DIA EDM Webinar eCTD Update - December 5, 2013 Mark Gray
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

DIA EDM Webinar
eCTD Update
December 5, 2013
Mark Gray
Director, Division of Data Management Services & Solutions
(CDER/OSP/OBI) 1
Disclaimer
• Views expressed in this presentation are those of the speaker and
not necessarily of the Food and Drug Administration.
• The views and opinions expressed in the following PowerPoint
slides are those of the individual presenter and should not be
attributed to Drug Information Association, Inc. (“DIA”), its directors,
officers, employees, volunteers, members, chapters, councils,
Special Interest Area Communities or affiliates, or any organization
with which the presenter is employed or affiliated.
• These PowerPoint slides are the intellectual property of the
individual presenter and are protected under the copyright laws of
the United States of America and other countries. Used by
permission. All rights reserved. Drug Information Association, DIA
and DIA logo are registered trademarks or trademarks of Drug
Information Association Inc. All other trademarks are the property of
their respective owners.
2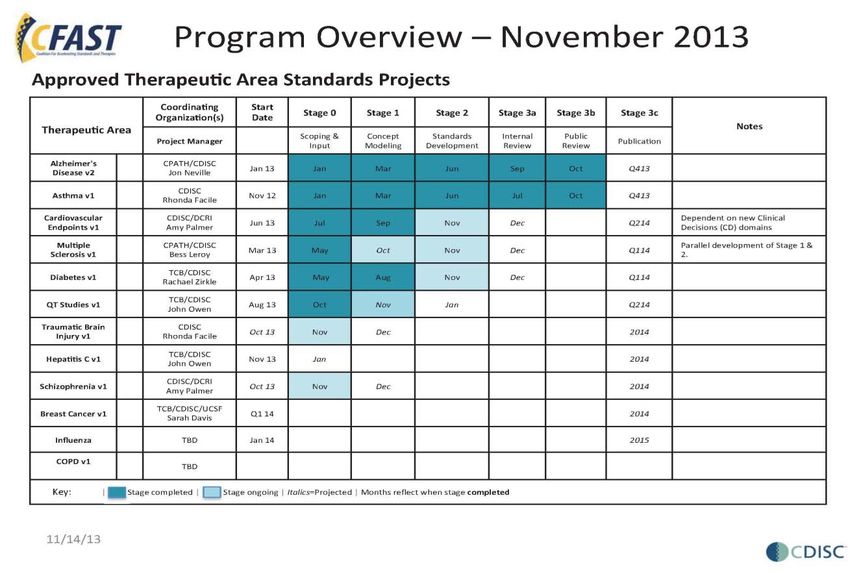
Agenda
• eCTD Guidance
• eCTD Metrics
• Module 1 Update
• eCTD v4.0
3
eCTD Guidance
• Food and Drug Administration Safety and
Innovation Act (FDASIA) of 2012
» Gives FDA the authority to require electronic
submissions for certain application types after
issuance of final guidance
» Reauthorizes PDUFA and established GDUFA
• PDUFA and GDUFA will require electronic
submissions in the eCTD format, after issuance
of final guidance
4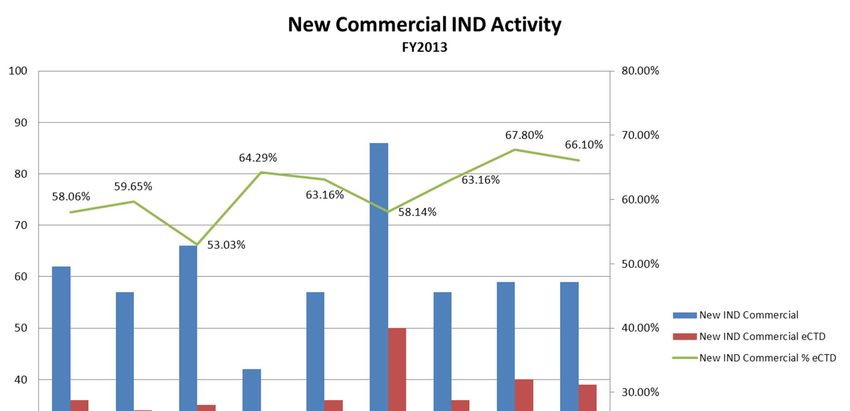
PDUFA V eCTD Guidance Process
• PDUFA Performance Goals
– Issue guidance for NDA, BLA, and IND submissions in the eCTD
format
• Issue Draft Guidance by December 31, 2012
• Based on eCTD v3.2.2
• Issue final eCTD guidance no later than 12 months after public
comment period
• Implementation
– NDA and BLA – 24 months after publication of final guidance
– Commercial INDs – 36 months after publication of final guidance
• NOTE: FDA will follow this process to meet the GDUFA
