by guest on November 4, 2021
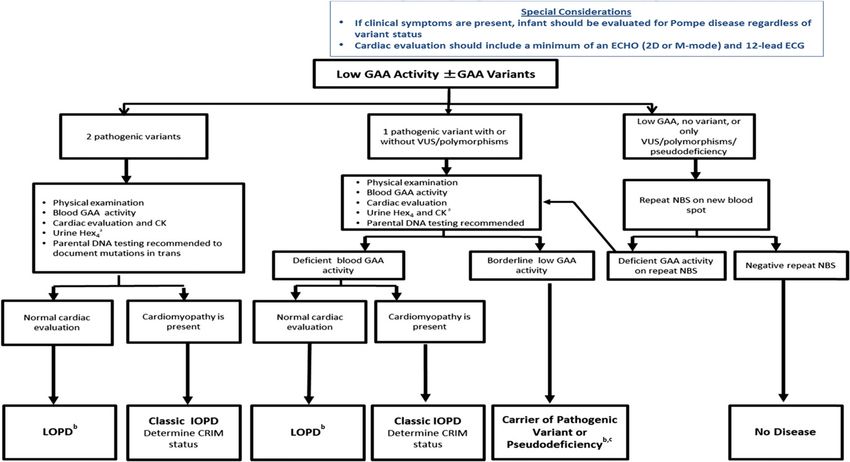
PEDIATRICS Volume 140, Number s1, July 2017 S15FIGURE 1
Diagnostic algorithm for Pompe disease with DNA sequencing as part of the NBS laboratory protocol. Modified from the New York Mid-Atlantic Consortium
for Genetic and Newborn Screening Services (NYMAC) Pompe Disease NBS Symposium 2013. a Obtain as a baseline for monitoring response to treatment;
can also be postponed until a definitive diagnosis is obtained. b The diagnosis of LOPD based on enzymatic and molecular analyses remains a clinical
challenge, because these patients by definition will be normal at the time of diagnosis. Patients will need to be followed closely for the development of
clinical signs and symptoms; however, currently, we do not know if all patients with enzymatic and molecular variants suggesting LOPD will actually go
on to develop disease. Patients with low GAA activity and molecular variants in trans previously identified in patients with LOPD will be at very high risk of
developing LOPD, although patients with low GAA activity and molecular variants not previously identified have less certain clinical outcomes. There are
also patients with low GAA activity above the range seen in previous LOPD patients with VUS (not a pseudodeficiency allele) who will need to be followed for
possible LOPD. In situations where greater ambiguity exists, analysis in another tissue, as noted, may help to more clearly delineate the patients’ disease
status. c Includes VUS with borderline low GAA activity.
evaluated when classifying diagnosed In patients with classic IOPD, there Wolff-Parkinson-White can develop
patients. is minimal variability in the clinical over time. The most prominent
phenotype as is seen and expected manifestation in LOPD is progressive
IOPD across the rest of the Pompe disease muscle weakness (neck, trunk,
spectrum.4,12 For most patients, arms, legs, and diaphragm).9,14,15
Throughout this article, classic IOPD follow-up testing will be done by DBS It must be recognized that there
will be used to describe infantile- or lymphocyte/leukocyte assay.13 are other clinical presentations for
onset patients with cardiomyopathy. Few will have a skin biopsy as patients with LOPD.16 Patients with
The signs and symptoms in patients follow-up testing.
LOPD generally have GAA enzyme
with symptom onset at ≤12 months
activity levels that are 2% to 40% of
of age typically include prominent LOPD normal values in skin fibroblasts.9,17,18
cardiac involvement (cardiomegaly/
Although unexpectedly low GAA
hypertrophic cardiomyopathy), In older children and adults (age of
activity levels 12 months),
and hypotonia, delays in motor significant cardiac involvement noted in adults with Pompe disease,17
development, respiratory distress, is usually not observed, and in GAA activity levels >1% generally
feeding problems, and failure to the vast majority, there is no have not been noted in patients with
thrive.4,8–11 Patients with classic cardiac involvement. However, classic IOPD. Some patients have
IOPD have absent or low GAA activity cardiac involvement in the form symptom onset at ≤12 months of age
in DBS or lymphocyte specimens. of cardiac hypertrophy and heart but without cardiomyopathy. These
In skin fibroblast assays, classic rhythm disturbances, such as patients are often referred to as
IOPD patients havediagnosis. Recommendations
for good laboratory practices for
biochemical genetic testing and NBS
have been published by the Centers
for Disease Control and Prevention
and should be consulted.25
Blood Assays
Advances in diagnostic capabilities
and laboratory methodology that
measure GAA activity in blood
(eg, enzyme assays by using DBSs,
purified lymphocytes, and mixed
leukocytes) have been developed.
These assays are less invasive, more
rapid, and easier to standardize
than diagnostic methods measuring
enzymatic activity in other tissues
that have been used in the past.
