CONFERENCE 79th PHYSICAL ELECTRONICS - University of Central Florida - PEC 2019
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
79th PHYSICAL ELECTRONICS CONFERENCE PROGRAM AND ABSTRACT BOOK University of Central Florida Harris Corporation Engineering Center June 2-5, 2019
2

79th ANNUAL PHYSICAL ELECTRONICS CONFERENCE WORKING PROGRAM June 2-5, 2019 Harris Corporation Engineering Center University of Central Florida, Orlando, FL SPONSORS AMERICAN VACUUM SOCIETY SCIENTA OMICRON SPECS SIGMA SURFACE SCIENCE LEYBOLD THERMO FISHER SCIENTIFIC CREATEC SENTYS ANCORP UCF OFFICE OF RESEARCH UCF COLLEGE OF SCIENCES UCF COLLEGE OF ENGINEERING AND COMPUTER SCIENCE UCF NANOSCIENCE AND TECHNOLOGY CENTER UCF DEPARTMENT OF PHYSICS UCF FACULTY CLUSTER INITIATIVE 3
PEC GENERAL ORGANIZING COMMITTEE Brian Swartzentruber (Chair) Sandia Laboratories Karsten Pohl (Treasurer) University of New Hampshire Carl Ventrice (co-Treasurer) CNSE, University of Albany, SUNY Daniel Dougherty North Carolina State University Jane Hinch Rutgers University Seth King University of Wisconsin-La Crosse Paul Thibado University of Arkansas, Fayetteville Nicholas Strandwitz Lehigh University PEC LOCAL ORGANIZING COMMITTEE Talat Rahman (Chair) University of Central Florida William Kaden University of Central Florida Mihai Vaida University of Central Florida Parag Banerjee University of Central Florida Laurene Tetard University of Central Florida Zahra Hooshmand University of Central Florida Duy Le University of Central Florida 4
SPONSORS We enthusiastically thank the following sponsors for supporting PEC 2019. NOTTINGHAM PRIZE SPONSOR POSTER PRIZE SPONSOR GENERAL SPONSORS OFFICE OF RESEARCH COLLEGE OF SCIENCES DEPARTMENT OF PHYSICS FACULTY CLUSTER INITIATIVE NANOSCIENCE & TECHNOLOGY CENTER COLLEGE OF ENGINEERING & COMPUTER SCIENCE 5

HISTORY OF PEC The 79th Physical Electronics Conference at the University of Central Florida follows in the tradition of annual PEC meetings held on university campuses and research labs in North America. This topical conference provides a yearly forum for the dissemination and discussion of novel and fundamental theoretical and experimental research in the physics, chemistry, biology, and engineering of surfaces and interfaces. It includes the prestigious Nottingham Prize competition for best presentation based on doctoral research. Over 75 years the emphases of the conference have changed, yet the meeting remains centered in its basis of fundamental science at material interfaces. Central themes now include the interfaces of metals, semiconductors, ionic conductors, dielectrics, insulators, fluids, porous materials, and the wealth of biomaterials. Physicists, chemists, and engineers, with interests in these fields, come together to present and discuss experimental and theoretical research on exposed (gas-solid or gas-liquid) surfaces, or buried (liquid-solid and solid-solid) interfaces. Representative topics include (but are not limited to) electronic, chemical, magnetic, and structural properties of interfaces; energetics, kinetics, and dynamics of physical and chemical transformations at surfaces; formation, assembly, structural and electronic properties, and modeling of nanoscale surface architectures; effects of electron correlation at surfaces, and topological insulators; interfacial interactions of biological materials; impacts of interface chirality; mechanisms of film growth and interface evolution; and transfer of energy, electrons, or ions across materials interfaces. New methods/techniques for interrogation of interfaces, novel devices or sensors, and new applications for structurally and chemically tailored interfaces also fall within the scope of this meeting. 6
PREVIOUS PEC HOSTS 1966 Massachusetts Institute of Technology 1993 Rensselaer Polytechnic Institute 1967 Massachusetts Institute of Technology 1994 University of Tennessee 1968 University of Minnesota 1995 Arizona State University 1969 Yale University 1996 Boston University 1970 University of Wisconsin - Milwaukee 1997 University of Oregon 1971 National Bureau of Standards 1998 Pennsylvania State University 1972 Sandia Laboratories 1999 University of California - Berkeley 1973 University of California - Berkeley 2000 Louisiana State University 1974 Bell Laboratories 2001 Sandia National Laboratories 1975 Pennsylvania State University 2002 Georgia Institute of Technology 1976 University of Wisconsin-Madison 2003 Cornell University 1977 Stanford University 2004 University of California – Davis 1978 Oak Ridge National Laboratory 2005 University of Wisconsin - Madison 1979 University of Maryland 2006 Princeton University 1980 Cornell University 2007 University of Illinois, Urbana-Champaign 1981 Montana State University 2008 University of California - Riverside 1982 Georgia Institute of Technology 2009 Rutgers University 1983 Sandia National Laboratories 2010 University of Wisconsin - Milwaukee 1984 Princeton University 2011 University at Albany 1985 University of Wisconsin - Milwaukee 2012 University of Texas - Dallas 1986 University of Texas - Austin 2013 North Carolina State University 1987 IBM Almaden Research 2014 University of Wisconsin - La Crosse 1988 Brookhaven National Laboratory 2015 Rutgers University - New Brunswick 1989 University of Washington 2016 University of Arkansas - Fayetteville 1990 National Institute of Standards 2017 Portland State University 1991 Rutgers University – Piscataway 2018 University of New Hampshire 1992 University of California - Irvine 7