eCTD submission requirements
5
eCTD Guidance Status
• Issued guidance for NDA, BLA, ANDA and IND requiring
submissions in the eCTD format
– FR Notice Published 1/3/2013
– Draft eCTD Guidance
– PDUFA V process: Issue final eCTD guidance no later than 12 months
after public comment period
• Comment period closed March 4, 2013
• Plan is to reissue the eCTD draft guidance
– Based on internal discussions and updated Interpretation of FDASIA
– Still based on eCTD v3.2.2
– Issued updated draft eCTD guidance in early 2014
• Updated implementation target – Mandatory eCTD submission
– NDA, BLA, and ANDA: Late 2016 – early 2017
– Commercial INDs: Late 2017 – early 2018
6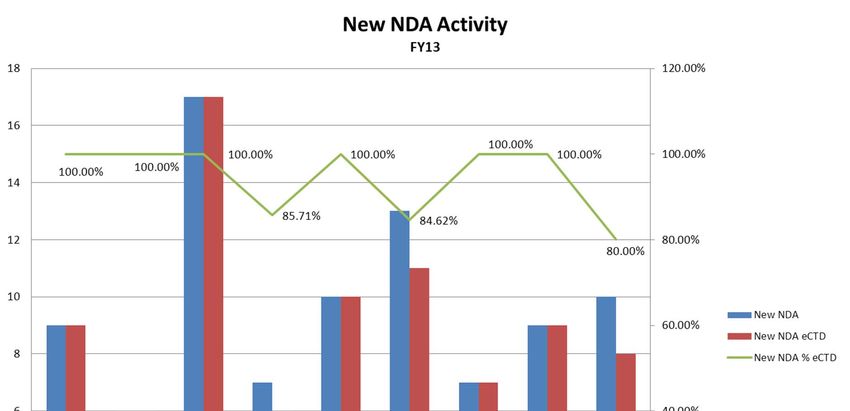
FDA eCTD Submissions
as of October 4th, 2013
Application Number of Number of
Type Applications Sequences
IND 5,582 264,813
NDA 2,507 85,856
ANDA 8,645 85,308
BLA 266 24,383
MF 1,931 9,782
FDA Internal 834 1,570
Total 19,771 471,705
7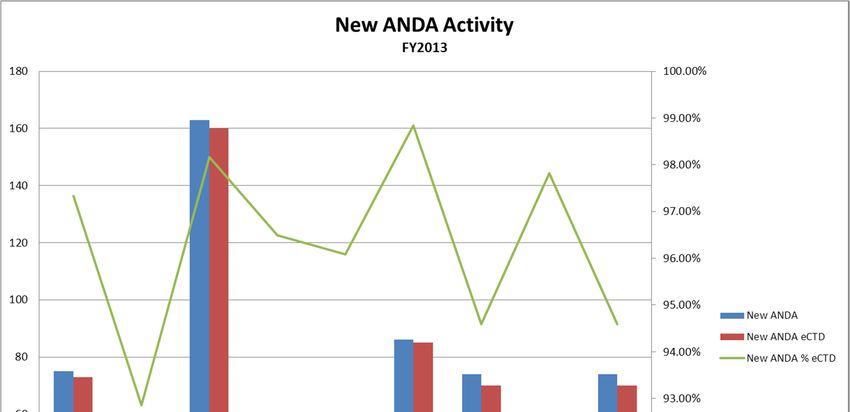
8
CDER Investigational New Drugs
FY2009 FY2010 FY2011 FY2012 FY2013
IND Research 12,863 14,816 16,039 14,767 15,176
IND Commercial 74,163 77,402 77,013 76,419 76,672
IND Total 87,026 92,218 93,052 91,186 91,848
IND Research Electronic 456 721 1,185 1,477 1,841
IND Commercial Electronic 24,913 36,794 48,116 55,108 60,722
IND Electronic Total 25,369 37,515 49,301 56,585 62,563
IND Electronic % 29.15% 40.68% 52.98% 62.05% 68.12%
IND Research eCTD 326 595 1,008 1,324 1,595
IND Commercial eCTD 24,448 36,219 47,564 54,677 60,259
IND eCTD 24,774 36,814 48,572 56,001 61,854
eCTD % of Total 28.47% 39.92% 52.20% 61.41% 67.34%
eCTD % of Electronic 97.66% 98.13% 98.52% 98.97% 98.87%
9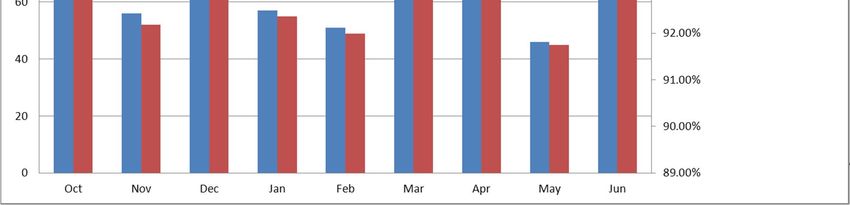
10
CDER New Drug Applications
FY2009 FY2010 FY2011 FY2012 FY2013
NDA Total 22,148 22,443 23,254 23,746 22,822
NDA Electronic 13,297 15,497 17,396 18,694 18,563
NDA Electronic % 60.04% 69.05% 74.81% 78.72% 81.34%
NDA eCTD 11,146 14,007 15,937 17,682 17,747
NDA eCTD % of Total 50.33% 62.41% 68.53% 74.46% 77.76%
NDA eCTD % of Electronic 83.82% 90.39% 91.61% 94.59% 95.60%
1112
CDER Abbreviated New Drug Applications
FY2009 FY2010 FY2011 FY2012 FY2013