DBS samples are more stable than
whole-blood samples. Measuring
FIGURE 2 GAA activity in DBS samples is
Diagnostic algorithm for Pompe disease without DNA sequencing as part of the NBS laboratory therefore valuable for the diagnosis
protocol. When pathogenic variants are detected, parental DNA analysis is recommended to confirm of Pompe disease.26 Recent results
presence of variants in trans. a Blood-based assays include DBSs, purified lymphocytes, and mixed have supported its reliability and
leukocyte assay methods. b Obtain as a baseline for monitoring response to treatment; can also
be postponed until a definitive diagnosis is obtained. c Need to ensure assay is being done in a sensitivity.18 Many laboratories offer
laboratory with appropriate enzyme assay experience and capabilities and that the patient has not lymphocyte assay of GAA from whole-
received a blood transfusion or other interventions that would result in normal GAA enzyme levels. blood specimens. The sampling,
shipping, and handling requirements
Cardiac involvement can occur, used to confirm a diagnosis of Pompe of the testing laboratory must be
however, in some of these cases.19 disease. These assays measure followed carefully for these samples.
Hypertrophic cardiomyopathy, GAA enzyme activity in blood, However, enzymatic analysis is not
once thought to be limited only to cultured skin fibroblasts, or muscle sufficient to confirm a diagnosis.2,27–29
patients with classic IOPD is being biopsy specimens.4,18,22–24 Each has A molecular test is needed to confirm
reported in patients who do not fall associated benefits and drawbacks. the diagnosis made by GAA
into the classic IOPD category.20,21 Because availability and standard activity.4,24,26,29,30 It is important
In this article, LOPD will be used for practice are variable across different to note that enzymatic analysis,
all patients diagnosed with Pompe geographic regions, not all methods for example, in lymphocytes, is
disease other than those with classic may be options for all laboratories. generally less specific than results
IOPD and includes patients with Factors that need to be considered obtained with skin fibroblasts, and
onset of symptoms at ≤12 months of when evaluating the results of GAA so there can be a “gray zone” of
age without cardiomyopathy or with enzymatic activity testing include the enzyme activity where one cannot
cardiac involvement (non-classic presence of a pseudodeficiency allele differentiate classic IOPD from LOPD.
infantile-onset disease) and juvenile that can alter the residual enzyme
and adult patients traditionally level in a screened infant and the Skin Fibroblast Assays
classified as LOPD. possibility that the assay conditions Measuring enzyme activity in skin
and procedures may not be optimal, fibroblasts has long been the gold
leading to false-positive results. It is standard for measuring GAA activity.
COMPONENTS OF SECOND-TIER therefore essential that molecular An advantage of the skin fibroblast
CONFIRMATORY TESTING genetic analysis be done to confirm assay is that it provides an accurate
or rule out a diagnosis of Pompe determination of residual enzyme
Enzyme-Based Assays
disease in the presence of low GAA activity. Also, with appropriate
Biochemical assays used to activity and to ensure that treatment informed consent, samples can
demonstrate deficient GAA enzyme with ERT is not started in patients be banked and made available for
activity are the standard methods without a confirmed molecular future use if and when needed.
Downloaded from www.aappublications.org/news by guest on November 4, 2021
PEDIATRICS Volume 140, Number s1, July 2017 S17Skin fibroblasts also allow for Disease Laboratory of the Duke Confirmation of variants in trans
determination of the cross-reactive University Biochemical Genetics requires parental testing to confirm
immunologic material (CRIM) Laboratory. Detailed information the phase of the variants. In cases
status.4,24,26 However, with the about sample requirements, of classic IOPD, prediction of CRIM
availability of molecular testing and testing, and shipping can be found status is possible with genotyping
its ability to predict CRIM status, the at https://pediatrics.duke.edu/ alone in close to 92% cases.4,5,24,37
skin fibroblast assay is not needed divisions/medical-genetics/
The nature and site of the 2 variants
in most cases. Skin biopsy should biochemical-genetics-laboratory/
in the GAA gene typically predict a
only be considered as a second-tier glycogen-storage-disease-laboratory.
patient’s clinical course. Sometimes,
diagnostic tool when indeterminate
only 1 previously identified
results have been obtained by other Non–Enzyme-based Confirmatory
Methods variant is found coupled with a
methods and when a patient has 1
novel, unclassified sequence VUS,
variant and low enzyme activity in
Genotyping/Variant Analysis thus complicating the diagnosis
blood and assay of enzyme activity in
of Pompe disease. More than 400
another tissue is needed to confirm Variant analysis of the GAA gene
pathogenic variants have been
a diagnosis. The time required to is essential for confirming the
identified, in addition to numerous
culture skin fibroblasts to obtain biochemical diagnosis of Pompe
polymorphisms and VUSs in the GAA
enough material for adequate disease. A diagnosis is usually
gene. Pseudodeficiency alleles have
enzyme assay can be 4 to 6 weeks, confirmed by documentation of
also been described. A database
which is too long to allow early 2 known pathogenic variants in
with a list of these variants (the
initiation of treatment in classic IOPD trans identified through full gene
Pompe Center Mutation Database,
and limits the usefulness of this test sequencing. In some instances,
Erasmus Medical Center, Rotterdam,
in the infantile setting. however, only 1 variant is detected
Netherlands) can be found at http://
because the second variant could be
Muscle Biopsy Assays cluster15.erasmusmc.nl/klgn/
a deletion or duplication or could
pompe/mutations.html. Additionally,
GAA activity can also be measured reside in a promoter region or deep
a database listing GAA gene variants
in fresh muscle tissue obtained in the intron. In these cases, it is
(Duke Children’s Hospital and Health
by biopsy. Histologic examination necessary to use other methods
Center) identified in CRIM-negative
may reveal the abnormal glycogen to confirm a diagnosis, such as
patients with Pompe disease is
accumulation that is typical of the measuring GAA enzymatic activity
available at https://pediatrics.duke.