NOTTINGHAM PRIZE The Nottingham Prize was originally established in 1966 from contributions given in memory of Professor Wayne B. Nottingham of the Massachusetts Institute of Technology by his many friends and associates. The prize, currently consisting of a certificate and $1,500, is awarded to the best student paper presented at the conference. This prize represents a seminal honor since many Nottingham winners have gone on to become leaders in the field of surface science. In addition, you and your advisor’s names are added to the Nottingham Wikipedia website. PREVIOUS NOTTINGHAM WINNERS Year Winner Institution PI 1966 L. F. Cordes University of Minnesota W. T. Peria 1967 D. Steiner Massachusetts Institute of Technology E. P. Gyftopoulos J.V. Hollweg Massachusetts Institute of Technology E. P. Gyftopoulos 1968 E. Ward Plummer Cornell University T. N. Rhodin 1969 John C. Tracy Cornell University J. M. Blakely 1970 J. M. Baker Cornell University J. M. Blakely 1971 D. P. Smith University of Minnesota W. T. Peria 1972 W. Henry Weinberg University of California, Berkeley R. Merrill 1973 J. R. Bower Bartol Research Foundation J. M. Chen 1974 N. J. Dionne Cornell University T. N. Rhodin T. Gustafsson Chalmers University of Technology P. O. Nillson 1975 L. C. Isett Cornell University J. M. Blakely 1976 J. A. Knapp Montana State University G. A. Lapeyre 1977 S.-L. Weng University of Pennsylvania E. W. Plummer 1978 Gwo-Ching Wang University of Wisconsin, Madison M. G. Lagally 1979 Wilson Ho University of Pennsylvania E. W. Plummer 1980 R. Di Foggio University of Chicago R. Gomer Harry J. Levinson University of Pennsylvania E. W. Plummer 1981 Ruud M. Tromp FOM Institute for Atomic & Molecular F. W. Saris Physics 1982 P. O. Hahn University of Hanover M. Henzler 1983 R. Raue University of Köln (Cologne) and KFA G. Güntherodt & M. Jülich Campagna 1984 M. Onellion Rice University G. K. Walters 1985 K. Gibson University of Chicago S. J. Sibener J. W. M. Frenken FOM Institute for Atomic & Molecular J. F. van der Veen Physics 1986 S. M. Yalisove University of Pennsylvania W. R. Graham 1987 John D. Beckerle Massachusetts Institute of Technology S. T. Ceyer 1988 Lee J. Richter Cornell University W. Ho 1989 J.-K. Zuo Rensselaer Polytechnic Institute C.-C. Wang 1990 Y.-W. Mo University of Wisconsin, Madison M. G. Lagally 1991 B. Swartzentruber University of Wisconsin, Madison M. B. Webb 8
1992 Thomas Michely KFA Jülich G. Comsa 1993 A. K. Swan Boston University M. El-Batanouny 1994 G. Rosenfeld KFA Jülich G. Comsa 1995 Marcus K. Weldon Harvard University C. Friend 1996 J. Carpinelli University of Tennessee E. W. Plummer B. Kohler Fritz Haber Institute M. Scheffler 1997 D. Gragson University of Oregon G. Richmond 1998 Barry C. Stipe Cornell University W. Ho M. S. Hoogeman FOM Institute & Leiden Univ. J. W. M. Frenken 1999 K. Pelhos Rutgers University T. E. Madey 2000 Lincoln Lauhon Cornell University W. Ho 2001 Gayle Thayer University of California, Davis & Sandia S. Chiang & R. Livermore Hwang 2002 Denis Potapenko Rutgers University B. J. Hinch 2003 John Pierce University of Tennessee E. W. Plummer & J. Shen 2004 Peter Wahl Max Planck Inst. for Solid-State Physics K. Kern 2005 Nathan Guisinger Northwestern University M. Hersam 2006 Mustafa M. Ozer University of Tennessee-Knoxville J. R. Thompson & H. H. Weitering Paul C. Snijders Delft University of Technology H. H. Weitering & T. M. Klapwijk 2007 P. Maksymovych University of Pittsburgh J. T. Yates, Jr. 2008 Brett Goldsmith University of California, Irvine P. G. Collins 2009 Alpha T. N' Diaye University of Köln (Cologne) T. Michely 2010 Heather Tierney Tufts University C. Sykes 2011 Tanza Lewis University of California, Irvine J. Hemminger & B. Winter 2012 Daniel Schwarz University of Twente B. Poelsema 2013 Benjamin A. Gray University of Arkansas - Fayetteville J. Chakhalian 2014 Donna A. Kunkel University of Nebraska - Lincoln A. Enders 2015 Christoph Große Max Planck Inst. for Solid State Physics K. Kern Amanda Larson University of New Hampshire K. Pohl 2016 Charlotte Herbig University of Köln (Cologne) T. Michely 2017 Erik S. Skibinski Cornell University M. A. Hines 2018 Felix Lüpke FZ Julich B. Voigtländer 9