ANDA Total 29,554 19,408 22,186 26,514 42,687
ANDA Electronic 11,045 11,637 16,554 20,639 29,521
ANDA Electronic % 37.38% 59.96% 74.61% 77.84% 69.16%
ANDA eCTD 6,341 8,113 12,915 16,314 23,295
ANDA eCTD % of Total 21.46% 41.80% 58.21% 61.53% 54.57%
ANDA eCTD % of Electronic 57.41% 69.72% 78.02% 79.04% 78.91%
1314
FDA eCTD M1 Update
• Update includes;
– Additional submission metadata to facilitate submission
processing
• Contact information (e.g., regulatory, technical)
• Application cross references
• Supplement effective date type (PAS, CBE-0, CBE-30)
– Submission Numbering
• Application Type – Application Number
• Submission Type – Submission Id
• Submission Sub-Type – Sequence Number
– Flexibility to reduce possibility of DTD changes
• Attribute values (e.g. submission-type, submission-sub-type)
– Functionality for grouped submissions
– Updated M1 Headings and Hierarchy
• Major changes to 1.15 Promotional Material
• Heading attributes for 1.1 Forms and 1.15
15FDA eCTD M1 Update
• Implementation
– CDER will accept eCTD promotional submissions when the M1 update
is implemented
– After transitioning an application to the new M1, that application must
continue to use the new M1 DTD
– Errata (4/2/2013) and FR Notice (8/26/2013)
• Additional attributes on 1.15.2.1 Material
– Material Id – Applicant’s id code
– Issue Date – Date of initial dissemination or publication
• Added promotional labeling contact type
– Target Implementation Update
• New implementation date is 4th quarter 2014
– Mitigate risk based on number of software/system updates required
– Will give 30 days advance notice to industry
• Impact on eCTD v3.2.2 electronic submission requirement
– Both the current M1 DTD (v2.01) and updated M1 DTD (v3.2) will be
accepted
16eCTD v4.0 Project
• Implementation of the Health Level Seven (HL7) Regulated Product
Submission (RPS) standard
– HL7 exchange standard that can be used for the submission of any
regulated product
– Medical Device (IMDRF) participation in the RPS project
• What does this mean?
– eCTD v4 will use the RPS exchange message
– eCTD v4 is a subset of RPS implemented specifically for human
pharmaceuticals
– Huh? Ah, what?