disease. Although it provides reliable in another tissue. This is especially
edu/sites/pediatrics.duke.edu/files/
results, muscle biopsy is no longer important in patients with LOPD.
field/attachments/GAA_mutation_
recommended for confirmatory Although current genetic testing is a
database.pdf. The lists continue to
testing because it is invasive and reliable means for identifying IOPD
expand each year as new variants are
adds unnecessary discomfort for patients, data for patients with LOPD
identified.
patients. The related risks associated or not affected by the disease are
with the need for anesthesia also less certain. Many new genotypes The role of gene sequencing in NBS
must be considered, particularly in are being identified through NBS continues to expand. One laboratory
infants who have classic IOPD.4,24,26 with unclear phenotypes, making it with DBS sequencing capabilities
Because of the variable and difficult to determine the percentage can now have target turnaround
heterogeneous muscle involvement, of cases not correctly identified times (TATs) of 2 to 3 days. Variant
muscle histology cannot be used as a through molecular genetic testing. analysis can be done at the same
sole method of diagnosis, particularly Only close and continued follow-up time as the DBS is retested for GAA
for the juvenile or adult patient will help to resolve these cases. activity measurement to serve
in whom the extent of glycogen Variant analysis is particularly as a second-tier screening test.
accumulation is less and more important in confirmatory testing for Consistency has been demonstrated
variable than in the patient with detecting a pseudodeficiency allele or between measurements of GAA
IOPD.26,29 If muscle biopsies are done, alleles, which can produce misleading activity in DBSs and gene sequencing
it is important that they are handled low levels of GAA activity and lead to results. The latter is important for
by laboratories with experience with false-positive results. Confirmatory identification of pseudodeficiency
Pompe disease and with capabilities testing is especially important among allele(s) and determination of CRIM
to perform appropriate histologic the Asian population in which the status so that appropriate follow-up
analyses and recognize potential GAA pseudodeficiency allele is seen can be planned and unnecessary
hallmarks of the disorder,31–34 at a higher frequency (up to 4%) screening avoided.38 A TAT of 2 to 3
such as the Glycogen Storage compared with white populations.35,36 days for sequencing is necessary to
Downloaded from www.aappublications.org/news by guest on November 4, 2021
S18 BURTON et alavoid delays in initiating treatment values do not exclude a diagnosis of assays. Increases in Hex4 are often
in cases of classic IOPD, for which LOPD39 because ∼5% of patients with noted to precede muscle weakness
hypertrophic cardiomyopathy is LOPD have normal CK levels.22,40,41 and, as such, Hex4 is a sensitive
usually present by birth. A fast TAT marker of disease activity in patients.
is especially important in states Other Serum Enzymes However, careful assessments by
where GAA sequencing is not part AST, ALT, and, to a lesser extent, a trained physical therapist can
of the NBS second-tier test. Parents lactate dehydrogenase serve as often capture subtle changes that
should provide informed consent for serum enzyme biomarkers of muscle are missed by motor milestone
testing because nonpaternity can be damage in Pompe disease. AST assessments. Patients with LOPD
detected through molecular genetic levels typically are higher than those who eventually need treatment tend
testing. Depending on the geographic of ALT. As is true of CK, levels of to have Hex4 levels at the upper level
location, written consent may be these enzymes occasionally may be of normal or above, suggesting that
required. within normal limits in patients with Hex4 levels, combined with other
LOPD.13 laboratory results (such as CK, AST,
Laboratory Analyses and ALT) and the patient’s clinical
In addition to measuring GAA
Urine Hexose Tetrasaccharide picture can be useful in cases of
enzyme activity and gene sequencing, Glc4 has been found to be elevated LOPD.