PROGRAM SUMMARY SUNDAY 4:00 – 6:00 PM Check In Libra Community Area Office 5:00 – 8:00 PM Welcome Reception Physical Sciences Building Atrium MONDAY 7:45 – 8:45 AM Breakfast, Registration, and HCEC Atrium Setup of Posters and Exhibits 8:45 – 9:00 AM Opening Remarks HCEC 101 9:00 – 10:00 AM Oral Presentations HCEC 101 10:00 – 10:20 AM Coffee Break HCEC Atrium 10:20 – 11:00 AM Keynote Presentation HCEC 101 Donna Chen, University of South Carolina 11:00 – 12:20 PM Oral Presentations HCEC 101 12:20 – 1:20 PM Lunch HCEC Atrium 1:20 – 2:00 PM Keynote Presentation HCEC 101 Vincent Smentkowski, General Electric 2:00 – 3:00 PM Oral Presentations HCEC 101 3:00 – 3:20 PM Coffee Break HCEC Atrium 3:20 – 4:00 PM Oral Presentations HCEC 101 4:00 – 6:00 PM Poster Session HCEC Atrium 6:30 – 8:30 PM Annual Picnic Rahman Residence 8:30 – 9:30 PM General Committee Meeting Rahman Residence TUESDAY 8:00 – 9:00 AM Breakfast HCEC Atrium 9:00 – 9:20 AM Oral Presentation HCEC 101 9:20 – 10:00 AM Keynote Presentation HCEC 101 Bjørk Hammer, Aarhus University 10:00 – 10:20 AM Coffee Break HCEC Atrium 10:20 – 12:00 PM Nottingham Presentations HCEC 101 12:00 – 1:00 PM Lunch HCEC Atrium 1:00 – 2:00 PM Lab Tours Details provided at meeting 2:00 – 3:15 PM Nottingham Presentations HCEC 101 3:15 – 3:35 PM Coffee Break HCEC Atrium 10
3:35 – 4:50 PM Nottingham Presentations HCEC 101 4:50 – 5:30 PM Student Involvement & Networking HCEC 101 6:30 – 8:30 PM Nottingham Banquet Details provided at meeting 8:30 – 10:30 PM Nottingham Celebration Details provided at meeting WEDNESDAY 7:00 – 9:00 AM Check Out Libra Community Area Office 8:00 – 8:50 AM Breakfast HCEC Atrium 8:50 – 9:30 AM Invited Presentation HCEC 101 Ludwig Bartels, UC – Riverside 9:30 – 10:10 AM Oral Presentations HCEC 101 10:10 – 10:30 AM Coffee Break HCEC Atrium 10:30 – 11:30 AM Oral Presentations HCEC 101 11:30 – 11:45 AM Closing remarks HCEC 101 11:45 AM Lunch HCEC Atrium 11
CONFERENCE AGENDA SUNDAY 4:00 – 6:00 PM Check In Libra Community Area Office 5:00 – 8:00 PM Welcome Reception Physical Sciences Building Atrium 12
MONDAY 7:45 – 8:45 AM Breakfast, Registration, and Harris Corporation Setup of Posters and Exhibits Engineering Center 8:45 – 9:00 AM Opening Remarks 9:00 – 10:00 AM Session 1: William Kaden, University of Central Florida 9:00 AM O.1 Sulfur-Metal Interactions on Ag(100) and Ag(110) at Low Coverages Peter Spurgeon 9:20 AM O.2 Defect-laden Hexagonal Boron Nitride for the Reduction of Carbon Dioxide Richard Blair 9:40 AM O.3 Electrochemical Catalysis at Grain-Boundary Surface Sites Xiaofeng Feng 10:00 – 10:20 AM Coffee Break 10:20 – 11:00 AM Keynote Presentation K.1 Designing New Heterogeneous Catalysts Through Metal-Metal Interactions Donna Chen, Department of Chemistry & Biochemistry, University of South Carolina 11:00 – 12:20 PM Session 2: Seth King, University of Wisconsin – La Crosse 11:00 AM O.4 Defect engineering of 2D-Transition Metal Dichalcogenides by Incorporation of Excess Metal Atoms into its Crystal Structure Paula Coelho 11:20 AM O.5 Atomically Precise Graphene Nanoribbons: From Synthesis to Devices Alexander Sinitskii 11:40 AM O.6 Fluctuation-Induced Transport from Freestanding Graphene Paul Thibado 12:00 PM O.7 Nitrogen vacancy on hexagonal boron nitride as single orbital catalyst: A first principles study Duy Le 12:20 – 1:20 PM Lunch 13
1:20 – 2:00 PM Keynote Presentation K.2 The Importance of Surface Modification and Analysis in the Industrial Sector Vincent Smentkowski, Structural Materials Characterization, General Electric Global Research Center 2:00 – 4:00 PM Session 3: Paul Thibado, Univ. of Arkansas, Fayetteville 2:00 PM O.8 Mechanistic understanding of hydrocarbon fuels formation via CO hydrogenation reaction on 2D catalytic materials Mihai Vaida 2:20 PM O.9 Crystal Structure Reconstruction on Surface of Quantum Spin Liquid Candidate -RuCl3 Zhongwei Dai 2:40 PM O.10 Nonvolatile voltage controlled molecular spin state switching Peter Dowben 3:00 – 3:20 PM Coffee Break 3:20 PM O.11 Defect Doping ZnO Thin-Films with - Radiation Seth King 3:40 PM O.12 Rational Computational Design of Efficient Electro-catalysts for Hydrogen Fuel Cell Cathodes Sergey Stolbov 4:00 – 6:00 PM Poster Session Atrium 6:30 – 8:30 PM Annual Picnic Rahman Residence 8:30 – 9:30 PM General Committee Meeting Rahman Residence 14
TUESDAY 8:00 – 9:00 AM Breakfast Harris Corporation Engineering Center 9:00 – 9:20 AM Oral Presentation O.13 Reduced Graphene Oxide-Mediated Gold/Cobalt Oxide Catalyst with Strong Metal-Support Interaction for CO Oxidation Shouhua Xie 9:20 – 10:00 AM Keynote Presentation K.3 Machine learning for atomistic structure search Bjørk Hammer, Department of Physics and Astronomy, Aarhus University 10:00 – 10:20 AM Coffee Break 10:20 – 12:00 PM Nottingham Session 1: Brian Swartzentruber, CINT/Sandia Nat’l Labs 10:20 AM N.1 Mechanistic Studies of Reducible Metal Oxides as Hydrodeoxygenation Catalysts Akbar Mahdavi-Shakib 10:45 AM N.2 Redox active Organic Chains on Au(111) for single site catalysis: A first- principles study Naseem Ud Din 11:10 AM N.3 Encapsulation of Metal Nanoparticles Near the Surface of Graphite Ann Lii-Rosales 11:35 AM N.4 Well-Defined Pt/CeO2 catalysts synthesized by Atomic Layer Deposition Bochuan Song 12:00 – 1:00 PM Lunch 1:00 – 2:00 PM Laboratory Tours 2:00 – 3:15 PM Nottingham Session 2: Carl Ventrice, SUNY Polytechnic Institute 2:00 PM N.5 Normal-Stress-Induced Mechanochemical Decomposition of Methyl Thiolates on Cu(100) Alejandro Boscoboinik 2:25 PM N.6 Symmetry Considerations for Interactions on MoS2 Prescott Evans 2:50 PM N.7 Distinct multiple fermionic states in a single topological metal M. Mofazzel Hosen 15