• The eCTD headings and hierarchy are not changing
• Think of it as a technology upgrade with some enhancements
17RPS Message Capabilities (Summary)
• Submission metadata (e.g, Application Type and Number)
– Has additional metadata to facilitate processing of the submissions
• Life-cycle functionality (active, inactive)
– Ability to life-cycle one to one, one to many, many to one
• Correct/modify Keywords
• Handles bundled/global/grouped submissions
• File Reuse – Submit file once and cross-reference
• Ability to identify file types (e.g, SDTM dataset) for additional processing
• Standardize submission format/structure by application type (e.g. NDA, DMF)
• Two-way communication
– The regulatory authority can use RPS to send correspondence to the submitter
• ICH and Regional requirements
– Additional product information
– Multi-regulator submissions
– ICH and regional requirements incorporated into one model 18eCTD v4.0 ICH Schedule
• 2012 / 2013 Accomplishments
– DSTU Ballot
– Test case development & testing
– Controlled vocabulary development
– Draft Implementation Guides
• Draft eCTD v4 ICH Implementation Guide
• Draft Regional Module 1 Implementation Guides
• ICH M8 Step 2 for Testing
– http://estri.ich.org/new-eCTD/index.htm
– Draft ICH Implementation Guide
– Lessons Learnt
– Draft ICH Code List
– Schema Files
– Links to Regional eCTD v4.0 web pages
19eCTD v4.0 ICH Schedule
• RPS Normative Ballot (September 2013)
– Reviewing HL7 RPS ballot comments
– Will require a re-ballot
– ISO approval process starts after a successful HL7 normative ballot
• Testing (January 2014 – June 2014)
• Update/Finalize Implementation Guides (May 2013 – November 2014)
• ICH Step 2 Signoff (November 2014)
• ICH Step 3 Comment & Reconciliation (November 2014 – June 2015)
• ICH Step 4 Signoff (November 2015)
– Update Step 2 Implementation Guide (June 2015 – November 2015)
• Based on current Implementation schedule: FDA begin receiving eCTD v4.0
submissions in 2016
– Most likely will start with a pilot
– Requires guidance update
• FDA is required to issue revised final guidance before mandating eCTD 20
v4.0eCTD Website
– eCTD Guidance
– eCTD Headings & Hierarchy
– Specifications
• ICH (Modules 2 – 5)
• FDA Module 1
• eCTD Validation Criteria
• Related Specifications (e.g., PDF, Transmission, Study Data)
– eCTD supportive files (DTD, stylesheet, valid values)
– Link to the eCTD Updated Module 1 information
eCTD Website address:
http://www.fda.gov/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirements/Elect
ronicSubmissions/ucm153574.htm
21Quality and Product Data
Standards
DIA EDM and ERS/eCTD Webinar
December 5, 2013
Jared Lantzy
Data Management Solutions Team
Division of Data Management Services and Solutions
22
(FDA/CDER/OSP/OBI)Disclaimer
• Views expressed in this presentation are those of the speaker and
not necessarily of the Food and Drug Administration.
• The views and opinions expressed in the following PowerPoint
slides are those of the individual presenter and should not be
attributed to Drug Information Association, Inc. (“DIA”), its directors,
officers, employees, volunteers, members, chapters, councils,
Special Interest Area Communities or affiliates, or any organization
with which the presenter is employed or affiliated.
• These PowerPoint slides are the intellectual property of the
individual presenter and are protected under the copyright laws of
the United States of America and other countries. Used by
permission. All rights reserved. Drug Information Association, DIA
and DIA logo are registered trademarks or trademarks of Drug
Information Association Inc. All other trademarks are the property of
their respective owners.
