Computational Module I - UZH Chemistry
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Computational Module I
Tutorial day-3
Department of Chemistry
University of Zurich, Switzerland, 2020
What is thermochemistry?
Frequency calculations
Conformational analysis
Modeling chemistry in solution (PCM)
What is thermochemistry?
Thermochemistry is the study of energy changes involved in chemical reactions.
Thermochemistry is used to predict whether a reaction is spontaneous or
non-spontaneous, favorable or unfavorable.
Thermodynamic Terms:
• Chemical system: open (energy and matter can move in or out), closed (only
energy can move in or out), or isolated (neither energy or matter can move in or
out)
• Reaction: exothermic (energy is released) or endothermic (energy is absorbed)
• Heat Capacity (cal/K): the amount of heat needed to raise the temperature of one
kilogram of mass by one kelvin.
• Enthalpy H (cal): a thermodynamic quantity equivalent to the total heat content of
a system. ∆H of a process is equivalent to its heat change at a constant pressure
(H = U + P*V)
• Entropy S (cal/K): a thermodynamic function, which can be viewed as a measure
of randomness or disorder, and describes the number of arrangements (position
and/or energy levels) that are available to a system existing in a given state.
• Gibbs free energy G (cal): it has no physical reality as a property of matter - the
sign of ∆G indicates the direction of a chemical reaction and determines if a
reaction is spontaneous or not (G = H - T*S)
1 / 15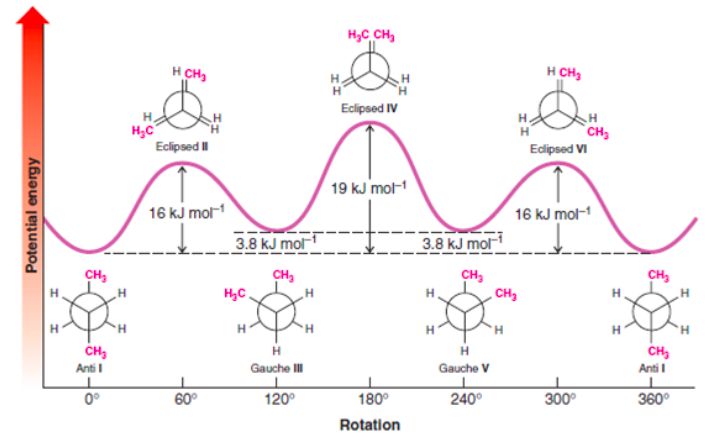
Examples
Figure: Exothermic reaction Figure: Exothermic process: rust
Figure: Endothermic reaction
Figure: Endothermic process: cooking an egg
2 / 15
Example of Gaussian energy output
Figure: Energy values obtained in output from a Gaussian calculation
• The zero-point energy (ZPE) is a correction to the electronic energy of the
molecule to account for the effects of molecular vibrations, that are present even
at 0 K. Final predicted energies must always include a scaled zero-point or
thermal energy correction.
• The thermal correction to the Energy includes the effects of molecular translation,
rotation and vibration at the specified temperature and pressure. It already
includes the zero-point energy: E = Eelectr + ZPE + Evib + Erot + Etrans
3 / 15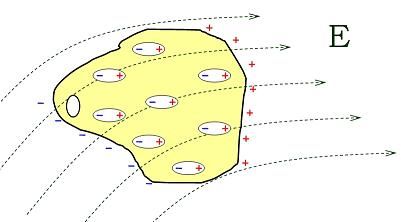
Frequency calculations
• Frequency calculation can provide many info: IR/Raman spectra, force constants,
stationary points, thermodynamic quantities (enthalpy, entropy, etc)
• A frequency calculation has to be performed on a previously optimised structure
(use the same basis-set)
• By default, the analysis is carried at 298.15 K and 1 atmosphere of pressure,
using the principal isotope for each element type
• Scaling factors are needed to eliminate known systematic errors in the calculated
frequencies (the scaling factors are basis-set dependent)
Figure: Recommended scaling factors for frequencies and zero-point energies, for different
calculation types
4 / 15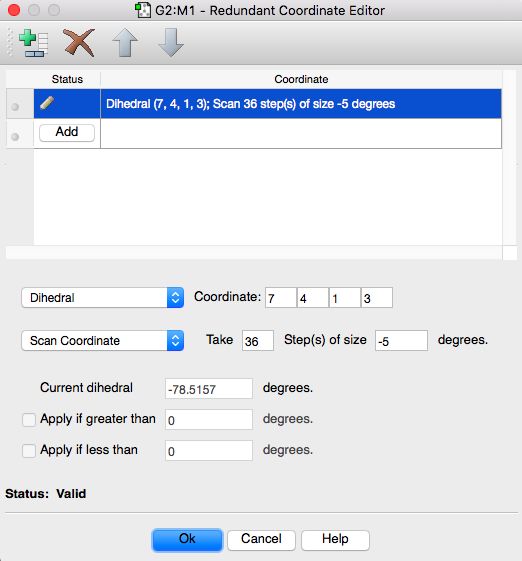
Stationary points I
• A stationary point can be characterised, in the output, from:
• the number of imaginary frequencies
• the normal mode corresponding to the imaginary frequency
• If any of the frequency values are less than zero, these frequencies are known as
imaginary frequencies
• By definition, a structure which has n imaginary frequencies is an nth order saddle
point. Thus, the minimum will have zero imaginary frequencies, and an ordinary
transition structure (TS) will have one imaginary frequency since it is a first order
saddle point
• Be careful: the found TS may not be related to the reactants and products of
interest. The nature of the saddle point can be understood looking at the normal
mode corresponding to the imaginary frequency (animated vibrations; IRC)
5 / 15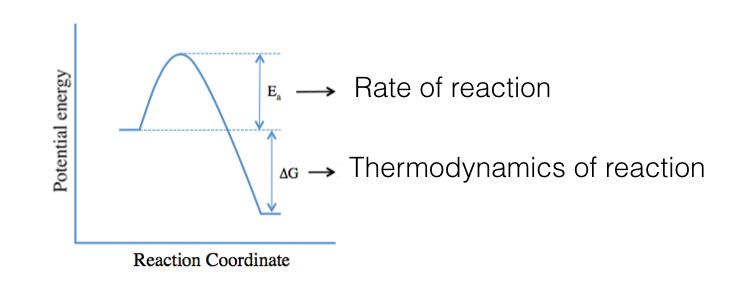
Stationary points II
6 / 15Conformational Analysis I - SCAN
• SCAN: PES scans allow you to explore a region of the potential energy surface,
corresponding to the process in which you are interested. PES scans do not
include a geometry optimisation
• Rigid SCAN: all coordinates are kept frozen, except for the particular coordinate
being scanned. A single point energy calculation is performed for each generated
structure
• Relaxed SCAN: the scan coordinates are kept frozen, while the others are
optimized. Each optimization locates the minimum energy geometry with the
scanned parameters set to specific values.