3:15 – 3:35 PM Coffee Break 3:35 – 4:50 PM Nottingham Session 3: Karsten Pohl, University of New Hampshire 3:35 PM N.8 Oxidation of Semiconductors and Semimetals by Supersonic Beams of O2 with Scanning Tunneling Microscopy Visualization Ross Edel 4:00 PM N.9 Growth and Characterization of Large-Area Ultrathin InSe Hadallia Bergeron 4:25 PM N.10 Borophene and its Functionalization Xiaolong Liu 4:50 – 5:30 PM Student Involvement & Networking Opportunity 6:30 – 8:30 PM Nottingham Banquet Details Provided at Meeting 8:30 – 10:30 PM Nottingham Celebration Details Provided at Meeting 16
WEDNESDAY 7:00 – 9:00 AM Check Out Libra Community Area Office 8:00 – 8:50 AM Breakfast Harris Corporation Engineering Center 8:50 – 9:30 AM Invited Presentation I.1 Preparation of Transition Metal Di- and Trichalcogenides: From vdW Semiconducting Films to vdW 1D Metallic Nanowires Ludwig Bartels, Department of Chemistry, University of California – Riverside 9:30 – 10:10 AM Session 4: Talat Rahman, University of Central Florida 9:30 AM O.14 3d Molecular assembly structure and tilt-angle evaluation with non- contact AFM Karsten Pohl 9:50 AM O.15 Development and characterization of MoN/Ru(0001) and Mo2N/Ag(100) thin-films for model-catalysis applications William Kaden 10:10 – 10:30 AM Coffee Break 10:30 – 11:30 AM Session 5: Mihai Vaida, University of Central Florida 10:30 AM O.16 Rubrene vs. fluorine-functionalized reubrene molecules on a metal surface: self-assembly, electronic structure and energy alignment of a monolayer of Ag(100) Jonathan Viereck 10:50 AM O.17 Electron Induced Disordering and Decomposition of SAMs Carl Ventrice 11:10 AM O.18 The molecule deprotonation and the excitation spectrum of Ag44(MNBA)2, Zahra Hooshmand 11:30 – 11:45 AM Closing Remarks 11:45 AM Boxed Lunch 17
ABSTRACT BOOK KEYNOTE PRESENTATIONS Pages K.1 – K.3 INVITED PRESENTATION Page I.1 NOTTINGHAM PRESENTATIONS Pages N.1 – N.10 ORAL PRESENTATIONS Pages O.1 – O.18 POSTER PRESENTATIONS Pages P.1 – P.18 18
KEYNOTE Designing New Heterogeneous Catalysts Through Metal-Metal Interactions Donna A. Chen, Amy J. Brandt, Thathsara D. Maddumapatabandi, Deependra M. Shakya, and Sharfa Farzandh Department of Chemistry and Biochemistry, University of South Carolina, 631 Sumter Street, Columbia, SC, 29208, USA Email: dachen@sc.edu Bimetallic catalysts often exhibit superior activity compared to their monometallic counterparts. The bimetallic Pt-Re system has been used for decades in industrial reforming reactions, and more recently Pt- Re catalysts have demonstrated enhanced activity for the water gas shift (WGS) reaction and aqueous phase reforming of polyols. However, the exact origins of the superior activity of Pt-Re over pure Pt are not fully understood; specifically little is known about the role of oxidized Re under reaction conditions. In order to understand this improved activity of Pt upon addition of Re, it is necessary to control bimetallic cluster composition, cluster sizes and oxidation states of the Re, as well as to probe Re oxidation states under reaction conditions. We have prepared and characterized Pt-Re bimetallic clusters on titania, which is the most active support for WGS. Experiments were carried out in an ultrahigh vacuum chamber directly coupled to a microreactor so that activity could be evaluated under catalytically relevant pressures while surfaces could be characterized using X-ray photoelectron spectroscopy and infrared absorption reflection spectroscopy before and after reaction. The greatest WGS activity was observed for surfaces consisting of Pt on Re. While the presence of rhenium oxide did not improve catalytic activity, Pt-Re interactions were found to decrease CO poisoning on the bimetallic catalyst. The role of metal-support interactions was investigated by depositing bimetallic clusters on a non-interacting graphite support and comparing with activity on titania. K.1
