Neurophysiological index as a biomarker for ALS progression: Validity of mixed effects models
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Amyotrophic Lateral Sclerosis, 2011; 12: 33–38
ORIGINAL ARTICLE
Neurophysiological index as a biomarker for ALS progression:
Validity of mixed effects models
BENJAMIN C. CHEAH1,2, STEVE VUCIC1, ARUN V. KRISHNAN1, ROBERT A. BOLAND1
& MATTHEW C. KIERNAN1,2
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
1Neuroscience
Research Australia, Sydney, New South Wales, and 2Prince of Wales Clinical School, University of
New South Wales, Sydney, New South Wales, Australia
Abstract
Our objective was to evaluate the neurophysiological index (NI) as a biomarker for amyotrophic lateral sclerosis (ALS)
and to assess the validity of linear mixed effects models for describing longitudinal changes. Functional assessment and
nerve conduction studies were undertaken in 58 ALS patients. Neurophysiological data were collected on four occasions
over 12 weeks (baseline, weeks 4, 8 and 12). The NI was calculated for the abductor digiti minimi and ulnar nerve at the
wrist. NI declined at a rate of 0.04 per week (S.E. 0.006, p 0.0001). Patients with bulbar-onset disease had 0.88 greater
NI than patients with upper limb-onset disease over the follow-up period (S.E. 0.39, p 0.03). There were no differences
For personal use only.
in the rates of decline among patients with different disease phenotypes. Rates of change in NI and functional impairment
were weakly correlated (Spearman's r 0.29, p 0.03). Linear mixed effects models were appropriate for detailing the
longitudinal changes in NI. The present findings support incorporation of NI as an outcome measure for ALS clinical
trials conducted over short time periods.
Key words: Amyotrophic lateral sclerosis, motor neuron disease, neurophysiology, clinical trial, mixed effects model
Introduction In the initial validation studies, assessment of NI
was undertaken every 3–6 months over an 18-month
There remains a critical need to devise objective bio-
period, with ALS patients grouped by their rate of
markers of disease progression in amyotrophic lat-
eral sclerosis (ALS) (1,2). The neurophysiological disease progression. A uniform finding across these
index (NI) was developed to quantify peripheral dis- studies was that the NI decreased by a substantially
ease burden in ALS patients (3). Unlike other esti- greater margin than clinical measures of disease
mates of peripheral nerve integrity (e.g. motor unit severity, including forced vital capacity, maximum
number estimation, MUNE) (4), the NI is relatively voluntary isometric contraction, and the ALS Func-
simple to measure, with the individual components tional Rating Scale-Revised (ALSFRS-R). In patients
obtainable using standard clinical neurophysiologi- with rapidly progressive disease, the NI declined by
cal equipment. The NI is principally recorded from as much as 50% over one year (7,8).
the abductor digiti minimi (ADM) and ulnar nerve, The present study aimed to systematically assess
although measurements have been made from the the utility of the NI in the phase II clinical trial setting,
abductor pollicis brevis and median nerve (5,6). and to specifically determine the responsiveness
Consistent with the progressive nature of ALS, ADM of the NI to disease progression when measured
strength is positively correlated with compound at four-weekly intervals. In addition, the validity of
muscle action potential (CMAP) and F-wave fre- linear mixed effects modelling was determined for
quency, and negatively correlated with distal motor the NI data, with specific comparisons to changes in
latency (DML; 3). ALSFRS-R.
Correspondence: M. C. Kiernan, Neuroscience Research Australia, Barker Street, Randwick, NSW 2031, Australia. Fax: 61 2 9382 2437.
E-mail: M.Kiernan@unsw.edu.au
(Received 18 July 2010; accepted 8 October 2010)
ISSN 1748-2968 print/ISSN 1471-180X online © 2011 Informa Healthcare
DOI: 10.3109/17482968.2010.53174234 B. C. Cheah et al.
