FISH and chips: the recipe for improved prognostication and outcomes for children with medulloblastoma
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Cancer Genetics 204 (2011) 577e588
REVIEW
FISH and chips: the recipe for improved
prognostication and outcomes for children
with medulloblastoma
Vijay Ramaswamy, Paul A. Northcott, Michael D. Taylor*
Developmental and Stem Cell Biology and Division of Neurosurgery, Arthur and Sonia Labatt Brain Tumour Research Centre,
The Hospital for Sick Children, Toronto, Ontario, Canada
Rapidly evolving genomic technologies have permitted progressively detailed studies of medul-
loblastoma biology in recent years. These data have increased our understanding of the molec-
ular pathogenesis of medulloblastoma, identified prognostic markers, and suggested future
avenues for targeted therapy. Although current randomized trials are still stratified based largely
on clinical variables, the use of molecular markers is approaching routine use in the clinic. In
particular, integrated genomics has uncovered that medulloblastoma comprises four distinct
molecular and clinical variants: WNT, sonic hedgehog (SHH), group 3, and group 4. Children with
WNT medulloblastoma have improved survival, whereas those with group 3 medulloblastoma
have a dismal prognosis. Additionally, integrated genomics has shown that adult medulloblas-
toma is molecularly and clinically distinct from the childhood variants. Prognostic and predictive
markers identified by genomics should drive changes in stratification of treatment protocols for
medulloblastoma patients on clinical trials once they can be demonstrated to be reliable, repro-
ducible, and practical. Cases with excellent prognoses (WNT cases) should be considered for
therapy de-escalation, whereas those with bleak prognoses (group 3 cases) should be prioritized
for experimental therapy. In this review, we will summarize the genomic data published over the
past decade and attempt to interpret its prognostic significance, relevance to the clinic, and use in
upcoming clinical trials.
Keywords Medulloblastoma, genomics, subgroups, copy-number alterations, prognosis
ª 2011 Elsevier Inc. All rights reserved.
Medulloblastoma is the most common malignant brain tumor children with average-risk medulloblastoma (comprising
in children, comprising approximately 13% of all pediatric children over the age of 3 y with residual primary-site tumor
brain tumors and 1.8% of young adult (ages 20e34 y) brain under 1.5 cm in diameter and no dissemination of disease
tumors (1). Outcomes after multimodal therapy are variable beyond the primary site) have an event-free survival rate of
and current treatment is based on a scheme of clinical risk up to 85% after receiving a combination of reduced-dose
stratification. Current risk stratification consists of i) extent of craniospinal irradiation (2340 cGy), posterior fossa or tumor
resection, ii) dissemination beyond the primary site, and iii) bed boost (5400e5580 cGy), and adjuvant chemotherapy
age less than 3 years; where presence of any of these three (5,6). Post-irradiation adjuvant chemotherapy is primarily
criteria is classified as high risk, and the remainder as cisplatin-based combination therapy with similar outcomes
average or standard risk (2). Some experts have suggested across various regimens, however pre-irradiation chemo-
that the large-cell/anaplastic histological variant of medullo- therapy resulting in delayed radiation may have a negative
blastoma should also be used as a marker of high risk effect on outcome (7). In patients with high risk disease,
disease (3,4). According to this risk-stratification scheme, 5-year event-free survival is approximately 70% with
a combination of high dose craniospinal irradiation (3600
cGy), posterior fossa or tumor bed boost (5400e5580 cGy),
Received October 14, 2011; received in revised form November and cisplatin-based adjuvant chemotherapy (4,8). Patients
3, 2011; accepted November 7, 2011. with frank disseminated disease receiving 2340 cGy cranio-
* Corresponding author. spinal irradiation have a dismal 5-year survival rate of below
E-mail address: mdtaylor@sickkids.ca 50% (5,7). Children under the age of 3 years are treated with
2210-7762/$ - see front matter ª 2011 Elsevier Inc. All rights reserved.
doi:10.1016/j.cancergen.2011.11.001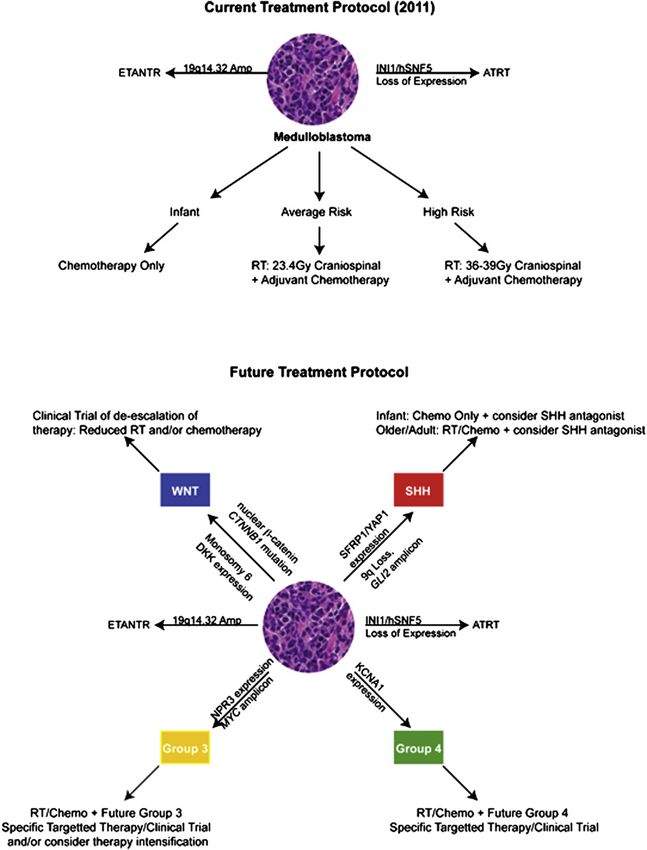
578 V. Ramaswamy et al.
surgery followed by intensive chemotherapy-only regimens specifically the identification of distinct subgroups of medul-
that frequently include high dose systemic methotrexate or loblastoma with distinct biology and clinical outcomes
intraventricular methotrexate, and often receive high dose (22e32). This review will attempt to summarize the tremen-
chemotherapy with autologous stem cell support (9e12). dous advances in the genomics of medulloblastoma over the
Children under the age of 3 years have event-free survivals past 20 years, focusing on the clinical application of inte-
of 38e58% with overall survival of 60e73%; however, infants grated genomic approaches.
with desmoplastic histology have excellent survival rates of
up to 90% with surgery and chemotherapy alone (10,12,13).
A group particularly prone to the side effects of radiation Early karyotyping, identification of common
comprises children between the ages of 3 and 5 years, and in chromosomal aberrations
many instances they are treated with chemotherapy-only
approaches (9). This current system of risk stratification Initial efforts focused on karyotyping of medulloblastoma,
along with advances in radiation and chemotherapy has with the first reports in 1965 identifying various chromosomal
resulted in significant improvements in survival over the past duplications and extra chromosomal DNA fragments (double
30 years, however significant challenges still exist (14). minutes) in patients with medulloblastoma (33,34). Over the
Although subsets of patients with medulloblastoma have next 20 years, advances in karyotyping and Giemsa-banding
excellent outcomes, there are significant neurocognitive and led to the observation that in cultured medulloblastoma cell
endocrine side effects as a result of therapy, particularly in lines, chromosomal aberrations were a common occurrence
those children under the age of 7 years (15). The current (35e37). The first insights into the potential clinical applica-
system of staging assumes all patients with medulloblastoma tion of profiling chromosomal aberrations were reported in
have similar biological behaviors, and as such, many children 1987, in which a report of 4 children identified aneuploidy as
with standard-risk medulloblastoma and excellent outcomes a marker of poor prognosis (38). Subsequent karyotype
may currently be over treated. Conversely, the 15% of studies of primary medulloblastomas identified other
patients with standard risk medulloblastoma who relapse common chromosomal aberrations such as deletion of 6q,
may be undertreated during the up-front treatment phase. deletion of 16q, isochromosome 17q, and deletion of chro-
Furthermore, a subset of patients with high risk medullo- mosome 17p (39e44). The presence of the TP53 gene on
blastoma may be over-treated with high dose craniospinal the short arm of chromosome 17 suggested that this finding
irradiation, leading to significant neurocognitive and endo- was not insignificant, and later studies have suggested that
crine impairment. loss of 17p and isochromosome 17q may be negative
The 2007 World Health Organization Classification of prognostic markers (45e48). However, the prognostic effects
Tumors of the Central Nervous System lists five major of a TP53 mutation remain controversial today (49,50).