KEYNOTE The Importance of Surface Modification and Analysis in the Industrial Sector Vincent Smentkowski General Electric – Research, Structured Materials - Characterization, 1 Research Circle, Building K1 1d41, Niskayuna, NY, 12309, USA Email: smentkow@ge.com The composition, microstructure, phase, chemical bonding, electronic states, and/or texture at the surfaces of material is often different than that of the bulk. The outer surface is where many material interactions/reactions take place [1,2,3]. Surface scientists and analysts are grouped into three different categories: (1) researchers that perform a complete characterization of clean, single crystal surfaces, (2) researchers that study the interaction of adsorbates onto clean surfaces under ultrahigh vacuum conditions, or (3) researchers that are analyzing as-received, real-life parts. The first two groups of scientists are often trying to understand fundamental phenomena under well controlled conditions and are often in academia. The later group of researchers, are often found in an industrial setting, and are usually classified as applied surface scientists/analysts. Often their samples contain multiple components and as the samples are transferred from ambient conditions into the surface analysis instrument, are not clean. The field of surface analysis rapidly grew in the late 1960’s and early 1970’s with the introduction of commercial surface analysis instrumentation [1,2, 3]. It is of interest and importance to note that the first surface analysis instruments such as Auger Electron Spectroscopy (AES) [4-6], X-ray Photoelectron Spectroscopy (XPS) [7], Ion Scattering Spectroscopy (ISS) [8] and Secondary Ion Mass Spectroscopy (SIMS) [9, 10] were developed at, or in collaboration with, industrial research laboratories such as General Electric[4-6], Hewlett-Packard [7], 3M [8], Knolls Atomic Power Laboratory-General Electric [9, 10], respectively. These first surface analysis instruments were designed and built to study practical phenomena in real material systems. My talk will summarize my career path, which started with studies of adsorbates on atomically clean single crystal substrates and developed into performing surface analysis on real life samples to support the development of next generation industrial materials and products. The roles both professional societies and collaborations (both internal within GE and external) had will be highlighted. I will discuss the characterization challenges faced in an industrial setting where the surface analyst is often provided non- ideal samples and is asked to provide both a rapid and complete analysis of the sample. The criticality of talking with the person that submits the sample to understand their issue(s) and what they expect from the analysis (as well as being certain they realize possible complications) will be highlighted via real samples. References: [1], C.B. Duke, J. Vac. Sci. Technol A 2(2) (1984) 139 [2] Introduction to Surface Chemistry and Catalysis, 2nd Ed., Gabor A Somorjai and Yimin Li Eds, John Wiley and Sons, 2010. [3] Surface Analysis and Techniques in Biology, Vincent S. Smentkowski, Editor, Springer, 2014. [4] L.A. Harris, General Electric Rept No 67-C-201 (1967); J. Appl Phys. 39 (1968) 1419 [5] L.A. Harris J. Vac. Sci. Technol 11 (1974) 23 [6] G. Gergely, Vacuum 45 (2/3) (1994) 311 [7] In cooperation with Siegbahn, a group of engineers from Hewlett-Packard in the USA (Mike Kelly, Charles Bryson, Lavier Faye, Robert Chaney) produced the first commercial monochromatic XPS instrument in 1969; http://en.wikipedia.org/wiki/X-ray_photoelectron_spectroscopy ; M. A. Kelly, J. Chem. Educ, 81(12) (2004) 1726. [8] D.P. Smith, J. Appl Phys 38 (1967) 340; P. S. Wang, W. E. Moddeman, L. D. Haws, T. N. Wittberg, J. A. Peters “Surface Studies of Plastic-Bonded E£TN and RDX by X-Ray Photoelectron Spectroscopy (XPS) and Ion-Scattering Spectroscopy (ISS)” http://www.osti.gov/bridge/servlets/purl/6254094/6254094.pdf [9] J.A. McHugh and J.C. Sheffield, J. Applied Physics 35 (3) (1964) 512. [10] J.A. McHugh and J.C. Sheffield Anal. Chem. 39(3) (1967) 377 K.2
KEYNOTE Machine learning for atomistic structure search Bjørk Hammer Department of Physics and Astronomy, Aarhus University, Ny Munkegade 120, Building 1520, DK-8000 Aarhus C, Denmark Email: hammer@phys.au.dk During structure search with Density Functional Theory (DFT) databases of structure-energy relations are established that may be used to direct and speed up the structure search itself. In this talk, I describe how machine learning methods such as clustering and regression may assist Monte Carlo [1] and evolutionary search methods [2-5] in finding the optimum structure of molecules and inorganic surface reconstructions. One key element of using machine learning methods in conjunction with DFT is the possibility of decomposing the total DFT energy into atomic contributions. Owing to the nature of the Kohn-Sham equations (the one-electron Schrödinger Equation) such a decomposition is ordinarily not supported. Nonetheless, our experience shows that the atomic energies are highly meaningful and lead to a speed up of the structure determination when used appropriately. As a final topic, I will demonstrate a search strategy in which the computer collects knowledge of atomistic interactions while searching, which eventually enables it to search in a rational way [6]. This approach combines image recognition and reinforcement learning strategies and follow the recent advancements by Google Deep Mind in their development of the Alpha Go Zero method for playing the board game Go. [1] Machine learning