23A Clarification
“Quality” Data Standards
Not referring to the condition or worth of the
data, but the standardization of data normally
found in review documents in Module 3
(Quality) of the eCTD
24Agenda
• The Problem
• The Solution
• Current CDER Projects
• Next Steps
25The Problem
• An immense amount of data is submitted
in each and every CDER application
– Unstructured, in the text of documents
– Module 1 – Regional
– Module 3 – Quality
26The Problem (2)
• Manual data entry into systems
• Multiple systems
– Same data, different functions
• Data is not linked in a useful way
27The Solution
• Data standards for Quality data
– Structured data that can be properly linked to
all its applications, sponsors, products,
substances, and facilities
• eSubmission of structured data with the
application
• Automated import into CDER’s master
data system
28Current CDER Projects
• CDER master data management
– New systems, processes, and procedures to
combine new and existing data from within
CDER to create and maintain a single,
accurate source of data for CDER systems to
reference and consume
– For example, provide the ability to quickly link
a specific drug substance to all applications
and products using that substance
29Current CDER Projects (2)
• Facility information
– 356h Form
– GDUFA Facility Self-ID (SPL)
– Standardized Establishment Information List
(SPL)
• Product and Substance Information
– Drug Registation and Listing (SPL)
– ISO IDMP Standards (SPL)
30Next Steps
• Standards development within CDER,
ICH, and ISO
– IDMP is early in the ISO standardization
process with industry partner support
31Next Steps (2)
• Writing and issuing draft guidance in
accordance with FDASIA
– Draft guidances are published for comment,
revised, made final, and then another 24
months before submission can be required
– FDA will be requiring certain types of
electronic submissions AND standardized
data within the application
32Industry Questions
• IDMP looking at the overarching standard, given the complexity
what is how mandated, how soon and realize the extensive impacts
on the sponsors, vendors and user community? Takeaway w/
standards and timelines
– Does FDA realize the implications to industry
• IDMP; would like to see the current timelines and know current
thinking of SPL technology and if there will be a link with IDMP
• Electronic application forms and filled out form, do you think FDA
will be moving towards that the information will be captured in the
Admin information in eCTD instead of the same information being
captured in the FDA PDF forms? Why have information on the
metadata being captured since it is duplicative
• What are people doing to ignore FDA forms security?
33Validation
• FDA Forms
– Use the FDA Forms as downloaded from our
forms website, without changing any settings
or adding or removing any additional security
features.
– Our validation tool will ignore PDF security
errors for the proper unmodified FDA Forms
located in the proper section of the eCTD
34Validation (2)
• FDA Forms (cont.)
– If you are unable to use digital signatures,
submit an unsigned fillable form with the form
number as the file name (e.g. 356h.pdf or
1571.pdf)
– Submit your signed scanned form as signed
form.pdf, ensuring you don’t inlcude the form
number in the file name
35Validation (3)
• PDF Errors
– Font embedding
• The intent is to ensure the review division can
access all the information in your submission
• If you stick to the “standard fonts” there is no need
to embed, these fonts are always available on
reviewer PCs
• If you use “non-standard fonts” you should fully
embed the font.
36Validation (4)
• PDF Errors
– Font embedding (cont.)
• We are not rejecting submissions with medium
error 5005 Non standard font (not embedded)
• If we are unable to access information because
your non-standard font is not embedded, we will
contact you to resubmit the affected document(s)
– This could affect a review clock if the scope of
documents is wide enough!
37Study Data Standards Update
Ron Fitzmartin, PhD, MBA
Office of Strategic Programs
Center for Drug Evaluation and Research
Food and Drug Administration
EDM and ERS Webinar:
FDA Update/Progress Report
December 5, 2013
38Disclaimer
• Views expressed in this presentation are those of the speaker and
not necessarily of the Food and Drug Administration.
• The views and opinions expressed in the following PowerPoint
slides are those of the individual presenter and should not be
attributed to Drug Information Association, Inc. (“DIA”), its directors,
officers, employees, volunteers, members, chapters, councils,
Special Interest Area Communities or affiliates, or any organization
with which the presenter is employed or affiliated.
• These PowerPoint slides are the intellectual property of the
individual presenter and are protected under the copyright laws of
the United States of America and other countries. Used by
permission. All rights reserved. Drug Information Association, DIA
and DIA logo are registered trademarks or trademarks of Drug
Information Association Inc. All other trademarks are the property of
their respective owners.
39Topics
• CDER-CBER Study Data Standards Statement
• Path to Required Data Standards
• Guidance and Notice Update
• Therapeutic Area Standards Development
40
40Study Data Standards
for Regulatory Submissions
41
41De-Constructing the Statement
“FDA recognizes the investment made by sponsors over the
past decade to develop the expertise and infrastructure to
utilize CDISC standards.”
Pharma’s challenges have never been greater. R&D spend
increases 5 percent annually, while output of NMEs
approved has dropped by ~22 percent.
--Accenture, 2013
Companies have invested staff, $$$ and time in processes,
technology and CDISC Standards.
42
4243
43De-Constructing the Statement
PDUFA V states “that FDA will develop guidance for industry
on the use of CDISC data standards for the electronic
submission of study data in applications.”