there are a number of serum enzyme in both urine and plasma in patients
biomarkers that are indicative of with Pompe disease. Measurements
tissue damage, including creatine in urine are more reliable and easier Cardiac Evaluation
kinase (CK), CK-MB, aspartate to obtain for follow-up. Glc4 can
aminotransferase (AST), and alanine also be measured in urine as the Assessments for cardiac
aminotransferase (ALT), and urine total hexose tetrasaccharide (Hex4) involvement (eg, cardiomegaly and
biomarkers, including urine glucose fraction, a breakdown product of cardiomyopathy) are necessary for
tetrasaccharide (Glc4), that can be glycogen. Here, Glc4 measurements diagnosis of patients with classic
helpful when trying to establish a will be reported as Hex4. Hex4 serves IOPD and for distinguishing them
diagnosis in a patient with a positive as a useful biomarker of glycogen from patients with LOPD (including
NBS result for Pompe disease.13,39 storage, which is helpful in assessing non-classic IOPD). Components of
However, clinicians need to bear severity and overall disease burden the cardiac evaluation should include
in mind that although helpful and may be useful as an adjunctive an electrocardiogram (ECG) and
diagnostically, these findings are diagnostic biomarker in infants with echocardiogram (ECHO). Where
not specific to Pompe disease and a positive newborn screen. Levels of cardiac evaluation is not immediately
individually have limited prognostic excretion are higher in infants and available, a chest radiograph
value. those with significant disease burden looking for an enlarged cardiac
and are correlated with muscle shadow may be a useful initial
Serum CK biopsy glycogen content. Hex4 is also evaluation. Although not typically
Serum CK is uniformly elevated useful for monitoring the clinical seen in patients with LOPD, cardiac
in patients with classic IOPD and response to treatment.13,43 Although involvement is starting to be found
in children and juveniles with also elevated in other glycogen more frequently in patients who do
symptomatic Pompe disease and storage disorders, diagnostically, not have classic IOPD.4,45,46 Typical
therefore is an important part of Hex4 is close to 100% sensitive in findings during cardiac evaluation
confirmatory testing. CK elevation identifying patients with classic for Pompe disease are summarized
is a sensitive although nonspecific IOPD. This was confirmed by Chien in Table 1. If cardiomyopathy is
marker of muscle damage.22,40,41 et al44 who reported baseline urinary present at the initial evaluation,
For example, in the NBS setting, CK Hex4 concentrations in patients this confirms the infant has classic
levels may be falsely elevated as a with classic IOPD, LOPD, and a IOPD, and CRIM status should be
result of trauma associated with pseudodeficiency allele identified determined as quickly as possible
birth. A more significant elevation is through NBS in Taiwan. Baseline before initiation of ERT. If the results
usually found in patients with classic Hex4 was not elevated in the LOPD of the cardiac evaluation are normal,
IOPD.42 Elevated CK levels may be and GAA pseudodeficiency cohorts then the diagnosis of LOPD is more
the only manifestation in patients but was elevated in the patients likely, although some patients with
with “asymptomatic” LOPD, but data with classic IOPD. Negative results non-classic IOPD have developed
on the age at which CK levels rise in therefore may be helpful in ruling out cardiomyopathy after several
presymptomatic patients are lacking the diagnosis of classic IOPD when months.19–21 Therefore, patients with
at this time. However, normal CK combined with results of enzyme low enzyme activity will have to be
Downloaded from www.aappublications.org/news by guest on November 4, 2021
PEDIATRICS Volume 140, Number s1, July 2017 S19TABLE 1 Cardiac Evaluations and Findings20,45,46 radiograph, ECG, and ECHO, must
Evaluation Characteristic Findings be performed to detect classic
Classic IOPD LOPD IOPD immediately. Where cardiac
evaluation capabilities are limited,
ECG Large and tall QRS complex Abnormalities, if present, less
severe than in IOPD valuable information can be obtained
Some have short PR interval from just a chest radiograph and
Delta wave due to WPW, or other aberrant ECG. Blood GAA activity needs to be
pathways measured to confirm GAA deficiency.
ECHO Cardiac involvement always detected. However, Cardiac involvement generally
in NBS, LVM/LVMI may be just above the ULN; not detected, and, if present,
therefore, if LVM/ LVMI is normal, but there is is much less severe than that
a high suspicion of involvement, a follow-up seen in IOPD SUMMARY
ECHO is needed
Evidence of significant hypertrophic Cardiomyopathy typically not The objective of the initial patient
cardiomyopathy evident in LOPD but LV/cardiac evaluation after a positive NBS
hypertrophy may be present in result is to establish or refute the
some patients, especially those diagnosis of Pompe disease and to
with non-classic IOPD
categorize patients accurately. Once
Reduced left ventricular function (ie, EF and SF) Increased LVMI
in later stages; hyperdynamic state usually these are established, it is then up to
seen in initial stages the clinicians to determine whether
Increased LVM/LVMI and LVOT obstruction Reduced left ventricular function the patient should be referred for
(ie, EF and SF) as cardiac immediate treatment with ERT or
disease progresses
should go into follow-up without
Chest radiograph Enlarged cardiac shadow Usually normal
Cardiomegaly treatment. Clearly patients with
classic IOPD should be referred
EF, ejection fraction; LVM, left ventricular mass; LVMI, left ventricular mass index; LVOT, left ventricular outflow tract; SF,
systolic fraction; ULN, upper limit of normal; WPW, Wolff-Parkinson-White. for treatment with ERT. All other
patients should be managed
managed closely for onset of cardiac POMPE DIAGNOSTIC ALGORITHM closely, especially by monitoring
involvement. WITHOUT DNA SEQUENCING AS PART the attainment of motor milestones
OF NBS LABORATORY PROTOCOL for any evidence of delays that may
signify the onset of clinical LOPD.