enhanced global optimization by clustering local environments to enable bundled atomic energies, S.A. Meldgaard, E.L. Kolsbjerg, B Hammer, The Journal of Chemical Physics 149, 134104 (2018). [2] Exploration versus exploitation in global atomistic structure optimization, M.S. Jørgensen, U.F. Larsen, K.W. Jacobsen, B. Hammer, The Journal of Physical Chemistry A 122, 1504-1509 (2018). [3] On-the-fly machine learning of atomic potential in density functional theory structure optimization, T.L. Jacobsen, M.S. Jørgensen, B. Hammer, Physical Review Letters 120, 026102 (2018). [4] Atomic energies from a convolutional neural network, X Chen, M.S. Jørgensen, J Li, B Hammer, Journal of Chemical Theory and Computation 14 (7), 3933-3942 (2018). [5] Neural-network-enhanced evolutionary algorithm applied to supported metal nanoparticles, E.L. Kolsbjerg, A.A. Peterson, B Hammer, Physical Review B 97 (19), 195424 (2018). [6] Atomistic Structure Learning, M.S. Jørgensen, H.L. Mortensen, S.A. Meldgaard, E.L. Kolsbjerg, T.L. Jacobsen, https://arxiv.org/abs/1902.10501. K.3
INVITED Preparation of Transition Metal Di- and Trichalcogenides: From vdW Semiconducting Films to vdW 1D Metallic Nanowires Ludwig Bartels Department of Chemistry, University of California, Riverside, 124 Pierce Annex, Riverside, CA, 92521, USA Email: bartels@ucr.edu The transition metal triselenide (TMT) TaSe3 consist of Ta-Se3-Ta-Se3- stacks in which the central Ta- atoms column is separated from adjacent ones by a vdW gap owing to the Se3 trimers, much similar to how the Mo-atoms plane in MoSe2 is separated from the adjacent ones by vdW gaps owing to the Se planes on each of its sides. TaSe3 is metallic; motivated by the presence of a vdW gap between neighboring wires, we explored the cross-section dependent conductivity of the material along the wire direction. We found it to be unaffected by the wire dimension even down to 7nm, the smallest wires we could contact successfully. This is much different than what is observed for bulk conductors like copper, where the wire resistance increases dramatically as the wire dimension is decreases below the electron mean free path. Moreover, the current density that the nanowires can endure are on the order of ~108 A/cm2 and an electromigration energy barrier more than twice that of copper is found. In this talk, I will present how the TaSe3 nanowires are synthesized via chemical vapor deposition (CVD) on a SiO2/Si substrates at a temperature compatible with industry-standard back-end-of-line (BEOL) processing. The results show the potential that these quasi 1D van der Waals TMTs have for downscaled interconnect applications. Time permitting, I will also speak about our recent achievement in process control in the deposition of TMD films leading to wafer scale material. I.1
NOTTINGHAM Mechanistic Studies of Reducible Metal Oxides as Hydrodeoxygenation Catalysts A. Mahdavi-Shakib1,2,3* and B. G. Frederick1,2,3 *Nottingham Contestant 1 Laboratory for Surface Science and Technology (LASST), 2 Forest Bioproducts Research Institute (FBRI), 3 Dept. of Chemistry, University of Maine, Orono, ME 04469 Email: akbar.mahdavi@maine.edu Hydrodeoxygenation (HDO) reactions are a class of reactions that remove oxygen from molecules as H2O using H2 gas and a catalyst. Ruthenium supported titania (Ru/TiO2) catalysts are excellent catalysts for HDO of phenol to benzene,[1] an important reaction for the conversion of lignin part of biomass to biofuels. In the proposed mechanism, [2] HDO reaction takes place at the interface of Ru and TiO2, surface hydroxyls with basic character are required, and water acts as a co-catalyst.[2] We showed that HDO rates and selectivities of Ru/TiO2 catalysts strongly vary depending on the support crystal structure and preparation conditions. Surface hydroxyls of different TiO2 samples (anatase, rutile, and pyrogenic) were identified using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) experiments performed on vigorously cleaned surfaces and a facet-specific assignment was proposed using density functional theory (DFT) calculations performed by our collaborators. Electronic structure of TiO2 samples were studied using the reaction of vigorously cleaned TiO2 samples with H2/D2. Our results are used to derive structure-activity relationships for the Ru/TiO2 catalyzed HDO reactions. References: [1] C. Newman, X. Zhou, B. Goundie, I.T. Ghampson, R.A. Pollock, Z. Ross, M.C. Wheeler, R.W. Meulenberg, R.N. Austin, B.G. Frederick, Effects of support identity and metal dispersion in supported ruthenium hydrodeoxygenation catalysts, Applied Catalysis A: General, 477 (2014) 64-74. [2] R.C. Nelson, B. Baek, P. Ruiz, B. Goundie, A. Brooks, M.C. Wheeler, B.G. Frederick, L.C. Grabow, R.N. Austin, Experimental and Theoretical Insights into the Hydrogen-Efficient Direct Hydrodeoxygenation Mechanism of Phenol over Ru/TiO2, ACS Catal., 5 (2015) 6509-6523. N.1
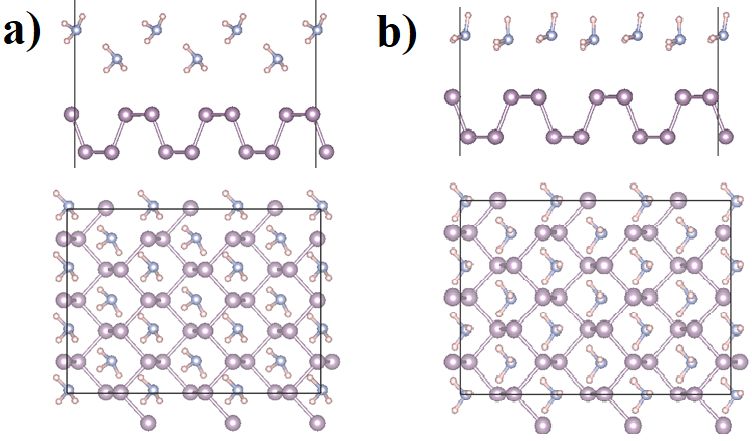