histological variants consisting of the classic histology,
medulloblastoma with extensive nodularity (MBEN), des-
moplastic, large-cell and anaplastic (16). Histological/ Early identification of putative oncogenes and
morphological classification schemes comprise three main insights from familial syndromes
histological subgroups, specifically desmoplastic/MBEN,
large-cell/anaplastic and classic histology (17). The des- Oncogenes such as MYC and ABL have been identified as
moplastic/MBEN histological subgroup tends to have an markers of poor outcome in other cancers such as leukemia,
improved prognosis in young children, whereas the large- where cytogenic markers are used routinely in risk stratifi-
cell/anaplastic histological subgroup has a worse prog- cation (21). A focus on chromosome 17 in medulloblastoma
nosis overall. Specifically, patients under age 4 years with led to the identification of other oncogenes, such as ERBB2
desmoplastic/MBEN histology have excellent outcomes and ABR, with expression of the ERBB2 gene being asso-
approaching 90% with chemotherapy alone; however, this ciated with reduced survival (51e53). Amplification of the
survival benefit does not extend to adults with desmoplastic MYC oncogene was identified in medulloblastoma cell lines
histology (13,18e20). However, histological subclassifica- and in a subset of primary tumors, possibly correlating with
tion suffers from several drawbacks. First, approximately poor outcome (53e56). Further studies have implicated
80% of medulloblastomas exhibit classic histology, and as amplification of the MYC gene in up to 8% of medulloblas-
such, a considerable proportion of classic histology medul- tomas, and more-recent studies have confirmed the poor
loblastomas has a poor prognosis. Specifically, patients with prognostic significance of amplification of the MYC gene
metastases on presentation are treated with more intensive (57,58). Putative markers of good clinical outcome, specifi-
chemotherapy regardless of pathology (17). Second, cases cally the neurotrophic receptor TrkC, were also identified
of clear-cut anaplasia have poor prognoses; however, there during this period; however, subsequent studies failed to
are considerable discrepancies in the determination of the confirm its significance (4,19,59e62). These early studies
extent of anaplasia in a classic histology patient, along with suggested that chromosomal abnormalities were common
considerable inter-observer variability. and oncogene amplifications existed in medulloblastoma.
As a result of the prognostic uncertainties underlying Insight from familial cancer syndromes has also identified
clinical risk stratification, there has been considerable interest genes and pathways important in the pathogenesis of
in developing molecular or cytogenic markers of prognosis, medulloblastoma. In Turcot syndrome type 2, 20% of patients
similar to risk-stratification schemes used in childhood develop colorectal carcinoma and a smaller percentage
leukemia (21). Over the past 10 years, immense knowledge develops medulloblastoma. These patients harbor germline
into the biological pathways promoting medulloblastoma has mutations in a component of the Wnt signaling pathway, the
been gained through integrated genomic approaches, Adenomatous polyposis coli (APC ) gene, which normallyMedulloblastoma genomics 579
promotes cytoplasmic b-catenin degradation (63,64). A worse outcome (71). The frequent involvement of chromo-
failure to degrade cytoplasmic b-catenin allows its entry into somes 7 and 17 was also noted in a subsequent study of 19
the nucleus, thus promoting specific gene expression. Further patients, which also suggested that high level amplification of
studies of mutations of the CTNNB1 gene (encoding b- MYCN and MYC were present in 20% of cases (44).
catenin) have identified that it is frequently associated with Although these early studies using CGH had limited resolu-
monosomy 6 and that it is a common but not essential feature tion, they suggested that genomic imbalances were common
of Wnt-activated medulloblastomas. Prognostically patients in medulloblastoma and that common areas of genomic
with becatenin mutations and activation of the Wnt pathway instability may contain oncogenes contributing to the patho-
have improved overall outcome. Several studies have genesis of the tumor.
confirmed that nuclear immunopositivity for becatenin
confers a considerable survival advantage (8,17,65).
Because immunohistochemistry to detect nuclear b-catenin, Initial expression array studies
and interphase fluorescence in situ hybridization (FISH) for
monosomy 6 use techniques available in most modern The emergence of microarray (chip) technology in the 1990s
neuropathology labs, they are entering routine use for the allowed the comprehensive oligonucleotide expressionearray
study of medulloblastoma cases in many centers in North profiling of large numbers of primary tumor samples. Based on
America and Europe. the successful application of oligonucleotide expression array
Gorlin syndrome (Nevoid basal-cell carcinoma syndrome) profiling in distinguishing acute lymphoblastic leukemia from
patients have mutations in the patched homologue 1 gene acute myeloid leukemia and the observation that metastatic
(PTCH1) in about 85% of cases, which lead to overactivity of melanoma could be distinguished from non-metastatic lesions,
the sonic hedgehog (SHH) signaling pathway (63). Patients efforts were undertaken to apply oligonucleotide expression
with Gorlin syndrome have a 3e5% lifetime risk of devel- profiling to medulloblastoma (72,73). The first study using
oping medulloblastoma, where the majority of cases are of expression profiling of medulloblastoma compared 10 meta-
the desmoplastic histology and occur in young children, and static primary tumors to 13 non-metastatic primary tumors and
the outcome is favorable. Seminal insights linking Gorlin found that expression of 85 genes differed significantly
syndrome to SHH signaling have resulted in a profusion of between the two, and that M0 and Mþ dissemination status
papers on SHH signaling in both cerebellar development and could be assigned based on expression profiling to an accu-
medulloblastoma, and the recognition that one of the four racy of 72% (74). This study also suggested the significant
subgroups of medulloblastoma is driven by SHH signaling. overexpression of platelet-derived growth factor receptor
Germline mutations in the SUFU gene, the downstream SHH a (PDGFRA) in metastatic versus non-metastatic tumors. An
signaling pathway mediator, have been reported in a number independent analysis of 27 different primary tumors, and re-
of infants with medulloblastoma, further strengthening the analysis of the oligonucleotide probe set used, suggested it
suggestion that medulloblastoma and SHH signaling are was platelet-derived growth factor receptor b, rather than
linked (66e69). PDGFRA, that was overexpressed in metastatic tumors (75).
Finally, the LieFraumeni syndrome is associated with A subsequent gene-expression profiling study by Pomeroy
inactivating germline mutations in the TP53 gene where up to et al. confirmed that PDGRFA, genes associated with MYC
5% of affected patients develop embryonal tumors (including and ERBB2, and other genes correlated with metastases in
medulloblastoma) and choroid plexus carcinoma (29). There both studies (76). Taken together, these three studies sug-
exists contradictory data concerning the role of mutation of gested a role for platelet-derived growth factor signaling in
TP53 as an independent marker of prognosis in sporadic metastatic medulloblastoma.
medulloblastoma, and as such its role in risk stratification The first expression array study to attempt to subclassify
remains unclear (49,50). medulloblastoma was a study by Pomeroy et al. where the
gene expression profiles of 99 primary medulloblastomas
were analyzed using hierarchical clustering. This study
Initial comparative genomic hybridization showed that desmoplastic medulloblastoma was molecularly
studies distinct from classic medulloblastoma and had differential
expression of the SHH receptor PTCH as well as differential
Although early studies identified areas of chromosomal expression of downstream targets of the SHH pathway. This
aberrations in medulloblastoma, specifically on chromosome suggested that, similar to desmoplastic medulloblastoma in
17, they were limited by poor resolution. The advent of Gorlin syndrome, sporadic desmoplastic medulloblastoma
comparative genomic hybridization (CGH) in the 1990s was characterized by activation of the SHH pathway (77).