The algorithm in Fig 2 provides
POMPE DISEASE DIAGNOSTIC recommendations for the stepwise
ALGORITHM WITH DNA SEQUENCING AS The Main Challenges
diagnostic evaluation that should
PART OF NBS LABORATORY PROTOCOL
be followed when variant analysis/ The recommendations for
The algorithm in Fig 1 provides DNA sequencing is not available as confirmatory testing and initial
recommendations for the stepwise part of the NBS laboratory protocol. evaluation provided in this article for
diagnostic evaluation that should be For NBS laboratories that do not a broad global audience are based on
followed when mutation analysis/ have DNA sequencing capabilities, the clinical experience and expertise
DNA sequencing is available as part the responsibility for obtaining of the members of the Pompe Disease
of the NBS laboratory protocol. sequencing of the GAA gene will fall Newborn Screening Working Group,
Recommended evaluations based on on the referral center. Typically, the who are involved in the screening
a combination of low GAA activity results of sequencing will not be and care of patients with Pompe
and the presence or absence of GAA available as quickly in this setting. disease and who are all coauthors
variants is provided. When followed, A TAT of 2 to 3 days for sequencing on sections in this NBS guidance
this algorithm will lead to 1 of 3 results is ideal. The clinicians supplement. However, the Working
diagnoses: responsible for the follow-up of Group recognizes that clinical
patients in these settings should practices, standards of care, and
• Classic IOPD consider contacting laboratories resource capabilities vary regionally
• LOPD (for all patients with a that have experience with GAA and by testing centers. Confirmatory
confirmed diagnosis who are not gene sequencing and pathogenic testing recommendations need to be
classified as having classic IOPD, variants to explore faster TATs in in harmony with state or regional
and includes patients with non- light of the crucial need for quick protocols, which are dependent
classic IOPD and with LOPD) results. The diagnosis of classic IOPD on local resources. Also, individual
cannot be delayed by waiting for patient needs and health status must
• No disease/not affected/carrier/ the sequencing result. Therefore, be considered along with local/
pseudodeficiency. cardiac evaluation, ideally by chest regional insurance reimbursement
Downloaded from www.aappublications.org/news by guest on November 4, 2021
S20 BURTON et alprograms and regulations. Therefore, [time of the study]) and Shire of Washington (Seattle, WA); and
the recommendations should serve (Lexington, MA [current affiliation]); Kathryn J. Swoboda, Massachusetts
as guidance rather than as set or Olaf Bodamer, MD, PhD, Boston General Hospital (Boston, MA).
standard practices. Understandably, Children’s Hospital (Boston, MA); We thank Zsuzsanna Devecseri, MD,
adaptations will be needed and Barbara K. Burton, MD, Northwestern MBA, Joan Keutzer, PhD, and Susan E.
expected based on region-specific University Feinberg School of Sparks, MD, PhD, of Sanofi Genzyme
practices and capabilities. Medicine, and Ann & Robert H. Lurie for critical review of the manuscript
Pompe disease is a continuum Children's Hospital (Chicago, IL); and Marianne B. Zajdel of Sanofi
of disease, and our knowledge Debra Day-Salvatore, MD, St Peter’s Genzyme for medical writing support.
about it continues to improve University Hospital (New Brunswick,
as new information becomes NJ); Roberto Giugliani, MD, PhD,
available in both the clinical and Hospital de Clinicas de Porto Alegre ABBREVIATIONS
laboratory settings. Therefore, the and Federal University of Rio Grande
ALT: alanine aminotransferase
recommendations provided here are do Sul (Porto Alegre, Brazil); Wuh-
AST: aspartate aminotransferase
based on our current knowledge and Liang Hwu, MD, PhD, National
CK: creatine kinase
experience. Because we are currently Taiwan University Hospital, and
CRIM: cross-reactive immuno-
in a dynamic phase in the field of NBS National Taiwan University College
logic material
and lysosomal storage disorders, of Medicine (Taipei, Taiwan); Simon
DBS: dried blood spot
recommendations will likely change A. Jones, MBChB, BSc, MRCPCH, St
ECG: electrocardiogram
as we learn more about Pompe Mary’s Hospital, Central Manchester
ECHO: echocardiogram
disease and as technology advances University Hospitals NHS Foundation
ERT: enzyme replacement
and innovations occur over time. Trust, Manchester Academic Health
therapy
Science Centre, University of
Thus, we expect the need to revisit GAA: acid α-glucosidase
and revise these recommendations as Manchester (Manchester, UK); Priya
Glc4: glucose tetrasaccharide
appropriate in the next 3 to 5 years. S. Kishnani, MD, Duke University
Hex4: hexose tetrasaccharide
(Durham, NC); David F. Kronn, MD,
IOPD: infantile-onset Pompe
New York Medical College, Valhalla,
ACKNOWLEDGMENTS disease
NY; Kimitoshi Nakamura, MD, PhD,
LOPD: late-onset Pompe disease
The members of the Pompe Disease Kumamoto University (Kumamoto,
NBS: newborn screening
Newborn Screening Working Group Japan); Torayuki Okuyama, MD, PhD,
TAT: turnaround time
(in alphabetical order) are as follows: National Center for Child Health
VUS: variant of unknown
Andrea Atherton, MS, CGC, Children’s and Development (Tokyo, Japan);
significance
Mercy Hospital (Kansas City, MO C. Ronald Scott, MD, University
FUNDING: Sanofi Genzyme (Cambridge, MA) facilitated and provided financial support for the meeting of the Pompe Disease Newborn Screening Working Group to
discuss and develop the recommendations provided in all articles comprising the “Newborn Screening, Diagnosis, and Treatment for Pompe Disease” guidance
supplement and also paid for editorial and writing support for this supplement. The recommendations and opinions expressed in this article and in all others in
the Supplement are those of the authors based on their clinical expertise and experience and do not necessarily reflect those of Sanofi Genzyme.