NOTTINGHAM Redox active Metal Organic Chains on Au(111) for single site catalysis: A first-principles study Naseem Ud Din*, Talat Rahman Department of Physics University of Central Florida Orlando 32826 *Nottingham Contestant Email: naseem.din@knights.ucf.edu A periodic network with uniform single metal active site, in coordination with redox-active organic ligands, is a promising class of materials for next generation single atom catalysts. Towards this quest and inspired by recent findings1, we have carried out first-principles density functional theory (DFT) based calculations of the geometrical and electronic structure and magnetic properties of several transition-metal-organic- chains (TM-C) both in gas phase as well as on Au(111) surface. Of particular interest are dipyridyltetrazine (DT), Bis-pyrimidinyl-tetrazine (BMTZ) and Bis-pyrimidine (BP) ligands used to design the TM-C with the single metal atoms (V, Cr, Mo, Fe, Co, Pt, Pd) as the coordination center. Our results suggest that TM-DT the TM centers form a fourfold coordination with nitrogen atoms of the ligand, making a TM-DT structure with high formation energy (~2.0 to 7.9 eV) suggesting that these chains can be stabilized. The charge analysis shows that TM centers loose charge while DT molecule gains charge which shows the oxidation of TM atoms and reduction of DT molecule. Moreover, by calculating the adsorption energies of CO, and O2 molecules on the TM atom sites of the chain we found that these molecules strongly bond to the TM atoms Mo, Cr, V, Fe and Co. The adsorption energies of CO, and O2 on theses metal centers ranges from 1.0 to 4.0 eV, while the adsorption energies of CO and O2 is very small on Pt and Pd center of chain. We analyzed the occupancy of d-orbitals of TM centers and found that TM atoms with partially occupied d-orbitals exhibit strong affinity, while the TM atoms with fully occupied d orbitals show weak affinity to the CO and O2 molecule. We investigated the oxidation state of these metal centers using core level shift. For Pt and V, the calculated +2 oxidation state is consistent with the experimentally reported value. The O2 molecule undergoes dissociation while adoption on V center and causes strong distortion to V-DT chain. In case of TM-BMTZ and TM-BP, the TM centers form a fourfold coordination with nitrogen atoms of the ligand, making a TM-BMTZ planar chain structure while TM-BP lead to a non-planar arrangement in gas phase. Bader charge analyses show that while the TM atom is oxidized in both formations, the effect of BMTZ is more reducing than that of BP. Spin polarized DFT calculations show that Fe-BP forms a ferromagnetic (FM) ground state while V-BP results in an anti-ferromagnetic (AFM) ground state. On the other hand, both Fe-BMTZ and V-BMTZ yield AFM ground state. Moreover, adsorption of atomic oxygen on the metal site distorts the planarity of TM-BMTZ and draws on average a charge of 0.65e ±0.05 from the metal center. Both Fe and V lose 0.24e and 0.4e respectively while forming the O-TM-BP complex, and 0.21e and 0.39e while forming O-TM-BMTZ complex. We further show that the Au(111) substrate draws electrons from V center of V-BP chain and altering the geometric structure and related properties. By analyzing the magnitude of charge transfer from V-BP to Au(111) substrate we found that charge redistribution is limited to the topmost layer of Au(111) surface. We discuss the implications of our findings on ongoing experiments. References: [1] C. D. Tempas, D. Skomski, B. J. Cook, D. Le, K. A. Smith, T. S. Rahman , K. G. Caulton, and S. L. Tait, Chemistry-A European Journal, 24 (2018) 15852-15858 N.2
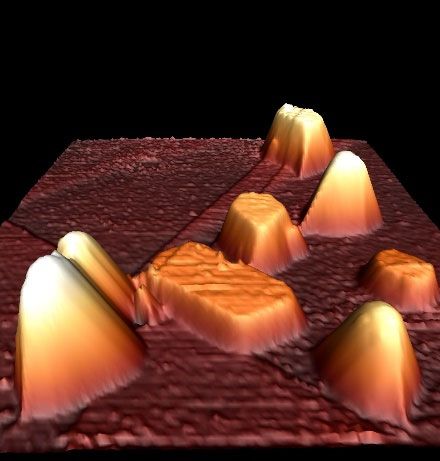
NOTTINGHAM Encapsulation of Metal Nanoparticles Near the Surface of Graphite Ann Lii-Rosales1,2* and Patricia A. Thiel1,2,3 1 The Ames Laboratory–USDOE, Ames, IA 50011, USA 2 Department of Chemistry, Iowa State University, Ames, IA 50011, USA 3 Department of Materials Science and Engineering, Iowa State University, Ames, IA 50011, USA *Nottingham Contestant Email: ylii@iastate.edu In this study we investigate the phenomenon of metal encapsulation near the surface of a layered material, graphite, by combining scanning tunneling microscopy (STM), X-ray photoelectron spectroscopy (XPS), and density functional theory (DFT) calculations. We develop a synthetic strategy that can encapsulate metals when two conditions are met: (i) defects must be generated on the graphite surface, via argon ion bombardment, and (ii) the graphite surface with ion-induced defects must be held at elevated temperature for metal deposition from the vapor phase. Our synthetic strategy is effective for a variety of metals, including rare earth metals Dy and Gd, transition and noble metals Cu, Fe, Ru and Pt. Characterization via STM shows that the metals form nanocrystalline islands composed of multiple metal layers. STM also reveals atomic features on top of the islands: A contiguous, carpet-like carbon lattice that drapes over the