allowed the identification of chromosomal aberrations across Although an unsupervised approach could not distinguish
the entire genome with higher resolution than that of Giemsa- biologically distinct subtypes of tumors or response to
banding. Reardon et al. conducted the first study of CGH in therapy, a learned approach using an eight-gene model was
medulloblastoma at St. Jude’s Children’s Research Hospital. able to accurately predict outcome with high accuracy, and
This study of 27 medulloblastomas revealed several genomic furthermore was more accurate than clinical based prediction
abnormalities: specifically, frequent losses on chromosomes models based on M status. Furthermore, this study also
10q, 16q, and 8p, frequent low-level gains on chromosomes showed that medulloblastoma was molecularly distinct from
17q and 7, and the presence of isochromosomes 17q (70). the atypical teratoid/rhabdoid tumors (AT/RT) characterized
A subsequent smaller study of six patients revealed frequent by mutations in SMARCB1/INI1/hSNF5 and supratentorial
gains on chromosomes 2, 7, and 17, and suggested that primitive neuroectodermal tumors, two other embryonal
higher numbers of chromosomal changes correlated with tumors (78). Subsequently, array CGH studies have been580 V. Ramaswamy et al.
used to define another distinct embryonal tumor termed the histology alone. Re-analysis of the Pomeroy et al. expression
embryonal tumor with abundant neuropil and true rosettes study from 2002 revealed that the genes associated with
(ETANTR, previously referred to as ependymoblastoma), treatment failure positively correlate with those up-regulated
which is defined by a focal amplification of a microRNA in group 3 (23,77). The fourth subgroup, renamed group 4
cluster at the 19q14.32 locus (79,80). Collectively, this (Table 1: group D in Northcott et al., group C/D in Kool et al.,
suggests that distinct biological subsets of medulloblastoma and group c2/c4 in Cho et al.) is characterized by neuronal or
exist and that gene expression analysis may provide a more glutaminergic signaling and reduced progression-free survival
accurate risk stratification schema than existing clinical compared with that of the WNT and SHH subgroups. Little is
methods. known about this subgroup, although it is common in all age
groups. It is now generally accepted that there are four broad
subgroups, with two principal non-WNT/SHH groups, one
Medulloblastoma comprises distinct characterized by poor outcome and components of photore-
molecular variants ceptor or g-aminobutyric acidemediated (GABAergic)
signaling (group 3) and one characterized by neuronal or
Advances in array technology over the past 10 years have glutaminergic genes (group 4). Data from Cho et al.suggest
allowed for integrative genomic approaches in the molecular that group 3 may be comprised of more than subtype (c1 and
subclassification of medulloblastoma. An initial study of 46 c5), and that patients with a MYC amplicon and a poor prog-
tumors using unsupervised clustering of gene expression nosis comprise a separate and distinct subtype of group 3. The
profiles revealed the presence of five distinct subgroups (25). full characterization of the two non-WNT/SHH subgroups is
Of these five groups, one group was characterized by up- underway, as are reproducible and simpler methods to identify
regulation of members of the WNT signaling pathway with patients in group 3 who have uniformly poor outcomes.
frequent mutations of the b-catenin gene (CTNNB1) and These studies have led to the emergence of new prediction
monosomy 6; another group was characterized by up- models that are more robust than existing clinical models of
regulation of members of the SHH signaling pathway with risk stratification. When adjusting for the molecular subgroup,
known activating mutations. Three subsequent studies using the presence of metastases is no longer a predictor of
integrated genomics in larger cohorts, similar to the cohort of outcome. For example, patients with WNT-group tumors and
Thompson et al., revealed a WNT subgroup, a SHH subgroup, metastases have an overall survival greater than 90% with no
and multiple non-SHH/WNT subgroups (23,58,81). Activating significant difference between the M0 and Mþ subgroups, and
mutations and genetic events specific to the WNT and SHH patients with SHH or group 4 tumors had no significant survival
subgroups were identified by all three studies; furthermore, differences in the presence of metastasis. Conversely,
consistent with the findings of previous studies, the WNT patients with group 3 tumors have a 5-year overall survival of
subgroup of tumors had a favorable prognosis (57,65). In less than 30% regardless of metastases, suggesting that the
multiple cohorts, molecular sub-grouping appears to be more subset of children with classic histology and poor outcome
specific than histology and Chang staging, as large-cell/ likely belong to this group (Figure 1D; 23). In support of this
anaplastic histology and metastases were found in all concept, a Bayesian model of relapse prediction using clinical
subgroups, including the WNT subgroup in which they have and genetic variables suggests that sub-classification of
a favorable prognosis (23,49,82). A simple prediction model patients using both gene expression and copy-number varia-
using monosomy 6, chromosome 17 status, and amplification tion models outperform the clinical method as well as other
of MYC suggests almost universal survival for those patients individual markers of risk stratification that are subtype inde-
with monosomy 6 (WNT subgroup) and very poor survival for pendent (83). Functional validation of these subgroups is
those with amplification of MYC (57). The WNT subgroup of ongoing; supporting the notion that these subgroups have
medulloblastoma also had a favorable prognosis despite distinct biological properties, a recent study demonstrated that
harboring frequent mutations of TP53, raising questions as to the WNT and SHH subgroups have distinct developmental
its validity as an independent prognostic marker (49). The SHH origins (84). Moreover, to facilitate routine and inexpensive
subgroup was more common in young children with desmo- subgroup identification, a simple four-antibody immunohisto-
plastic histology and in adults, and known SHH mutations such chemical assay (DKK1 for WNT, SFRP1 for SHH, NPR3 for
as those of the PTCH and SUFU genes were present exclu- group 3, and KCNA1 for group 4) can be used to determine the
sively in the SHH group (69). Common genetic events in the subgroup in paraffin-embedded tissue to an accuracy of
WNT and SHH subgroups were replicated in all four studies. w98% (23). A separate study suggested nuclear b-catenin
The three studies by Northcott et al., Cho et al., and Kool immunoreactivity and GAB1 immunoreactivity accurately
et al. had varying numbers of non-WNT/SHH subgroups identified the WNT and SHH subgroups, respectively, and the
(23,58,81). However, despite the differences in the number of absence of filamin A and YAP1 immunoreactivity identified the
non-WNT/SHH subgroups, all three studies revealed the non-WNT/SHH group (85). Efforts are underway to further
presence of a subgroup characterized by poor outcome, more validate the immunohistochemical methods of subgroup
frequent metastases and frequent amplification of MYC (group determination for future use in clinical trials.
C in Northcott et al., group c1/c5 in Cho et al., and group E in
Kool et al.) This subgroup, renamed group 3 by a working
committee, is clinically characterized by male preponderance Integrated genomics provide insight into
and younger age, and is rare in adults. Furthermore, children adult-onset medulloblastoma
with group 3 tumors have poor clinical outcomes regardless of
metastases or histology, suggesting that tumor class is a more Adult medulloblastoma is a relatively small but poorly
potent marker of reduced survival than metastases or understood subset of medulloblastoma. Many adultMedulloblastoma genomics 581
Table 1 Clinical and molecular characteristics of medulloblastoma subgroups
Subgroup WNT SHH Group 3 Group 4
Kool A B E CD
Northcott WNT SHH Group C Group D
Cho c6 c3 c1/c5 c2/c4
Histology Classic (rarely LCA) Desmoplastic/MBEN/ Classic/LCA Classic/LCA
Classic/LCA
Immunohistochemical Nuclear b-catenin, GAB1, SFRP1, Filamin A, NPR3 reactive, absence KCNA1 reactive,
markers DKK1, Filamin A, YAP1, GLI1 reactive of Filamin A and YAP1 absence of Filamin A
YAP1 reactive and YAP1
Age Children and rarely adult Infant/Adult, infrequently Infant and children Children and adult
children
Sex FZM FZM M>F M>F
Metastases Rare Uncommon Very common Common
Specific genetic Frequent 6q-, 9q-, 20p-, 3qþ, 2þ, 14q-, MYC amp, 10q-, 1qþ, i(17q), MYCN amp
events in children CTNNB1 mut 10q-, GLI2 amp, MYCN 5q-, i(17q),
amp PTCH/SMO/
SUFU mutations
Specific genetic CTNNB1 mut, 17p-, 6q- 9q-, 2qþ, 10q-, CDK6 MYC amp CDK6 amp
events in adults amp, 6q-
Expression WNT/ b-catenin SHH signalling Phototransduction and Neuronal development
characteristics signalling glutamate signalling genes
microRNA miR-193a and miR-224, Up: miR-17/92, miR-199b, miR-183w96w182 miR-592, miR-183w
upregulation miR-23w27w24 miR-378, miR-28, 96w182
miR-95, miR-625
Down: miR-135a/b,
miR-124, miR-138
Prognosis Excellent in children, Very good in infants, Very poor Intermediate across
intermediate in adults intermediate in other all ages
ages
medulloblastomas occur in the lateral cerebellar hemisphere cases compared with 100% of pediatric cases. Moreover,
and have desmoplastic histologies. Most childhood studies of amplifications of the CDK6 gene were more common in adult
adjuvant chemotherapy have excluded adults from enroll- medulloblasotmas, compared with amplifications of the MYC
ment, and therefore treatment in adults is not uniform and the gene being more common in medulloblastomas in children, in
role of adjuvant chemotherapy is unclear, leading to many addition to gains of 3q, 4, and 19 being more common in those
patients being treated with craniospinal irradiation only. of children. Furthermore, monosomy 17 is exclusive to adult
Recent studies have suggested that adult medulloblastoma medulloblastoma.