Methods Yij ( b0 bi )( b1 ci ) X1ij b2 X 2i b3 X 3i eij
In total, 58 consecutive ALS patients (24 upper
limb-onset, 17 lower limb-onset, and 17 bulbar- where Yij represented the value of NI for patient
onset) were included in the present study. Patients i on study visit j (measured in weeks from baseline;
fulfilled the revised El Escorial criteria for a diag- j 1, 2, 3, 4), X1ij was the number of weeks from
nosis of ALS (9). In order to determine the utility baseline, and eij denoted the residual for the ith
of the NI compared to more generally accepted patient at the jth study visit. The residuals were
measures of disease progression, the ALSFRS-R calculated by subtracting the model-predicted NI
(10) was administered on each occasion, such that values from the observed values and, as such, repre-
both NI and ALSFRS-R were measured on four sented the variation not explained by the predictor
occasions (i.e. at baseline and weeks 4, 8, and 12). variables and random effects. Residuals should be
Only patients with baseline values of NI greater normally distributed with a mean of zero for a mixed
than zero were included. The study was approved effects model to be valid.
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
by the South Eastern Sydney Area Health Service The fixed effects comprised an intercept (i.e. the
Human Research Ethics Committee (Eastern Sec- mean NI at baseline for patients with upper limb-
tion). All patients and healthy control subjects pro- onset disease), mean rate of decline in NI per week
vided informed written consent to participate in (b1), difference in NI between patients with upper
this study. limb-onset disease and lower limb-onset disease
Standard nerve conduction studies were under- (X2i 1, X3i 0) over the entire study (b2), and dif-
taken, such that the ulnar nerve was stimulated at ference in NI between patients with upper limb-onset
the wrist with the resultant CMAP recorded over the and bulbar-onset disease (X2i 0, X3i 1) over the
ADM muscle (Synergy Version 12.0; Viasys Health- entire study (b3). Patients with upper limb-onset dis-
care, Surrey, UK). The ulnar nerve-ADM system ease thereby served as the referent category (X2i
was studied in preference to other nerve-muscle sys- 0, X3i 0).
tems principally because it remains intact until late The mixed effects model also incorporated indi-
in the course of disease, thereby enabling long-term vidual-specific random effects (i.e. deviations from
For personal use only.
follow-up (11). Skin temperature of the examined the estimated population mean) with respect to the
limb was maintained above 30°C. The ulnar nerve intercept (bi) and rate of decline in NI per week (ci)
was stimulated with a bipolar electrode at the wrist, for each patient. Accounting for random effects was
5–6 cm proximal to the recording electrode, which essential, given that each patient had unique baseline
was placed over the motor endplate of the patient's NI and different rates of decline in NI. The mixed
stronger ADM muscle, assessed using the Medical effects model assumed that the random effects were
Research Council scale. Therefore, only one hand normally distributed with means of zero: the baseline
was studied on each occasion. NI among the present cohort of patients was nor-
F-wave studies were undertaken using a sequence mally distributed, as well as the rate of decline in NI.
of 20 supramaximal stimuli (25% above maximal, at Therefore, patients with positive baseline random
0.5 Hz) that were applied to the ulnar nerve at the effects (bi) had ‘above average’ baseline NI, and those
wrist. A pulse width of 0.5 ms and an amplifier gain with negative baseline random effects (bi) had ‘below
of 0.1–0.5 mV were used. Only negative deflections average’ baseline NI. Patients with rate-of-decline
with amplitudes of at least 40 mV were accepted as random effects of zero had rates of decline that
F-waves in order to differentiate them from back- matched the estimated population mean rate of
ground noise. The NI was calculated according to decline. Patients with positive rate-of-decline ran-
the equation: dom effects (ci) deteriorated at a ‘below average’ rate
(i.e. they progressed more slowly than the popula-
CMAP (mV) F wave Frequency tion mean; the mean rate of decline represented a
NI
DML (ms) negative value); and patients with negative rate-of-
decline random effects (ci) deteriorated at an ‘above
average’ rate (i.e. they progressed more rapidly than
Statistical analysis
the estimated population mean rate of decline).
Neurophysiological and functional impairment data Validity of the linear mixed effects models in
were modelled using linear mixed effects models describing the longitudinal changes in NI was
(12). Mixed effects models have been previously assessed through visual inspection of the residual
applied in ALS to analyse the rate of change in ALS- and random effects distributions. Shapiro-Wilk test-
FRS-R in previous clinical trials (13–15). However, ing for normality was also undertaken. In keeping
the mixed effects approach has not been used to with previous ALS clinical trials, ALSFRS-R data
model longitudinal changes in neurophysiological were also modelled using a linear mixed effects
data. In the present study, linear mixed effects enabled model, with time from baseline as the sole indepen-
efficient estimation of each patient's rates of decline dent variable. In order to investigate whether there
in NI and ALSFRS-R. The model was of the form: was a relationship between the rate of change in NIMixed effects modelling of the neurophysiological index 35
and global disease progression, the rate-of-decline of muscle weakness in these phenotypes. There were
random effects from both mixed effects models were no significant differences in the rate of decline in NI
examined using Spearman's rank correlation coeffi- between patients with upper limb-onset disease and
cient. Statistical analyses were performed with R bulbar-onset disease, and between patients with upper
version 2.11.0 for Windows (16) using the mixed limb-onset disease and lower limb- onset disease.