POTENTIAL CONFLICT OF INTEREST: Barbara Burton, MD, received funding for clinical trials from Sanofi Genzyme, Biomarin, Shire, Synageva, and Ultragenyx, and
honoraria and/or consulting fees from Sanofi Genzyme, Biomarin, Shire, Hyperion, Alexion, and ReGenX Bio; David Kronn MD, is a member of the speakers bureau
of Sanofi Genzyme, and an investigator for the ADVANCE study, sponsored by Sanofi Genzyme; Wuh-Liang Hwu, MD, PhD, received research grants and consultation
fees from Sanofi Genzyme; and Priya Kishnani, MD, received consulting fees, honoraria, and/or research funding from Sanofi Genzyme, Amicus Therapeutics,
Baebies, Shire Pharmaceuticals, Alexion, and the Lysosomal Disease Network and is a member of the Pompe and Gaucher Disease Registry Advisory Boards for
Sanofi Genzyme.
REFERENCES
1. Kishnani PS, Amartino HM, Lindberg newborn screening is feasible: results and treated since birth. J Pediatr.
C, Miller TM, Wilson A, Keutzer J; from the Taiwan screening program. 2015;166(4):985–991.e2
Pompe Registry Boards of Advisors. Pediatrics. 2008;122(1). Available at: 4. Kishnani PS, Steiner RD, Bali D,
Timing of diagnosis of patients www.pediatrics.org/cgi/content/full/ et al. Pompe disease diagnosis
with Pompe disease: data from the 122/1/e39 and management guideline.
Pompe Registry. Am J Med Genet A. 3. Chien YH, Lee NC, Chen CA, et al. Genet Med. 2006;8(5):
2013;161A(10):2431–2443 Long-term prognosis of patients 267–288
2. Chien YH, Chiang SC, Zhang XK, et al. with infantile-onset Pompe disease 5. Bali DS, Goldstein JL, Banugaria
Early detection of Pompe disease by diagnosed by newborn screening S, et al. Predicting cross-reactive
Downloaded from www.aappublications.org/news by guest on November 4, 2021
PEDIATRICS Volume 140, Number s1, July 2017 S21immunological material (CRIM) status Ploeg AT. Disease severity in children 24. Reuser A, Verheijen F, Kroos M, et al.
in Pompe disease using GAA mutations: and adults with Pompe disease Enzymatic and molecular strategies to
lessons learned from 10 years of related to age and disease duration. diagnose Pompe disease. Expert Opin
clinical laboratory testing experience. Neurology. 2005;64(12):2139–2141 Med Diagn. 2010;4(1):79–89
Am J Med Genet C Semin Med Genet. 15. Winkel LP, Hagemans ML, van Doorn 25. Centers for Disease Control and
2012;160C(1):40–49 PA, et al. The natural course of non- Prevention (CDC). Good laboratory
6. Güngör D, Reuser AJ. How to classic Pompe’s disease; a review practices for biochemical genetic
describe the clinical spectrum in of 225 published cases. J Neurol. testing and newborn screening for
Pompe disease? Am J Med Genet A. 2005;252(8):875–884 inherited metabolic disorders. MMWR
2013;161A(2):399–400 Recomm Rep. 2012;61(RR-2):1–44
16. Schüller A, Wenninger S, Strigl-Pill N,
7. Kishnani PS, Beckemeyer AA, Schoser B. Toward deconstructing 26. Winchester B, Bali D, Bodamer OA,
Mendelsohn NJ. The new era of Pompe the phenotype of late-onset Pompe et al; Pompe Disease Diagnostic
disease: advances in the detection, disease. Am J Med Genet C Semin Med Working Group. Methods for a prompt
understanding of the phenotypic Genet. 2012;160C(1):80–88 and reliable laboratory diagnosis
spectrum, pathophysiology, and of Pompe disease: report from an
17. Bali DS, Tolun AA, Goldstein JL, Dai
management. Am J Med Genet C Semin international consensus meeting. Mol
J, Kishnani PS. Molecular analysis
Med Genet. 2012;160C(1):1–7 Genet Metab. 2008;93(3):275–281
and protein processing in late-onset
8. Kishnani PS, Howell RR. Pompe disease Pompe disease patients with low levels 27. Chamoles NA, Niizawa G, Blanco M,
in infants and children. J Pediatr. of acid α-glucosidase activity. Muscle Gaggioli D, Casentini C. Glycogen
2004;144(suppl 5):S35–S43 Nerve. 2011;43(5):665–670 storage disease type II: enzymatic
screening in dried blood spots
9. Hirschhorn R, Reuser AJJ. 18. Kallwass H, Carr C, Gerrein J, et al.
on filter paper. Clin Chim Acta.