(defined as age >16 y) has poor survival rates compared with The sub-grouping of adult medulloblastomas by combined
that of childhood medulloblastoma, particularly regarding the expression arrays and DNA copy-number analysis reveals
frequent occurrence of late relapses more than 4 years after that it comprises three major molecular variants (90). Unsu-
initial therapy (20,86e88). Until recently, it has not been clear pervised hierarchical clustering analysis revealed that
whether adult medulloblastoma is molecularly similar to the approximately 50% are enriched with SHH signaling genes,
childhood-onset disease. A study of combined array-based and 25% each of the WNT pathway and group 4. No group 3
CGH and medulloblastoma tissue microarrays revealed tumor was identified in this study, which is consistent with the
distinct genomic aberrations: specifically, CDK6 amplifica- studies by Northcott et al. and Cho et al. (23,58). Group 4
tion, 10q loss, and 17q gain were powerful predictors of poor and WNT tumors had a worse prognosis in adults than in
outcome (89). No adult with a tumor harboring a 10q deletion pediatric cohorts. Moreover, monosomy 6q in this study was
combined with a 17q gain was alive at 5 years compared with not exclusively associated with WNT signaling as determined
44% of adults with tumors with either mutation in isolation and by pathognomonic CTNNB1 gene mutations and b-catenin
92% for adults with medulloblastomas without 17q gain or 10q nuclear accumulation. A follow-up study comparing adult and
loss. This study was also surprising in that neither monosomy infant SHH-driven tumors revealed several genomic and
6 nor nuclear b-catenin accumulation was a predictor of good clinical differences (91). Specifically, adult and infant SHH-
outcomes in adults, and monosomy 6 in adults was accom- driven tumors had distinct transcriptional profiles using
panied by nuclear b-catenin mutations in only 50% of adult unsupervised non-negative matrix factorization and had582 V. Ramaswamy et al.
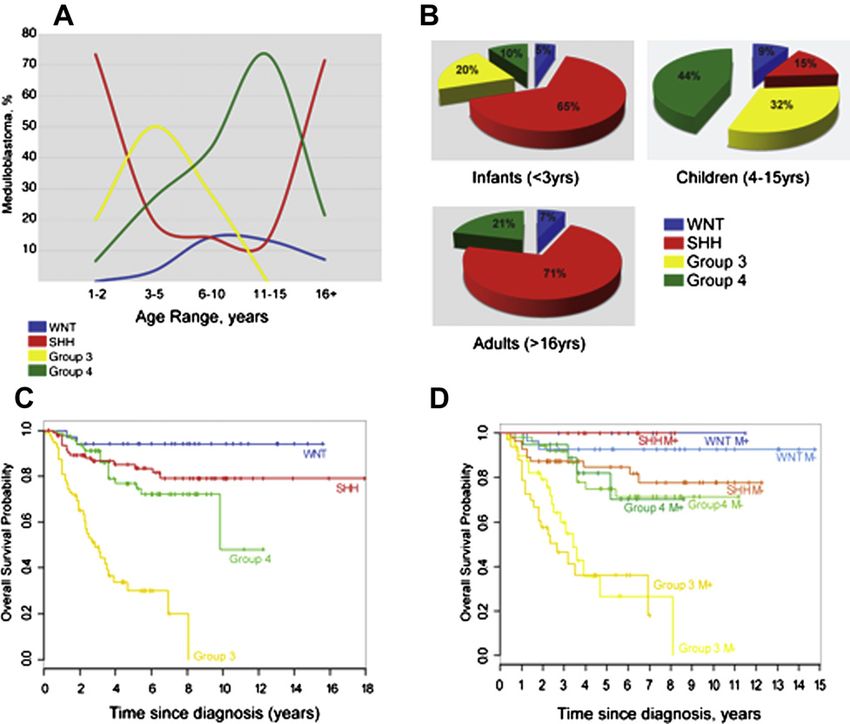
Figure 1 The age distribution and outcome in medulloblastoma. (A) Age at diagnosis by medulloblastoma subgroup. y-axis label:
Medulloblastomas, %; x-axis label: Age range, years. (B) Pie type chart of the frequency of subtypes in the infant (16 y) groups. (C) KaplaneMeier analysis showing overall survival (OS) of combined tissue microarray cohorts
from both DKFZ/Heidelberg and Johns Hopkins University (n Z 287) separated by subgroup. (D) KaplaneMeier analysis from
Figure 1C showing subgroups separated by metastatic status. (Adapted/Reprinted with permission. ª 2011 American Society of Clinical
Oncology. All rights reserved. Northcott PA et al, J Clin Oncol; 29(11), 2011:1408e1414, (23).)
distinct patterns of gene enrichment. Clinically, desmoplasia adult medulloblastoma is distinct from that of pediatric-onset
was not a predictor of good outcome in adults as it is in disease and may warrant a separate classification and risk-
infants, and, unlike in infants with desmoplastic histology, stratification paradigm.
metastatic dissemination in adults conferred a poor prog-
nosis (91,92). Amplification of the GLI2 gene was also
associated with poor outcome in adult cases compared with Epigenetics of medulloblastoma
outcomes for infant SHH-medulloblastomas.