effects model package (17). Results are presented as
mean standard deviation (SD) or median and the
Utility of linear mixed effects model for longitudinal
respective inter-quartile range, depending on whether
changes in NI
the data were normally distributed.
The validity of linear mixed effects models to analyse
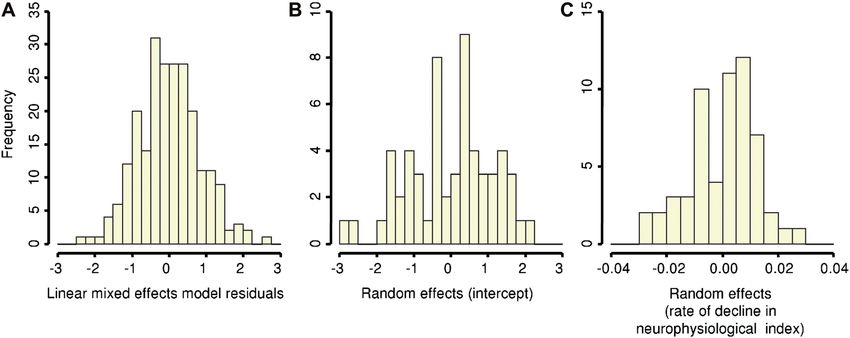
longitudinal changes in NI was confirmed by the
Results
normal distribution of residuals (Figure 3A; Shapiro-
The mean age of the present cohort of ALS patients Wilk test: p 0.90, p 0.05 being indicative of
at baseline was 54.9 9.3 years (36 males, 62.1%; departure from normality). There was no correlation
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
22 females, 37.9%), consistent with a representa- (r 0.02) between the random effects associated
tive sample (18). A total of 230 NI recordings with baseline NI (bi) and rate of decline (ci), indicat-
and 230 ALSFRS-R measurements were obtained. ing that baseline disease severity was independent of
The reduction in CMAP was consistent with a the rate of deterioration in NI.
progressive loss of motor units in ALS (Table I). The importance of accounting for subject-spe-
Longitudinal decline of F-wave frequency reflected cific random effects was highlighted by the presence
diminishing numbers of excitable motor neurons of spread about a mean of zero in the random effects
in the spinal cord. In addition, F-wave studies associated with the intercept (bi; Figure 3B) and rate
demonstrated distinct morphological changes over of decline (ci; Figure 3C) – standard deviations
the study duration, consistent with a progressive of 1.20 and 0.02, respectively. Shapiro-Wilk testing
reduction in lower motor neuron pools over the for normality, as well as histograms of the random
course of disease (Figure 1). The mild prolongation effects associated with the intercept and slope coef-
in DML probably reflected a combination of slow- ficient indicated that the random effects were nor-
For personal use only.
ing of conduction along unmyelinated collaterals mally distributed about a mean of zero (bi: p 0.39;
emerging from surviving motor axons that subse- ci: p 0.28; Figure 3). In total, such findings sug-
quently reinnervate denervated muscle fascicles gested that a mixed effects model was valid for
(19,20) and the selective loss of large calibre motor describing longitudinal changes in NI in the present
axons (21). cohort of ALS patients.