Glycogen storage disease type II: Rapid diagnosis of late-onset Pompe
2004;347(1–2):97–102
acid α-glucosidase (acid maltase disease by fluorometric assay of
deficiency). In: Scriver CR, Beaudet AL, alpha-glucosidase activities in 28. Zhang H, Kallwass H, Young SP, et al.
Sly WS, et al, eds. The Metabolic and dried blood spots. Mol Genet Metab. Comparison of maltose and
Molecular Bases of Inherited Disease. 2007;90(4):449–452 acarbose as inhibitors of maltase-
8th ed. New York, NY: McGraw-Hill; 2001 glucoamylase activity in assaying
19. Leslie N, Tinkle BT. Glycogen storage
acid alpha-glucosidase activity in
10. Kishnani PS, Hwu WL, Mandel H, disease type II (Pompe disease). In:
dried blood spots for the diagnosis of
Nicolino M, Yong F, Corzo D; Infantile- Pagon RA, Adam MP, Ardinger HH, et al,
infantile Pompe disease. Genet Med.
Onset Pompe Disease Natural eds. GeneReviews [Internet]. Seattle,
2006;8(5):302–306
History Study Group. A retrospective, WA: University of Washington, Seattle;
multinational, multicenter study 2007:1993–2015. Available at: www. 29. Vissing J, Lukacs Z, Straub V. Diagnosis
on the natural history of infantile- ncbi.nlm.nih.gov/books/NBK1261/. of Pompe disease: muscle biopsy vs
onset Pompe disease. J Pediatr. Updated May 9, 2013 blood-based assays. JAMA Neurol.
2006;148(5):671–676 2013;70(7):923–927
20. Lee DH, Qiu WJ, Lee J, Chien YH, Hwu
11. van den Hout HM, Hop W, van WL. Hypertrophic cardiomyopathy in 30. American Association of
Diggelen OP, et al. The natural course pompe disease is not limited to the Neuromuscular & Electrodiagnostic
of infantile Pompe’s disease: 20 classic infantile-onset phenotype. Medicine. Diagnostic criteria for
original cases compared with 133 JIMD Rep. 2014;17:71–75 late-onset (childhood and adult)
cases from the literature. Pediatrics. Pompe disease. Muscle Nerve.
21. Montagnese F, Barca E, Musumeci O,
2003;112(2):332–340 2009;40(1):149–160
et al. Clinical and molecular aspects
12. Smith WE, Sullivan-Saarela JA, Li JS, of 30 patients with late-onset Pompe 31. Anderson G, Smith VV, Malone M,
et al. Sibling phenotype concordance disease (LOPD): unusual features Sebire NJ. Blood film examination
in classical infantile Pompe and response to treatment. J Neurol. for vacuolated lymphocytes in the
disease. Am J Med Genet A. 2015;262(4):968–978 diagnosis of metabolic disorders;
2007;143A(21):2493–2501 retrospective experience of more than
22. Ausems MG, Lochman P, van Diggelen
13. Wang RY, Bodamer OA, Watson MS, OP, Ploos van Amstel HK, Reuser AJ, 2,500 cases from a single centre. J Clin
Wilcox WR; ACMG Work Group on Wokke JH. A diagnostic protocol for Pathol. 2005;58(12):1305–1310
Diagnostic Confirmation of Lysosomal adult-onset glycogen storage disease 32. Hagemans ML, Stigter RL, van Capelle
Storage Diseases. Lysosomal type II. Neurology. 1999;52(4):851–853 CI, et al. PAS-positive lymphocyte
storage diseases: diagnostic vacuoles can be used as diagnostic
23. Goldstein JL, Young SP, Changela M,
confirmation and management of screening test for Pompe disease. J
et al. Screening for Pompe disease
presymptomatic individuals. Genet Inherit Metab Dis. 2010;33(2):133–139
using a rapid dried blood spot
Med. 2011;13(5):457–484
method: experience of a clinical 33. Tsuburaya RS, Monma K, Oya Y, et al.
14. Hagemans ML, Winkel LP, Hop WC, diagnostic laboratory. Muscle Nerve. Acid phosphatase-positive globular
Reuser AJ, Van Doorn PA, Van der 2009;40(1):32–36 inclusions is a good diagnostic
Downloaded from www.aappublications.org/news by guest on November 4, 2021
S22 BURTON et almarker for two patients with adult- storage disease type II. J Inherit Metab 42. Bodamer OA, Hung CY. The diagnostic
onset Pompe disease lacking disease Dis. 2008;31(suppl 2):S261–S265 path to Pompe disease. Eur Neurol Rev.
specific pathology. Neuromuscul 38. Goldstein JL, Dickerson G, Kishnani 2014;9(1):83–86
Disord. 2012;22(5):389–393 PS, Rehder C, Bali DS. Blood-based 43. Young SP, Piraud M, Goldstein JL, et al.