These genomic studies specific to adult medulloblastoma Epigenetic events appear to play emerging roles in the
may have significant implications regarding the use of pathogenesis of medulloblastoma. A study of 212 primary
emerging therapies, specifically the use of smoothened medulloblastomas using high resolution single nucleotide
inhibitors such as GDC-0449, LDE225, and LEQ506X201 polymorphism genotyping identified genetic events converging
(93e98). A report of a single adult patient with extra-neural on lysine 9 of histone H3: specifically, focal events in two
metastatic medulloblastoma having a profound but transient histone lysine methyltransferases (EHMT1 and SMYD4), three
response to GDC-0449 suggests some potential utility to genes of the polycomb family of chromatin remodeling
SHH inhibition in the treatment of medulloblastoma (98). (L3MBTL2, L3MBTL3, and SCML2), two histone lysine
However, this patient developed resistance to smoothened demethylases (JMJD2C and JMJD2B) and one histone lysine
inhibition through a secondary SMO mutation that blocked the acetyltransferase (MYST3; 100). A subsequent study of 22
binding of the drug (97). Phase 2 clinical trials of smoothened medulloblastomas using comprehensive sequence analysis
inhibitors for relapsed medulloblastoma are underway, and revealed common inactivating mutations of the histone lysine
the application of integrated genomics to this group of methyltransferase genes MLL2 or MLL3 in 16% of tumors
patients offers the potential for targeted individualized thera- (101). Focal events and mutations in histone modifiers appear
pies. Notably, tumors with mutations in the SHH pathway that to occur across all medulloblastoma subgroups. This suggests
are downstream of the SMO protein (i.e., amplification of that perturbations in the histone code play an important role
GLI2 or mutation of SUFU ) are unlikely to be responsive to in the pathogenesis of medulloblastoma, and the emergence of
SMO protein inhibitors (91,99). Taken together, the genomic inhibitors of histone lysine methylation and acetylation provides
studies of adult medulloblastoma suggest that the biology of a new drug-targeting pathway to be explored (102e105).Medulloblastoma genomics 583
Figure 2 A proposed application of molecular sub-classification in future clinical trials for medulloblastoma. Top panel: The current
treatment protocol for newly diagnosed medulloblastoma. Following surgery, patients with medulloblastoma over age 3 y are stratified
into average-risk and high risk groups based on dissemination beyond the primary site and presence of residual tumor. Patients under
age 3 y are classified as infants. Bottom panel: Proposed treatments based on molecular sub-classification. Following surgery,
patients with medulloblastoma are classified into the four subgroups (WNT, SHH, group 3, group 4) based on a combination of
transcriptional profiling, immunohistochemistry, or FISH. Abbreviations: AT/RT, atypical teratoid/rhabdoid tumor; RT, radiation
therapy. Expression is determined by either mRNA expression or immunohistochemistry.
The role of hypermethylation of promoter-associated CpG response to alkylating agents in gliomas. Specifically, meth-
islands leading to transcriptional silencing has emerged as ylation of the O6-methylguanine methyltransferase promoter
a potential mechanism of inactivation of tumor suppressor (MGMT ) predicts a favorable response to alkylating agents
genes in several cancers, including medulloblastoma such as temozolomide and nitrosoureas (116,117). There is
(106e115). There is some evidence that the methylation some emerging in vitro data to suggest a correlation between
status of specific genes may provide clues into biological response to alkylators and MGMT promoter methylation in
sub-classification of medulloblastoma. Indeed, the tumor medulloblastoma; however, this requires further study in
suppressor gene SFRP1 is silenced by promoter region prospective cohorts (118).
methylation in WNT, group 3, and group 4 medulloblastomas,
but not in SHH tumors, where it remains highly expressed
(113). Another study suggested that promoter region meth- The role of microRNAs in medulloblastoma
ylation of COL1A2 can distinguish infant desmoplastic
medulloblastoma from other subtypes (112). Methylation of Small noncoding RNAs, particularly microRNAs (miRNAs),
specific genes has been shown to be predictive of treatment are known to play an important role in the pathogenesis of584 V. Ramaswamy et al.
many cancers through regulation of multiple target genes reproducible assays are required for clinical application
(119). Several studies have profiled miRNA-expression of genomic data. Assays that can be performed on
patterns in medulloblastoma, often to determine if specific paraffin-embedded tissue, specifically immunohistochem-
miRNAs are associated with specific subgroups. Alterations ical methods, represent the most reliable and cost-
in expression of specific miRNAs are associated with the effective method to identify subgroups for the purpose of
SHH (miR-17/92) and WNT subgroups (miR-193a and miR- clinical trials. Other methods, such as rapid cytogenetic
224), and specific miRNAs are associated with amplification identification of subgroups in cerebrospinal fluid, may also
of MYC (120e131). It remains to be determined whether be useful in sub-classification, particularly in those patients
miRNA profiling contributes to the risk stratification of who do not undergo re-resection at the time of relapse.
medulloblastoma compared with other methods, however Next-generation sequencing will also provide a tremen-
preliminary studies suggest miRNA patterns exist that are dous amount of data, which will provide further insights into
specific to each molecular subgroup (58). Efforts to profile the dysregulation of specific molecular signaling pathways in
the full extent of miRNA aberrations in medulloblastoma are medulloblastoma samples. Identifying these driver mutations,
ongoing (101,132). and distinguishing them from passenger mutations, will allow
an understanding of what drives tumorigenesis and help lead
to the development of accurate preclinical models, which can
Clinical translation and future challenges be used to develop new and novel therapies. A detailed
understanding of these driver events as well as subgroup-
Over the past 10 years, tremendous volumes of data have specific expression patterns should eventually lead to the
been generated through integrated genomic approaches to identification of the cells of origin for specific subgroups of
the molecular sub-classification of medulloblastoma. These medulloblastoma, allowing the creation of subgroup-
efforts have significantly advanced our understanding of the appropriate animal models of medulloblastoma, which are
pathogenesis of disease, and have delineated distinct critical for preclinical testing of novel agents. The further
molecular subtypes of medulloblastoma. These distinct profiling of both transcriptomes and the genetic events of
molecular subtypes are genetically, transcriptionally, and larger cohorts of tumors at increased depth and the use of
clinically separate, and therefore medulloblastoma likely unbiased techniques such as next-generation sequencing
comprises at least four different diseases. Further charac- platforms should lead to the identification and characteriza-
terization of these distinct molecular and clinical subtypes tion of all the homogeneous subgroups of medulloblastoma,
and identification of reliable and validated biomarkers will as well the driver events important in their initiation, mainte-
allow for targeted clinical trials where those children with nance, and progression. These various elements should
poor outcomes can be identified at surgery, and those chil- coalesce to form the preliminary data that are necessary for
dren with a favorable prognosis can be identified for reduc- the development of more effective, less toxic therapies for
tion of treatment intensity. For example, children with medulloblastoma.
WNT-subgroup medulloblastomas belong to a group that
may potentially benefit from reducing radiation doses to
1800 cGy of craniospinal irradiation or reduction of ototoxic
Acknowledgments
chemotherapy specifically cisplatin. The optimal mechanism
for identifying a WNT-subgroup medulloblastoma in a clin- M. D. T. is supported by a Clinician-Scientist Phase II Award
ical trial is not yet clear but could include FISH for monosomy from the Canadian Institutes of Health Research; the Pedi-
6, immunohistochemistry for b-catenin, sequencing of the atric Brain Tumor Foundation; and the National Institute of
CTNNB1 gene, immunohistochemistry for DKK1, or tran- Health (R01CA48699). V. R. is supported by fellowships
scriptional profiling using expression microarrays. Working from the Canadian Institutes of Health Research and the
out the optimal marker or group of markers for each Alberta Heritage Foundation for Medical Research/Alberta
subgroup represents a significant challenge for the imme- Innovates-Health Solutions.
diate future, and is necessary before subgroup-specific
clinical trials can move forward (Figure 2). References
Identification of distinct molecular subgroups also allows
selection of those patients who might benefit from targeted 1. CBTRUS. CBTRUS Statistical Report: Primary Brain and
therapies. To that effect, ongoing studies may help identify Central Nervous System Tumors Diagnosed in the United
those patients in the SHH subgroup who are most likely to States in 2004-2007. Hinsdale, IL: Central Brain Tumor
benefit from SMO inhibitors. Moreover, further molecular Registry of the United States; 2011.
sub-classification of the two non-WNT/SHH subgroups, 2. Packer RJ, Rood BR, MacDonald TJ. Medulloblastoma:
specifically group 3 tumors, will allow identification of those present concepts of stratification into risk groups. Pediatr
patients predicted to have a poor prognosis and potentially Neurosurg 2003;39:60e67.
lead to their upfront enrollment in clinical trials of dose- 3. Eberhart CG, Kratz J, Wang Y, et al. Histopathological and
intensified therapy or addition of new therapies. Long-term, molecular prognostic markers in medulloblastoma: c-myc, N-
myc, TrkC, and anaplasia. J Neuropathol Exp Neurol 2004;63:
a more thorough understanding and more in-depth sub-
441e449.
classification of the group 3 tumors will allow for the devel- 4. Gajjar A. Clinical, histopathologic, and molecular markers of
opment novel agents that target specific molecular prognosis: toward a new disease risk stratification system
pathways. for medulloblastoma. J Clin Oncol 2004;22:984e993.