• SCAN calculations provide insights into the structure of the PES, but they do not
define the lowest energy path between two structures, that need to be obtained
from intrinsic reaction coordinate (IRC) calculations
7 / 15Conformational Analysis: examples
Figure: Conformation analysis of Butane
8 / 15Intrinsic Reaction Coordinate (IRC)
• More precise method to determine which points on a potential energy surface
(PES) are connected by a certain transition structure (TS)
• An IRC calculation starts at the saddle point and follows the path in both directions
from the TS, optimizing the geometry of the system along the way. In this way two
minima on the PES are surely connected by a path passing through the TS
• Be careful: two minima on the PES can have more than one reaction paths that
connects them, with different TS through which the reaction evolves
• Once you have understood which minima the TS connects, you can go on
calculating the activation energy of the reaction, comparing the (zero-point
corrected) energies of the reactants and of the TS
• In Gaussian, the following types of calculations have to be done, in order:
optimize -> frequency -> IRC
9 / 15Modelling systems in solution
• System: solution (a solute is a substance dissolved in another substance, known
as a solvent)
• As in classical electrostatics (P is the polarization function of the medium, is the
permittivity):
−1
P= E
4π
• Electric polarization: slight relative shift of positive and negative electric charge in
opposite directions within an insulator, or dielectric, induced by an external electric
field
10 / 15Quantum Mechanical Continuum Solvation Models
• Continuum model: model in which many degrees of freedom of the constituent
particles are described in a continuous way (usually with a distribution function)
• Focused model: focused part + remainder -> there is no need to get a detailed
description of the solvent - a good description of the interaction is enough
• Model: solute (one or more molecules in a cavity) + solvent (mimicked by a
continuous dielectric medium with dielectric constant )
• Cavity: it should exclude the solvent and contain within its boundaries the largest
possible part of the solute charge distribution
• The interaction between solute/solvent is mainly electrostatic (mutual
polarization), formulated mathematically in terms of apparent charges at the
solute/solvent interface (electrostatic interaction solved self-consistently)
11 / 15The electrostatic problem
We are looking for the solution of a classical electrostatic problem (Poisson), within a
QM framework.
The charge distribution ρM of the solute, inside the cavity, polarizes the dielectric
continuum, which in turn polarizes the solute charge distribution (self-consistent
process). The general Poisson equation:
−∇[ε∇V (r )] = 4πρM (r )
can be simplified to:
−∇2 V (r ) = 4πρM (r ) within C
−∇2 V (r ) = 0 outside C
where C is the portion of space occupied by the cavity. V is the sum of the electrostatic
potential generated by the charge distribution and the reaction potential generated by
the polarization of the dielectric medium.
12 / 15Solutions of the electrostatic problem
Self-consistent reaction fields methods (SCRF)
• Self-consistent reaction fields (SCRF) methods have different approaches with
different definition of the cavity and of the reaction field
• Solutions implemented in Gaussian:
• The Onsager model is the simplest one
• The iso density PCM defines the cavity as a surface at constant electronic density
• The self-consistent Isodensity polarized continuum model includes the effect of
solvation, accounting for the full coupling between the cavity and the electron
density
13 / 15Integral Equation Formalism: IEF PCM (1997)
• IEFPCM is the default PCM formulation in Gaussian (from Gaussian G03)
14 / 15Bibliography I
James B. Foresman and Aeleen Frisch
Exploring Chemistry with Electronic Structure Methods - Second Edition
Gaussian, 1993
James B. Foresman and Aeleen Frisch
Exploring Chemistry with Electronic Structure Methods - Third Edition
Gaussian, 2015
Tomasi, J., Mennucci, B. and Cammi, R.
Quantum Mechanical Continuum Solvation Models
Chemical Reviews, 105(8):2999–3094, 2005
Alecu, I. M. and Zheng, Jingjing and Zhao, Yan and Truhlar, Donald G.
Computational Thermochemistry: Scale Factor Databases and Scale Factors for
Vibrational Frequencies Obtained from Electronic Model Chemistries
Journal of Chemical Theory and Computation, 6(9):287–2887, 2010
15 / 15You can also read

















