• Draft Guidance on Providing Regulatory Submissions in
Electronic Format: Standardized Study Data
• Draft Study Data Technical Conformance Guide
• Draft Therapeutic Area Data Standards Initiative Plan
• Notice on Pilot Evaluation of CDISC SDS XML
44
44De-Constructing the Statement
“FDA envisions a semantically interoperable and sustainable
submission environment that serves both regulated clinical
research and health care.”
“Shared Health And Clinical Research Electronic Library
(SHARE) is expected to dramatically improve integration
among CDISC foundational standards and controlled
terminologies, and support greater interoperability with
healthcare.”
--CDISC, October, 2013
45
45De-Constructing the Statement
“FDA does not foresee the replacement of CDISC
standards for study data and will not implement new
approaches without public input on the cost and utility of
those approaches.”
It has taken decades for Industry & FDA to
get to this point with study data standards,
specifically CDISC standards!
46
46FDASIA* Reauthorizes PDUFA V
“…develop “… periodically publish
standardized clinical final guidance specifying
PDUFA V
data terminology Performance the completed data
Goals- XII
through open standards, formats, and
standards terminologies that
development sponsors must use to
organizations (i.e., submit data in
CDISC)” applications.”
47
47
*FDA Safety And Innovation Act-2012Path to Required Study Data Standards (1)
FDASIA Statute
FDASIA
eGuidance
Electronic Submission
Electronic Submission
Guidance Electronic Submission
Electronic Regulatory
Guidance Guidance
Electronic Standardized
Submission (eCTD)
Study Data Guidance
Guidance
Binding and Non-Binding Binding and Non-Binding
Guidance Requiring eSubs in Guidance Requiring
eCTD Format Standardized Study Data 48
48Path to Required Study Data Standards (2)
Regulation FDASIA Statute
Binding FDASIA
Guidance eGuidance
Non-Binding & Electronic Standardized
Binding Study Data Guidance
Guidance
Supported & Study Data
Data Standards Technical
Required Technical
Catalog Conformance Guide
Resources
Standards
49
49Path to Required Study Data Standards (3)
Data Standards
Catalog Data Standards Catalog
• Catalog to include supported standards, and
timing for required standards.
• Data Standards webpage being re-designed.
– Easier navigation / access to information.
– Redundant information removed.
– Expected to be available when guidances are published.
• Aligned with Technical Conformance Guide.
50
http://www.fda.gov/forindustry/datastandards/studydatastandards/default.htmPath to Required Study Data Standards (4)
Study Data Technical
Conformance Guide Study Data Technical Conformance Guide
• Provides recommendations for submission of standardized
study data.
• Complements and assists in the interaction between
sponsors and divisions.
• It will not replace the need for sponsors to communicate with review
divisions.
• When final, will replace the Common Data Standards
Issues and Study Data Specifications documents.
51Path to Required Study Data Standards (6)
• Submission of standardized study data will be
required
– According to a phased-in schedule, but not before…
• Federal Register publication of the…
1. Draft FDASIA & Revised draft eStudy Data guidances, for public
comment.