Since advanced molecular diagnostic methods such as 5. Packer RJ, Gajjar A, Vezina G, et al. Phase III study of cra-
array technology are not universally available, simple and niospinal radiation therapy followed by adjuvant chemotherapyMedulloblastoma genomics 585
for newly diagnosed average-risk medulloblastoma. J Clin 26. Ng D, Stavrou T, Liu L, et al. Retrospective family study of
Oncol 2006;24:4202e4208. childhood medulloblastoma. Am J Med Genet A 2005;134:
6. Polkinghorn WR, Dunkel IJ, Souweidane MM, et al. Disease 399e403.
control and ototoxicity using intensity-modulated radiation 27. Yokota N, Mainprize TG, Taylor MD, et al. Identification of
therapy tumor-bed boost for medulloblastoma. Int J Radiat differentially expressed and developmentally regulated genes
Oncol Biol Phys 2011;81:e15ee20. in medulloblastoma using suppression subtraction hybridiza-
7. Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in tion. Oncogene 2004;23:3444e3453.
childhood: new biological advances. Lancet Neurol 2007;6: 28. Park PC, Taylor MD, Mainprize TG, et al. Transcriptional
1073e1085. profiling of medulloblastoma in children. J Neurosurg 2003;99:
8. Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted 534e541.
craniospinal radiotherapy followed by high-dose chemotherapy 29. Taylor MD, Mainprize TG, Rutka JT. Molecular insight into
and stem-cell rescue in children with newly diagnosed medul- medulloblastoma and central nervous system primitive neuro-
loblastoma (St Jude Medulloblastoma-96): long-term results ectodermal tumor biology from hereditary syndromes: a review.
from a prospective, multicentre trial. Lancet Oncol 2006;7: Neurosurgery 2000;47:888e901.
813e820. 30. Tamber MS, Bansal K, Liang ML, et al. Current concepts in
9. Grill J, Sainte-Rose C, Jouvet A, et al. Treatment of medullo- the molecular genetics of pediatric brain tumors: implications
blastoma with postoperative chemotherapy alone: an SFOP for emerging therapies. Childs Nerv Syst 2006;22:1379e
prospective trial in young children. Lancet Oncol 2005;6: 1394.
573e580. 31. Pfister SM, Korshunov A, Kool M, et al. Molecular diagnostics
10. Rutkowski S, Bode U, Deinlein F, et al. Treatment of early of CNS embryonal tumors. Acta Neuropathol 2010;120:
childhood medulloblastoma by postoperative chemotherapy 553e566.
alone. N Engl J Med 2005;352:978e986. 32. Dubuc AM, Northcott PA, Mack S, et al. The genetics of pediatric
11. Mason WP, Grovas A, Halpern S, et al. Intensive chemotherapy brain tumors. Curr Neurol Neurosci Rep 2010;10:215e223.
and bone marrow rescue for young children with newly diag- 33. Lubs HA, Salmon JH. The chromosomal complement of human
nosed malignant brain tumors. J Clin Oncol 1998;16:210e221. solid tumors. II. Karyotypes of glial tumors. J Neurosurg 1965;
12. Rutkowski S, Cohen B, Finlay J, et al. Medulloblastoma in 22:160e168.
young children. Pediatr Blood Cancer 2010;54:635e637. 34. Cox D, Yuncken C, Spriggs AI. Minute chromatin bodies in
13. von Bueren AO, von Hoff K, Pietsch T, et al. Treatment of malignant tumours of childhood. Lancet 1965;1:55e58.
young children with localized medulloblastoma by chemo- 35. Friedman HS, Bigner SH, McComb RD, et al. A model for
therapy alone: results of the prospective, multicenter trial HIT human medulloblastoma. Growth, morphology, and chromo-
2000 confirming the prognostic impact of histology. Neuro somal analysis in vitro and in athymic mice. J Neuropathol Exp
Oncol 2011;13:669e679. Neurol 1983;42:485e503.
14. Packer RJ, Vezina G. Management of and prognosis with 36. McAllister RM, Isaacs H, Rongey R, et al. Establishment of
medulloblastoma: therapy at a crossroads. Arch Neurol 2008; a human medulloblastoma cell line. Int J Cancer 1977;20:
65:1419e1424. 206e212.
15. Mulhern RK, Merchant TE, Gajjar A, et al. Late neurocognitive 37. Jacobsen PF, Jenkyn DJ, Papadimitriou JM. Establishment of
sequelae in survivors of brain tumours in childhood. Lancet a human medulloblastoma cell line and its hetero-
Oncol 2004;5:399e408. transplantation into nude mice. J Neuropathol Exp Neurol 1985;
16. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO clas- 44:472e485.
sification of tumours of the central nervous system. Acta Neu- 38. Latimer FR, AlSaadi AA, Robbins TO. Cytogenetic studies of
ropathol 2007;114:97e109. human brain tumors and their clinical significance. 1. Medullo-
17. Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk blastoma. J Neurooncol 1987;4:287e291.
stratification groups in childhood medulloblastoma using 39. Griffin CA, Hawkins AL, Packer RJ, et al. Chromosome
combined clinical, pathologic, and molecular variables. J Clin abnormalities in pediatric brain tumors. Cancer Res 1988;48:
Oncol 2011;29:1400e1407. 175e180.
18. Pietsch T, Taylor MD, Rutka JT. Molecular pathogenesis of 40. Bigner SH, Mark J, Friedman HS, et al. Structural chromosomal
childhood brain tumors. J Neurooncol 2004;70:203e215. abnormalities in human medulloblastoma. Cancer Genet
19. Ellison D. Classifying the medulloblastoma: insights from Cytogenet 1988;30:91e101.
morphology and molecular genetics. Neuropathol Appl Neuro- 41. Cogen PH, Daneshvar L, Metzger AK, et al. Deletion mapping
biol 2002;28:257e282. of the medulloblastoma locus on chromosome 17p. Genomics
20. Abacioglu U, Uzel O, Sengoz M, et al. Medulloblastoma in 1990;8:279e285.
adults: treatment results and prognostic factors. Int J Radiat 42. Cogen PH, Daneshvar L, Metzger AK, et al. Involvement of
Oncol Biol Phys 2002;54:855e860. multiple chromosome 17p loci in medulloblastoma tumorigen-
21. Swedow S, Campo E, Harris N, et al. WHO classification of esis: prognostic significance of molecular genetic markers in
tumours of haematopoietic and lymphoid tissues. Lyon: IARC childhood brain tumors: deletion mapping of the medulloblas-
Press; 2008. toma locus on chromosome 17p. Am J Hum Genet 1992;50:
22. Remke M, Hielscher T, Korshunov A, et al. FSTL5 is a marker 584e589.
of poor prognosis in non-WNT/Non-SHH medulloblastoma. 43. Thomas GA, Raffel C. Loss of heterozygosity on 6q, 16q, and
J Clin Oncol 2011;29:3852e3861. 17p in human central nervous system primitive neuro-
23. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma ectodermal tumors. Cancer Res 1991;51:639e643.
comprises four distinct molecular variants. J Clin Oncol 2011; 44. Bayani J, Zielenska M, Marrano P, et al. Molecular cytogenetic
29:1408e1414. analysis of medulloblastomas and supratentorial primitive
24. Northcott PA, Rutka JT, Taylor MD. Genomics of medullo- neuroectodermal tumors with conventional banding, compara-
blastoma: from Giemsa-banding to next-generation sequencing tive genomic hybridization, and spectral karyotyping.
in 20 years. Neurosurg Focus 2010;28:E6. J Neurosurg 2000;93:437e448.
25. Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies 45. Emadian SM, McDonald JD, Gerken SC, et al. Correlation of
medulloblastoma subgroups that are enriched for specific chromosome 17p loss with clinical outcome in medulloblas-
genetic alterations. J Clin Oncol 2006;24:1924e1931. toma. Clin Cancer Res 1996;2:1559e1564.586 V. Ramaswamy et al.