34. Kishnani PS, Amartino HM, Lindberg C, diagnostic testing for Pompe disease: Assessing disease severity in Pompe
Miller TM, Wilson A, Keutzer J. Methods consistency between GAA enzyme disease: the roles of a urinary glucose
of diagnosis of patients with Pompe activity in dried blood spots and GAA tetrasaccharide biomarker and
disease: data from the Pompe Registry. gene sequencing results. Muscle imaging techniques. Am J Med Genet C
Mol Genet Metab. 2014;113(1–2):84–91 Nerve. 2014;49(5):775–776 Semin Med Genet. 2012;160C(1):50–58
35. Kumamoto S, Katafuchi T, Nakamura 39. Toscano A, Montagnese F, Musumeci 44. Chien YH, Goldstein JL, Hwu WL,
K, et al. High frequency of acid O. Early is better? A new algorithm et al. Baseline urinary glucose
alpha-glucosidase pseudodeficiency for early diagnosis in late onset tetrasaccharide concentrations in
complicates newborn screening for Pompe disease (LOPD). Acta Myol. patients with infantile- and late-onset
glycogen storage disease type II in the 2013;32(2):78–81 Pompe disease identified by newborn
Japanese population. Mol Genet Metab. 40. Drummond LM. Creatine screening. JIMD Rep. 2015;19:67–73
2009;97(3):190–195 phosphokinase levels in the newborn 45. Forsha D, Li JS, Smith PB, van der
36. Shigeto S, Katafuchi T, Okada Y, et al. and their use in screening for Ploeg AT, Kishnani P, Pasquali SK; Late-
Improved assay for differential Duchenne muscular dystrophy. Arch Onset Treatment Study Investigators.
diagnosis between Pompe disease and Dis Child. 1979;54(5):362–366 Cardiovascular abnormalities in late-
acid α-glucosidase pseudodeficiency 41. Moat SJ, Bradley DM, Salmon R, Clarke onset Pompe disease and response to
on dried blood spots. Mol Genet Metab. A, Hartley L. Newborn bloodspot enzyme replacement therapy. Genet
2011;103(1):12–17 screening for Duchenne muscular Med. 2011;13(7):625–631
37. Joshi PR, Gläser D, Schmidt S, et al. dystrophy: 21 years experience 46. Limongelli G, Fratta F. S1.4
Molecular diagnosis of German in Wales (UK). Eur J Hum Genet. Cardiovascular involvement in Pompe
patients with late-onset glycogen 2013;21(10):1049–1053 disease. Acta Myol. 2011;30(3):202–203
Downloaded from www.aappublications.org/news by guest on November 4, 2021
PEDIATRICS Volume 140, Number s1, July 2017 S23The Initial Evaluation of Patients After Positive Newborn Screening:
Recommended Algorithms Leading to a Confirmed Diagnosis of Pompe Disease
Barbara K. Burton, David F. Kronn, Wuh-Liang Hwu, Priya S. Kishnani and on behalf
of the Pompe Disease Newborn Screening Working Group
Pediatrics 2017;140;S14
DOI: 10.1542/peds.2016-0280D
Updated Information & including high resolution figures, can be found at:
Services http://pediatrics.aappublications.org/content/140/Supplement_1/S14
References This article cites 44 articles, 5 of which you can access for free at:
http://pediatrics.aappublications.org/content/140/Supplement_1/S14#
BIBL
Permissions & Licensing Information about reproducing this article in parts (figures, tables) or
in its entirety can be found online at:
http://www.aappublications.org/site/misc/Permissions.xhtml
Reprints Information about ordering reprints can be found online:
http://www.aappublications.org/site/misc/reprints.xhtml
Downloaded from www.aappublications.org/news by guest on November 4, 2021The Initial Evaluation of Patients After Positive Newborn Screening:
Recommended Algorithms Leading to a Confirmed Diagnosis of Pompe Disease
Barbara K. Burton, David F. Kronn, Wuh-Liang Hwu, Priya S. Kishnani and on behalf
of the Pompe Disease Newborn Screening Working Group
Pediatrics 2017;140;S14
DOI: 10.1542/peds.2016-0280D
The online version of this article, along with updated information and services, is
located on the World Wide Web at:
http://pediatrics.aappublications.org/content/140/Supplement_1/S14
Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it
has been published continuously since 1948. Pediatrics is owned, published, and trademarked by
the American Academy of Pediatrics, 345 Park Avenue, Itasca, Illinois, 60143. Copyright © 2017
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.
Downloaded from www.aappublications.org/news by guest on November 4, 2021You can also read

















