2. Final FDASIA guidance & Final eStudy Data guidance.
– Final eStudy Data Guidance will specify a phased-in
schedule
• No earlier than 24 months after final guidance for certain NDAs,
BLAs, ANDAs. 52Guidance / Notice Update (1)
• Draft in Development
Draft FDASIA Guidance • Anticipate FR Publish: FY14
Revised • Draft Published February 2012
Draft eStudy Data Guidance • Revised Draft in Clearance
• Anticipate FR Publish: FY14
Study Data Technical • Draft in Clearance
Conformance Guide • Anticipate FR Publish: FY14
53
53Guidance / Notice Update (2)
FR Notice: Therapeutic Area • Published - October 2013
Standards Initiative Project Plan
FR Notice: Pilot Project to
• Published November 27, 2013
Evaluate Alternative for Study
Data Transport
54
54Collaboration to Develop
Therapeutic Area Data Standards
Public* Private
American College of
Cardiology
Alzheimer’s Association joint Biopharma
Bill & Melinda Gates partnership
Foundation
Industry
PKD Foundation
One Mind
*Sample list
Academia* Government*
National Institutes of
Duke University Health
active National Cancer Institute
University of Wisconsin participants
National Institute of
University of Pittsburgh Neurological Disorders
and Stroke
Wake Forest University Office of National
Coordinator
*Sample list Coalition for Accelerating Standards & Therapies *Sample list
55CFAST Project Stages & FDA’s Role
• Scientific & • FDA • FDA • FDA Testing /
Technical Divisio Divisio Acceptance
Input n n • Guidance
Expert Expert
• Planning / Review Review
Prioritization
56
• Initial Expert
ReviewFDA Process for TA Requirements
Requirements TA Standard
Plan & Guidance /
Review & Acceptance Policy
Scoping Acceptance Testing
• Interviews • Requirement • Evaluate • Update
• Review s report acceptability of technical
Internal review and TA standards documents,
documents acceptance for use in as needed
• Requirements: submissions • Issue
-Primary, • Ensure Federal
reviewers’ Register
-Secondary, readiness to Notice
-Covariates, use
-Exploratory standardized
data
http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirem 57
ents/ElectronicSubmissions/UCM297093.pdfFDA and CFAST Progress to Date
%
~59 TAs
% of TAs published & in CFAST
proposal, planning & development
stages
58
http://www.cdisc.org/therapeutic 58Thank You
Ron Fitzmartin
ronald.fitzmartin@fda.hhs.gov
Data Standards Questions
cder-edata@fda.hhs.gov
cber.cdisc@fda.hhs.gov
59DIA EDM Webinar
Top Ten Issues with Data
Douglas Warfield, Ph.D.
Technical Team Lead
Interdisciplinary Scientist
(CDER/OSP/OBI)
60Disclaimer
• Views expressed in this presentation are those of the speaker and
not necessarily of the Food and Drug Administration.
• The views and opinions expressed in the following PowerPoint
slides are those of the individual presenter and should not be
attributed to Drug Information Association, Inc. (“DIA”), its directors,
officers, employees, volunteers, members, chapters, councils,
Special Interest Area Communities or affiliates, or any organization
with which the presenter is employed or affiliated.
• These PowerPoint slides are the intellectual property of the
individual presenter and are protected under the copyright laws of
the United States of America and other countries. Used by
permission. All rights reserved. Drug Information Association, DIA
and DIA logo are registered trademarks or trademarks of Drug
Information Association Inc. All other trademarks are the property of
their respective owners.
61Data Top 10 - Two Categories
What and Where Content
1. Location/Name 1. USUBJID – DM
2. Define - XML & PDF duplicates
3. ADaM - no. 2. XPTs and File Size
columns 3. Splitting Datasets
4. Traceability 4. Units of Measure
5. Legacy and 5. Legacy and
Standardized Standardized
62What and Where
Naming
Important for
Automated
Processing!
Exact
spelling
of folder
names
Ref: Study Data Specifications
63What and Where
Placement
Important for
Automated
Processing!
Exact
placemen
t of
folders
Ref: Study Data Specifications
64What and Where
Data Definitions - Defines
Important for
Exact Reviewer’s
placemen
t of
Content
defines Navigation
and
Automated
Ref: Study Data Specifications
Processing!
65What and Where
ADaM - Too Many Columns!
Too many variables, can
1. Data Definitions of
affect usability. 50 variables
domain columns max.
2. Columns relevant to Some definitions irrelevant
the domain to the domain/analyses.
3. Domain dataset file More variables per domain,
size larger dataset file size.
4. Traceability of Tracing domain variables
complexity increases as no.
content of variables increases.
Ref: Electronic Regulatory Submissions and Review
Helpful Links, Study Data Standards Resources 66What and Where
Traceability!
eCRF Creating Analysis datasets from a source
(standardized) other than SDTM, when standardized data
(SDTM) is submitted, is problematic for
some review activities.
Raw Data*
(not submitted)
Analysis
Standardized Analyses datasets
should originate from SDTM datasets.
SDTM
Ref: Electronic Regulatory Submissions and Review
* Raw Data – research data collected in original Helpful Links, Study Data Standards Resources
tabular electronic form. 67What and Where
Legacy and Standardized!
1. Can sponsors Yes, consult with review
submit both? division prior to submission.
2. Where? Supported SDS structure supports
by specifications? both concurrently for study.
3. Traceability during Sponsor should plan quick
transition. transitions (regs. - PDUFA.)
4. Integrating Sponsor integrated/pooled
(pooled, ISS, ISE, non-standardized and
etc.) standardized dataset
placement is problematic.