46. Biegel JA, Janss AJ, Raffel C, et al. Prognostic significance of 66. Slade I, Murray A, Hanks S, et al. Heterogeneity of familial
chromosome 17p deletions in childhood primitive neuro- medulloblastoma and contribution of germline PTCH1 and
ectodermal tumors (medulloblastomas) of the central nervous SUFU mutations to sporadic medulloblastoma. Fam Cancer
system. Clin Cancer Res 1997;3:473e478. 2011;10:337e342.
47. McCabe MG, Backlund LM, Leong HS, et al. Chromosome 17 67. Brugieres L, Pierron G, Chompret A, et al. Incomplete pene-
alterations identify good-risk and poor-risk tumors indepen- trance of the predisposition to medulloblastoma associated with
dently of clinical factors in medulloblastoma. Neuro Oncol germ-line SUFU mutations. J Med Genet 2010;47:142e144.
2011;13:376e383. 68. Taylor MD, Zhang X, Liu L, et al. Failure of a medulloblastoma-
48. Pan E, Pellarin M, Holmes E, et al. Isochromosome 17q is derived mutant of SUFU to suppress WNT signaling. Oncogene
a negative prognostic factor in poor-risk childhood med- 2004;23:4577e4583.
ulloblastoma patients. Clin Cancer Res 2005;11:4733e4740. 69. Taylor MD, Liu L, Raffel C, et al. Mutations in SUFU predispose
49. Pfaff E, Remke M, Sturm D, et al. TP53 mutation is frequently to medulloblastoma. Nat Genet 2002;31:306e310.
associated with ctnnb1 mutation or mycn amplification and is 70. Reardon DA, Michalkiewicz E, Boyett JM, et al. Extensive
compatible with long-term survival in medulloblastoma. J Clin genomic abnormalities in childhood medulloblastoma by
Oncol 2010;28:5188e5196. comparative genomic hybridization. Cancer Res 1997;57:
50. Tabori U, Baskin B, Shago M, et al. Universal poor survival in 4042e4047.
children with medulloblastoma harboring somatic TP53 muta- 71. Nishizaki T, Harada K, Kubota H, et al. Genetic alterations in
tions. J Clin Oncol 2010;28:1345e1350. pediatric medulloblastomas detected by comparative genomic
51. Gilbertson RJ, Pearson AD, Perry RH, et al. Prognostic hybridization. Pediatric Neurosurg 1999;31:27e32.
significance of the c-erbB-2 oncogene product in childhood 72. Bittner M, Meltzer P, Chen Y, et al. Molecular classification of
medulloblastoma. Br J Cancer 1995;71:473e477. cutaneous malignant melanoma by gene expression profiling.
52. McDonald JD, Daneshvar L, Willert JR, et al. Physical mapping Nature 2000;406:536e540.
of chromosome 17p13.3 in the region of a putative tumor 73. Golub TR, Slonim DK, Tamayo P, et al. Molecular classification
suppressor gene important in medulloblastoma. Genomics of cancer: class discovery and class prediction by gene
1994;23:229e232. expression monitoring. Science 1999;286:531e537.
53. Gilbertson R, Wickramasinghe C, Hernan R, et al. Clinical and 74. MacDonald TJ, Brown KM, LaFleur B, et al. Expression
molecular stratification of disease risk in medulloblastoma. Br J profiling of medulloblastoma: PDGFRA and the RAS/MAPK
Cancer 2001;85:705e712. pathway as therapeutic targets for metastatic disease. Nat
54. Batra SK, McLendon RE, Koo JS, et al. Prognostic implications Genet 2001;29:143e152.
of chromosome 17p deletions in human medulloblastomas. 75. Gilbertson RJ, Clifford SC. PDGFRB is overexpressed in
J Neurooncol 1995;24:39e45. metastatic medulloblastoma. Nat Genet 2003;35:197e198.
55. Friedman HS, Burger PC, Bigner SH, et al. Phenotypic and 76. Chopra A, Brown KM, Rood BR, et al. The use of gene expression
genotypic analysis of a human medulloblastoma cell line and analysis to gain insights into signaling mechanisms of metastatic
transplantable xenograft (D341 Med) demonstrating amplifica- medulloblastoma. Pediatr Neurosurg 2003;39:68e74.
tion of c-myc. Am J Pathol 1988;130:472e484. 77. Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of
56. Bigner SH, Friedman HS, Vogelstein B, et al. Amplification of central nervous system embryonal tumour outcome based on
the c-myc gene in human medulloblastoma cell lines and gene expression. Nature 2002;415:436e442.
xenografts. Cancer Res 1990;50:2347e2350. 78. Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired
57. Pfister S, Remke M, Benner A, et al. Outcome prediction in mutations of INI1 in atypical teratoid and rhabdoid tumors.
pediatric medulloblastoma based on dna copy-number aber- Cancer Res 1999;59:74e79.
rations of chromosomes 6q and 17q and the MYC and MYCN 79. Korshunov A, Remke M, Gessi M, et al. Focal genomic
loci. J Clin Oncol 2009;27:1627e1636. amplification at 19q13.42 comprises a powerful diagnostic
58. Cho Y-J, Tsherniak A, Tamayo P, et al. Integrative genomic marker for embryonal tumors with ependymoblastic rosettes.
analysis of medulloblastoma identifies a molecular subgroup that Acta Neuropathol 2010;120:253e260.
drives poor clinical outcome. J Clin Oncol 2011;29:1424e1430. 80. Pfister S, Remke M, Castoldi M, et al. Novel genomic amplifi-
59. Grotzer MA, Hogarty MD, Janss AJ, et al. MYC messenger cation targeting the microRNA cluster at 19q13.42 in a pediatric
RNA expression predicts survival outcome in childhood primi- embryonal tumor with abundant neuropil and true rosettes.
tive neuroectodermal tumor/medulloblastoma. Clin Cancer Res Acta Neuropathol 2009;117:457e464.
2001;7:2425e2433. 81. Kool M, Koster J, Bunt J, et al. Integrated genomics identifies
60. Grotzer MA, Janss AJ, Fung K, et al. TrkC expression predicts five medulloblastoma subtypes with distinct genetic profiles,
good clinical outcome in primitive neuroectodermal brain pathway signatures and clinicopathological features. PLoS
tumors. J Clin Oncol 2000;18:1027e1035. ONE 2008;3:e3088.
61. Grotzer MA, Janss AJ, Phillips PC, et al. Neurotrophin receptor 82. Ellison DW, Onilude OE, Lindsey JC, et al. beta-Catenin status
TrkC predicts good clinical outcome in medulloblastoma and predicts a favorable outcome in childhood medulloblastoma:
other primitive neuroectodermal brain tumors. Klin Padiatr the United Kingdom Children’s Cancer Study Group Brain
2000;212:196e199. Tumour Committee. J Clin Oncol 2005;23:7951e7957.
62. Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the 83. Tamayo P, Cho Y-J, Tsherniak A, et al. Predicting Relapse in
neurotrophin receptor TrkC is linked to a favorable outcome in Patients With Medulloblastoma by Integrating Evidence From
medulloblastoma. Proc Natl Acad Sci U S A 1994;91: Clinical and Genomic Features. J Clin Oncol 2011;29:1415e1423.
12867e12871. 84. Gibson P, Tong Y, Robinson G, et al. Subtypes of medullo-
63. Hottinger AF, Khakoo Y. Neurooncology of familial cancer blastoma have distinct developmental origins. Nature 2010;
syndromes. J Child Neurol 2009;24:1526e1535. 468:1095e1099.
64. Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of 85. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clini-
Turcot’s syndrome. N Engl J Med 1995;332:839e847. copathological correlates of SHH, WNT, and non-SHH/WNT
65. Clifford SC, Lusher ME, Lindsey JC, et al. Wnt/Wingless molecular subgroups. Acta Neuropathol 2011;121:381e396.
pathway activation and chromosome 6 loss characterize 86. Smoll NR. Relative survival of childhood and adult medullo-
a distinct molecular subgroup of medulloblastomas associated blastomas and primitive neuroectodermal tumors (PNETs).
with a favorable prognosis. Cell Cycle 2006;5:2666e2670. Cancer. [Epub ahead of print].Medulloblastoma genomics 587
87. Padovani L, Sunyach MP, Perol D, et al. Common strategy for tumor biology and potential clinical utility. Oncogene 2001;20:
adult and pediatric medulloblastoma: a multicenter series of 5033e5042.