Neurophysiological index across clinical phenotypes Comparison with functional rating scales
At baseline, the mean NI was 2.79 (SD, 1.24). After At baseline, ALS patients had a mean ALSFRS-R
approximately 12 weeks of follow-up, the mean NI score of 41.6 3.0, consistent with a moderate level
decayed to 2.28 1.28, representing an 18.3% of functional impairment. Patient functional capac-
reduction (p 0.0001). The linear mixed effects ity diminished to a score of 39.7 3.3 at 12 weeks
model revealed that the mean rate of decline in NI (p 0.0001), a 4.5% change in functional impair-
was 0.04 per week (S.E. 0.006, p 0.0001; 95% CI ment. According to the mixed effects model, the
0.03–0.05; Figure 2). There was a non-significant ALSFRS-R declined at a rate of 0.16 units per week
difference of 0.51 between patients with upper limb- (95% CI 0.11–0.21, p 0.0001), which was associ-
onset disease and lower limb-onset disease over the ated with a substantially smaller standard error of
entire follow-up period (b2 0.51; S.E. 0.39, p 0.02. As such, ALSFRS-R may be considered to
0.19; 95% CI –1.29–0.27). The NI was 0.88 (b3) have a greater rate of decline compared to the vari-
higher in patients with bulbar-onset disease than in ance of decline.
patients with upper limb-onset disease over the entire Overall, the mixed effects model was a valid tool
duration of the study (S.E. 0.39, p 0.03; 95% for assessing longitudinal changes in ALSFRS-R,
CI 0.10–1.65), reflecting the differential evolution because of relative symmetry in the residual and
Table I. Changes in conventional neurophysiological variables at baseline and after 12 weeks follow-up. The overall changes were
consistent with lower motor neuron degeneration. Results are expressed as mean standard deviation or median (inter-quartile
range).
Variable Baseline Follow-up % change p-value
CMAP (mV) 8.2 2.7 7.4 2.5 –10.1 0.0001
F-wave frequency 0.97 (0.8 – 1.0) 0.90 (0.63 – 1.0) –7.2 0.002
DML (ms) 2.76 (2.4 – 3.0) 2.8 (2.45 – 3.0) 1.4 0.09
NI 2.79 1.24 2.28 1.28 –18.3 0.000136 B. C. Cheah et al.
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
Figure 1. F-wave studies recorded from an ALS patient, highlighting the longitudinal changes that occurred with disease progression
(A – early disease → D – severe disease). F-wave frequency is initially high (i.e. 1). With disease progression, the F-wave frequency
decreases with decline in the number of motor units. F-wave amplitude also increases, while latency increases due to conduction
along slowly-conducting unmyelinated fibres. Repeat (or identical) F-waves become more prominent, as the same motor units are
repeatedly activated with each stimulus (C). This occurs until a complete absence of F-waves becomes apparent, indicating the
absence of excitable motor neuron pools (D).
random effects distributions. In addition, there was weeks in ALS patients, thereby increasing the utility
a weak positive correlation between the random effects of this measure in a clinical trial setting. Further-
of both mixed effects models, indicating that the rate more, linear mixed effects modelling was success-
For personal use only.
of change in NI moderately reflected the clinical rate fully applied and determined to be an appropriate
of disease progression, as measured using ALSFRS-R tool for modelling neurophysiological data. Although
(Spearman's r 0.29, p 0.03). NI declined at a greater rate than ALSFRS-R, there
was correlation between these measures of disease
severity. While the frequency of recordings was not
Discussion
stressed in previous studies, the present study has
The present study has established that the NI established the utility of NI to enable longitudinal
detected deterioration occurring over as little as four studies to be conducted over shorter periods of time,
Figure 2. Spaghetti plot of individual NI traces, stratified by site of disease of onset, demonstrating longitudinal decline. In addition,
the model-predicted mean neurophysiological index for each disease phenotype is indicated by a thick line. Patients with bulbar-onset
disease had higher NI than patients with upper limb-onset disease. In contrast, there was no significant difference between patients
with upper limb-onset and lower limb-onset disease.Mixed effects modelling of the neurophysiological index 37
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
Figure 3. (A) Frequency histogram indicating that the residuals were normally distributed, confirming suitability of a linear mixed
effects model for modelling the longitudinal changes in neurophysiological index. Plots of the random effects of the intercept (B; bi)
and weekly rate of decline in neurophysiological index (C; c i) which, like the residuals, should conform to a normal distribution. The
random effects represented deviations from the estimated population mean baseline value and rate of decline.
with responsiveness to disease progression. As marginal model approaches, which do not contain
such, it is proposed that NI could be appropriately random effects, mixed effects models are able to
incorporated as an endpoint in ALS clinical trials, account for inter-subject differences in baseline mea-
particularly for phase II investigations. surements and rates of change through incorporat-
ing subject-specific random effects. Mixed effects
models are also better able to cope with missing data.
Application of the NI in ALS clinical trials
This may be particularly beneficial in the setting of
For personal use only.