68Content
USUBJID – DM duplicates
• Demographics datasets (Legacy or Standardized) where the unique
subject identifier is NOT unique within the dataset, result in
failures to “load” data for review in several automated review tools.
• Sponsors should expect review divisions to request sponsors
submit all demographics datasets compliant with the unique
subject identifier requirement.
• Sponsors and standards organizations should implement protocol
designs, data collection, and tabulations to ensure the result of
unique subject identifiers in the demographics datasets submitted
for review.
Ref: Electronic Regulatory Submissions and Review
Helpful Links, Study Data Standards Resources
69Content
XPTs and File Size
• Datasets’ size remain a major concern with data submitted.
• However, more sponsors are resizing submissions using the
maximum column required in the datasets submitted algorithm,
based on pre-review analyses of submissions.
• Sponsors should expect review divisions to request sponsors to
resize datasets, when larger datasets (> 1 gigabyte) are submitted
and resizing does not appear to have been to used.
• Sponsors and standards organizations should develop and
promote resizing techniques to ensure reduction of datasets
submitted for review.
Ref: Electronic Regulatory Submissions and Review
Helpful Links, Study Data Standards Resources
70Content
Splitting Datasets
• Split datasets’ remains a top issue...
• Review tools rely on specific naming of split datasets to
automatically combine during “load” processing. (e.g. lb01.xpt,
lb02.xpt, lb03.xpt are combined to lb.xpt in some review tools).
• Sponsors and standards organizations should develop and
promote splitting techniques to allow direct review based on
stratified datasets (e.g. labs by test type), while supporting
concatenation (identical variables structure) for combined
analyses.
Ref: Electronic Regulatory Submissions and Review
Helpful Links, Study Data Standards Resources
71Content
Units of Measure
• Units of Measure for all datasets with measurement values (e.g.
Labs, Vital Signs) remains a top issue with data submitted, related
to the variability received in units of measure indicated for
measurements in observations.
• Greater specificity by review divisions is required for measurement
types and the standardized measurement unit for the submission
of data for the type.
• Sponsors and standards organizations should develop and
promote reduction of measurement types and standardized units
of measure for the review.
Ref: Electronic Regulatory Submissions and Review
Helpful Links, Study Data Standards Resources
72Content
Units of Measure - Variability
Count of
LBORRESU and Total % by
Dataset LBTESTCD LBORRESU LBSTRESU Percent of Total
LBSTRRESU Test
Combinations
Lab BILI mg/dL umol/L 464 19.31%
Lab BILI umol/L umol/L 372 15.48%
Lab BILI mg/dL mg/dL 257 10.70% 45.49%
Lab GLUC mg/dL mmol/L 569 20.29%
Lab GLUC mmol/L mmol/L 461 16.44%
Lab GLUC mg/dL mg/dL 372 13.26% 49.98%
Lab WBC /HPF /HPF 353 4.74%
Lab WBC x10^3/uL giga/L 240 3.22% Variabilit
Lab WBC G/L giga/L 168 2.25%
Lab WBC giga/L giga/L 162 2.17% 12.38%
y
Vital Signs TEMP C C 896 55.24%
Vital Signs TEMP F C 321 19.79%
Vital Signs TEMP F F 116 7.15% 82.18%
Vital Signs WEIGHT kg kg 1456 60.27%
Vital Signs WEIGHT LB kg 240 9.93%
Vital Signs WEIGHT KG KG 149 6.17% 76.37%
Vital Signs SYSBP mmHg mmHg 1531 84.96% 84.96%
Vital Signs DIABP mmHg mmHg 1531 85.06% 85.06%
Note: Source: ~115 NDAs with ~920 different studies
73Content
Legacy and Standardized
Data submission issue.
1. Conversion to Sponsor should provide
standardized data? rationale and process used.
2. Review of legacy Limited review processes and
content. tools using less automation in
contrast to standardized
IT review data .
processes and
3. Review of
tools using automation
standardized
based on standardized
content. data. Plan for new regs.(
4. Traceability: legacy PDUFA.)
Standardization (CDISC)
vs. standardized includes traceable content
by specifications.
74Top Ten Issues with Data
Topics to Remember
Data - What and
Where
Data- Content
Importance for 21 Century
Review!
75You can also read

















