253 adults. Int J Radiat Oncol Biol Phys 2007;68:433e440. 108. Fruhwald MC, O’Dorisio MS, Smith L, et al. [Hypermethylation
88. Ang C, Hauerstock D, Guiot MC, et al. Characteristics and as a potential prognostic factor and a clue to a better under-
outcomes of medulloblastoma in adults. Pediatr Blood Cancer standing of the molecular pathogenesis of medullo-
2008;51:603e607. blastomaeresults of a genomewide methylation scan]. Klin
89. Korshunov A, Remke M, Werft W, et al. Adult and pediatric Padiatr 2001;213:197e203.
medulloblastomas are genetically distinct and require different 109. Lindsey JC, Lusher ME, Anderton JA, et al. Epigenetic
algorithms for molecular risk stratification. J Clin Oncol 2010; deregulation of multiple S100 gene family members by differ-
28:3054e3060. ential hypomethylation and hypermethylation events in medul-
90. Remke M, Hielscher T, Northcott PA, et al. Adult medullo- loblastoma. Br J Cancer 2007;97:267e274.
blastoma comprises three major molecular variants. J Clin 110. Muhlisch J, Bajanowski T, Rickert CH, et al. Frequent but
Oncol 2011;29:2717e2723. borderline methylation of p16 (INK4a) and TIMP3 in medullo-
91. Northcott PA, Hielscher T, Dubuc A, et al. Pediatric and adult blastoma and sPNET revealed by quantitative analyses.
sonic hedgehog medulloblastomas are clinically and molecu- J Neurooncol 2007;83:17e29.
larly distinct. Acta Neuropathol 2011;122:231e240. 111. Scott DK, Straughton D, Cole M, et al. Identification and
92. Al-Halabi H, Nantel A, Klekner A, et al. Preponderance of sonic analysis of tumor suppressor loci at chromosome 10q23.3-
hedgehog pathway activation characterizes adult medullo- 10q25.3 in medulloblastoma. Cell Cycle 2006;5:2381e2389.
blastoma. Acta Neuropathol 2011;121:229e239. 112. Anderton JA, Lindsey JC, Lusher ME, et al. Global analysis of
93. Buonamici S, Williams J, Morrissey M, et al. Interfering with the medulloblastoma epigenome identifies disease-subgroup-
resistance to smoothened antagonists by inhibition of the PI3K specific inactivation of COL1A2. Neuro-oncology 2008;10:
pathway in medulloblastoma. Sci Transl Med 2010;2:51ra70. 981e994.
94. LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of 113. Kongkham PN, Northcott PA, Croul SE, et al. The SFRP family
hedgehog pathway inhibitor vismodegib (GDC-0449) in of WNT inhibitors function as novel tumor suppressor genes
patients with refractory, locally advanced or metastatic solid epigenetically silenced in medulloblastoma. Oncogene 2010;
tumors. Clin Cancer Res 2011;17:2502e2511. 29:3017e3024.
95. Skvara H, Kalthoff F, Meingassner JG, et al. Topical treatment 114. Nakahara Y, Northcott PA, Li M, et al. Genetic and epigenetic
of basal cell carcinomas in nevoid basal cell carcinoma inactivation of Kruppel-like factor 4 in medulloblastoma.
syndrome with a smoothened inhibitor. J Invest Dermatol 2011; Neoplasia 2010;12:20e27.
131:1735e1744. 115. Kongkham PN, Northcott PA, Ra YS, et al. An epigenetic
96. Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the genome-wide screen identifies spint2 as a novel tumor
hedgehog pathway in advanced basal-cell carcinoma. N Engl J suppressor gene in pediatric medulloblastoma. Cancer Res
Med 2009;361:1164e1172. 2008;68:9945e9953.
97. Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation 116. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing
confers resistance to a Hedgehog pathway inhibitor in medul- and benefit from temozolomide in glioblastoma. N Engl J Med
loblastoma. Science 2009;326:572e574. 2005;352:997e1003.
98. Rudin CM, Hann CL, Laterra J, et al. Treatment of medullo- 117. Hegi ME, Liu L, Herman JG, et al. Correlation of
blastoma with hedgehog pathway inhibitor GDC-0449. N Engl J O6-methylguanine methyltransferase (MGMT) promoter
Med 2009;361:1173e1178. methylation with clinical outcomes in glioblastoma and clinical
99. Schreck KC, Taylor P, Marchionni L, et al. The notch target strategies to modulate MGMT activity. J Clin Oncol 2008;26:
Hes1 directly modulates Gli1 expression and hedgehog 4189e4199.
signaling: a potential mechanism of therapeutic resistance. Clin 118. Faoro D, von Bueren AO, Shalaby T, et al. Expression of
Cancer Res 2010;16:6060e6070. O-methylguanine-DNA methyltransferase in childhood medul-
100. Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent loblastoma. J Neurooncol 2011;103:59e69.
genetic events converge on control of histone lysine methyla- 119. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu
tion in medulloblastoma. Nat Genet 2009;41:465e472. Rev Med 2009;60:167e179.
101. Parsons DW, Li M, Zhang X, et al. The genetic landscape of the 120. Ajeawung NF, Li B, Kamnasaran D. Translational applications
childhood cancer medulloblastoma. Science 2011;331:435e439. of microRNA genes in medulloblastomas. Clin Invest Med
102. Lim SP, Neilsen P, Kumar R, et al. The application of delivery 2010;33:E223eE233.
systems for DNA methyltransferase inhibitors. BioDrugs 2011; 121. Birks DK, Barton VN, Donson AM, et al. Survey of microRNA
25:227e242. expression in pediatric brain tumors. Pediatr Blood Cancer
103. Miller CP, Singh MM, Rivera-Del Valle N, et al. Therapeutic 2011;56:211e216.
strategies to enhance the anticancer efficacy of histone deace- 122. Ferretti E, De Smaele E, Miele E, et al. Concerted microRNA
tylase inhibitors. J Biomed Biotechnol 2011;2011:514261. control of Hedgehog signalling in cerebellar neuronal progen-
104. Mummery A, Narendran A, Lee KY. Targeting epigenetics itor and tumour cells. EMBO J 2008;27:2616e2627.
through histone deacetylase inhibitors in acute lymphoblastic 123. Ferretti E, De Smaele E, Po A, et al. MicroRNA profiling in
leukemia. Curr Cancer Drug Targets [Epub ahead of print]. human medulloblastoma. Int J Cancer 2009;124:568e577.
105. Rahman R, Grundy R. Histone deacetylase inhibition as an 124. Garzia L, Andolfo I, Cusanelli E, et al. MicroRNA-199b-5p
anticancer telomerase-targeting strategy. Int J Cancer 2011; impairs cancer stem cells through negative regulation of HES1
129:2765e2774. in medulloblastoma. PLoS ONE 2009;4:e4998.
106. Fruhwald MC, O’Dorisio MS, Dai Z, et al. Aberrant hyper- 125. Gokhale A, Kunder R, Goel A, et al. Distinctive microRNA
methylation of the major breakpoint cluster region in 17p11.2 in signature of medulloblastomas associated with the WNT
medulloblastomas but not supratentorial PNETs. Genes signaling pathway. J Cancer Res Ther 2010;6:521e529.
Chromosomes Cancer 2001;30:38e47. 126. Liu W, Gong YH, Chao TF, et al. Identification of differentially
107. Fruhwald MC, O’Dorisio MS, Dai Z, et al. Aberrant promoter expressed microRNAs by microarray: a possible role for
methylation of previously unidentified target genes is microRNAs gene in medulloblastomas. Chin Med J (Engl)
a common abnormality in medulloblastomasdimplications for 2009;122:2405e2411.You can also read

















