In the present study, disease progression in ALS ALS clinical trials, where smaller sample sizes may
patients was detectable over a four-week period using be required.
the NI. In contrast, previous studies recorded the NI
at 3–6-monthly intervals. Given that the NI declined
Limitations of the NI and the present study
by a greater proportion than ALSFRS-R in the pres-
ent study, use of NI may expedite the completion of Use of the NI in ALS clinical trials may be associated
future phase II trials by shortening trial duration with limitations. Specifically, if baseline F-wave fre-
(22). Trials of short duration may appeal more to quency is low (i.e. 0.1–0.4), and depending on the
ALS patients, facilitating recruitment and minimiz- rate of disease progression, the NI may reach a ‘floor
ing attrition. The finding that the rate of reduction effect’ and assume a value of zero (due to complete
for NI correlated with the global rate of functional absence of F-waves) sooner than desired (5,6,23).
decline is another compelling reason to consider Such a predicament may not permit sufficient long-
use of the NI as a measure of disease progression in term follow-up; this may be avoided by studying
ALS trials, particularly since longitudinal changes in both hands initially to determine which ADM has
NI reflect the underlying pathological process of the greater baseline NI. Indeed, baseline F-wave fre-
progressive motor unit loss. quencies of zero excluded seven patients from con-
In terms of alternative approaches, MUNE has tributing data to the present study. Nevertheless, it
been utilized in clinical trials on ALS patients. For appeared that the progressive reduction in CMAP
comparison, the NI declined at a similar rate to was the greatest contributing factor towards decline
MUNE after three months follow-up (8). Although in NI in the present study, as CMAP underwent the
NI and MUNE performed similarly in that study, a greatest reduction compared to F-wave frequency
major benefit of NI over MUNE in a clinical trial and DML.
setting relates to the fact that NI is generally easier Conversely, measuring NI from limbs that are
to implement, using standard neurophysiological unaffected by disease may also be problematic, as no
equipment and protocols for recordings, many of deterioration may occur over the initial follow-up
which would form part of an electrodiagnostic work- period. In patients with intact upper limb muscles
up for ALS patients. In contrast, the various MUNE and lower limb weakness, an alternative approach
techniques require specialized software and specific may be recording the NI from abductor hallucis in
training. the lower limbs. The abductor hallucis (innervated
The present study has emphasized that linear by the tibial nerve) normally has an F-wave fre-
mixed effects models represent a useful tool for quency close to 1 (24). However, evaluation of NI
monitoring longitudinal changes in neurophysiolog- in the lower limbs of ALS patients as a measure of
ical data obtained from ALS patients. In contrast to disease progression has not been validated.38 B. C. Cheah et al.
Conclusions 6. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcit-
ability may precede the onset of familial amyotrophic lateral
The present study has highlighted that the NI was sclerosis. Brain. 2008;131:1540–50.
responsive to ALS disease progression over a short 7. de Carvalho M, Scotto M, Lopes A, Swash M. Clinical and
measurement period. Furthermore, the decline in neurophysiological evaluation of progression in amyotrophic
lateral sclerosis. Muscle Nerve. 2003;28:630–3.
NI occurred more rapidly than in conventional clin- 8. de Carvalho M, Scotto M, Lopes A, Swash M. Quantitating
ical measures of disease progression, such as the progression in ALS. Neurology. 2005;64:1783–5.
ALSFRS-R over the same period. The rate of decline 9. Brooks BR, Miller RG, Swash M, Munsat TL, World Fede
in neurophysiological variables was moderately ration of Neurology Research Group on Motor Neuron D.
correlated with functional deterioration. Given that El Escorial revisited: revised criteria for the diagnosis of
amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other
changes were noticeable over a short time frame, Motor Neuron Disord. 2000;1:293–9.
employing NI as a primary endpoint in future phase 10. Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D,
II clinical trials may enable shorter trial duration. Thurmond B, et al. The ALSFRS-R: a revised ALS functional
Finally, the present study has validated the use of rating scale that incorporates assessments of respiratory
function. BDNF ALS Study Group (Phase III). J Neurol Sci.
Amyotroph Lateral Scler Downloaded from informahealthcare.com by University of New South Wales on 07/12/12
linear mixed effects models to describe longitudinal
1999;169:13–21.
changes in NI. 11. de Carvalho M, Lopes A, Scotto M, Swash M. Reproduc-
ibility of neurophysiological and myometric measurement
Declaration of interest: Support from the in the ulnar nerve-abductor digiti minimi system. Muscle
Nerve. 2001;24:1391–5.
Australian Rotary Health Research Amyotrophic
12. Laird NM, Ware JH. Random effects models for longitudinal
Lateral Sclerosis Fund (Mary Jane Douglass Award) data. Biometrics. 1982;38:963–74.
and the National Health and Medical Research 13. Gordon PH, Cheung YK, Levin B, Andrew H, Doorish C,
Council of Australia is gratefully acknowledged. Macarthur RB, et al. A novel, efficient, randomized selection
Benjamin Cheah was awarded the University of trial comparing combinations of drug therapy for ALS.
Amyotroph Lateral Scler. 2008;9:212–22.
New South Wales BrainSciences PhD scholarship
14. Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde
and the 2010 Pfizer Biostatistics Collaboration of JL, Doorish C, et al. Efficacy of minocycline in patients with
Australia Award for Excellence for this study. Steve amyotrophic lateral sclerosis: a phase III randomized trial.
Vucic received funding from Clive and Vera Rama- Lancet Neurol. 2007;6:1045–53.
For personal use only.
ciotti Establishment grant and Sylvia and Charles 15. Moore DH, Miller RG. Improving efficiency of ALS clinical
trials using lead-in designs. Amyotroph Lateral Scler Other
Viertel Charitable Foundation Clinical Investiga-
Motor Neuron Disord. 2004;5:S57–60.
torship. Robert Boland receives grant support from 16. R Development Core Team. R: a language and environment
the New South Wales Office of Science and Medi- for statistical computing. 2.11.0 ed. Vienna, Austria; 2010.
cine Research. Matthew Kiernan has received grant 17. Pinheiro J, Bates D, DebRoy S, Sarkar D, Team RC. nlme:
support from the National Health and Medical linear and non-linear mixed effects models. In: R package
version 3.1-96 ed; 2009.
Research Council of Australia and the Motor
18. Eisen A. Amyotrophic lateral sclerosis: a 40-year personal
Neurone Disease Research Institute of Australia. perspective. J Clin Neurosci. 2009;16:505–12.
19. Wohlfart G. Collateral regeneration from residual motor
nerve fibres in amyotrophic lateral sclerosis. Neurology.
References
1957;7:124–34.
1. Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers 20. Hansen S, Ballantyne JP. A quantitative electrophysiologi-
in amyotrophic lateral sclerosis. Lancet Neurol. 2009;8: cal study of motor neuron disease. J Neurol Neurosurg
94–109. Psychiatry. 1978;41:773–83.
2. Winhammar JMC, Rowe DB, Henderson RD, Kiernan MC. 21. Sobue G, Matsuoka Y, Mukai E, Takayanagi T, Sobue I.
Assessment of disease progression in motor neuron disease. Pathology of myelinated fibres in cervical and lumbar ventral
Lancet Neurol. 2005;4:229–38. spinal roots in amyotrophic lateral sclerosis. J Neurol Sci.
3. de Carvalho M, Swash M. Nerve conduction studies in 1981;50:413–21.
amyotrophic lateral sclerosis. Muscle Nerve. 2000;23: 22. Aggarwal S, Cudkowicz M. ALS drug development: reflec-
344–52. tions from the past and a way forward. Neurotherapeutics.
4. Bromberg MB, Brownell AA. Motor unit number estimation 2008;5:516–27.
in the assessment of performance and function in motor 23. Vucic S, Kiernan MC. Abnormalities in cortical and periph-
neuron disease. Phys Med Rehabil Clin N Am. 2008;19: eral excitability in flail arm variant amyotrophic lateral scle-
509–32. rosis. J Neurol Neurosurg Psychiatry. 2007;78:849–52.
5. Vucic S, Kiernan MC. Novel threshold tracking techniques 24. Buschbacher RM. Tibial nerve F-waves recorded from
suggest that cortical hyperexcitability is an early feature of the abductor hallucis. Am J Phys Med Rehabil. 1999;78:
motor neuron disease. Brain. 2006;129:2436–46. S43–7.You can also read

















