islands, which serves as firm evidence of encapsulation. Figure 1 showcases encapsulated Cu islands. In addition, XPS shows that the metal islands are metallic. Furthermore, Ru islands remain metallic even after long-term exposure to ambient conditions. Despite the strategy’s considerable robustness, it is ineffective Figure 1. STM image of for Ag and Au. This is initially a prediction drawn from DFT results and encapsulated Cu islands. Inset then later attested by experiments. We find that the relative energetics of shows a schematic of a metal a single metal atom – adsorbed on the graphite surface vs. encapsulated between graphene layers – are of key significance. This shows that cluster embedded beneath a predicative capability can develop when coupling theoretical insights graphene overlayer. Image with the synthetic strategy. size is 750 nm x 750 nm. Our work opens up the possibility to fabricate arrays of nanoscale heat sinks (Cu) or nanomagnets (Fe), while the metals are protected from oxidation. We also envision that our synthetic strategy can be extended to other layered materials to create interesting nanostructures encapsulated near their surface. N.3
NOTTINGHAM Well-Defined Pt/CeO2 catalysts synthesized by Atomic Layer Deposition Bochuan Song1* and Helena E. Hagelin Weaver1 *Nottingham Contestant 1 Chemical Engineering, University of Florida, Gainesville, FL 32611, USA Email: bsong94@ufl.edu Using Atomic Layer Deposition (ALD) to synthesize heterogeneous catalysts with atomic level precision has gained increasing interest in recent years. In our laboratory we have synthesized well-defined heterogeneous catalysts by depositing Pt onto various CeO2 nanoshapes using ALD. The benefit of using CeO2 nanoshapes is that the surface facets of each shape are well defined, as the CeO2 octahedra have (111) surface terminations, the CeO2 cubes expose (100) surface facets, while the CeO2 rods consist of (100) and (110) surface facets. Therefore, this allows us to determine the influence of the support oxide surface structure on the active metal and ultimately on the probe reactions. These catalysts can also facilitate identifying structure-activity relationships, as well as comparisons to theoretical studies and ultra-high vacuum surface science studies on single crystals. By using ALD during catalyst synthesis we obtain better control over the active metal deposition, compared with conventional catalyst preparation techniques. We have shown that by tuning the ALD conditions, such as precursor temperature, deposition temperature and dose length, we can carefully control the Pt loading on the CeO2 nanoshapes. The Pt loading and the CeO2 surface facet have significant effects on the CO oxidation, which we have used as our initial probe reaction. N.4
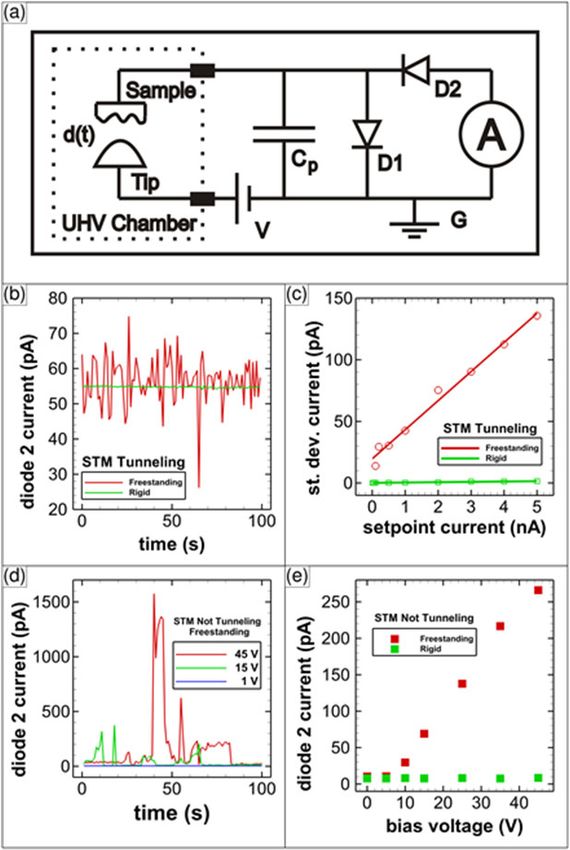
NOTTINGHAM Normal-Stress-Induced Mechanochemical Decomposition of Methyl Thiolates on Cu(100) A. M. Boscoboini1* and W. T. Tysoe1 *Nottingham Contestant 1 Department of Chemistry & Biochemistry, University of Wisconsin Milwaukee, 3210 N Cramer St, Milwaukee, WI 5321, USA Email: boscobo2@uwm.edu Improving lubricant additives and identifying how they react at surfaces is important for reducing macroscopic and microscopic friction (e.g. cars and micromachines)[1]. The mechanically induced rate of a chemical reaction is described by the Bell model [2] where the rate constant for a reaction under an applied stress is given by 0exp Δ ‡/ BT , here B is the Boltzmann constant, 0 is the reaction rate in the absence of stress, Δ ‡ is known as the activation volume and is the absolute temperature. While exponential increases in reaction rates with stress have been reported, there are currently no quantitative measurements. This is addressed by using contact-mode AFM to measure the kinetics of mechanochemically induced C-S bond cleavage methyl thiolate on Cu(100) in ultrahigh vacuum. The experimental rate of methyl thiolate decomposition increases exponentially with stress (Fig. 1, ■) with an activation volume of 37.6 ± 0.9 Å3, but this value does not agree quantitatively with the predictions of the Bell model (‒‒). However, the results of DFT calculations [3] of the energy barrier for methyl thiolate decomposition under the influence of a normal force (‒‒) provide excellent agreement with experiment. An analysis of the results indicates that the effect of a normal stress is to both Fig. 1. Comparison of experiment and theory. destabilize the initial state and to lower the energy of Pressure-induced methyl thiolate decomposition the transition state. These results will enable the on Cu(100) in ultrahigh vacuum. description of mechanochemical reactions to be placed on a firm theoretical footing that will ultimately enable them to be quantitatively predicted. References: [1] O.M. Braun, A.G. Naumovets, Surf. Sci. Rep. 60, 79–158 (2006). [2] G. I. Bell, Cell Biophys. 1, 133 (1979). [3] H. L. Adams, UWM Theses/Dissertations. 1566, 145, (2017). N.5
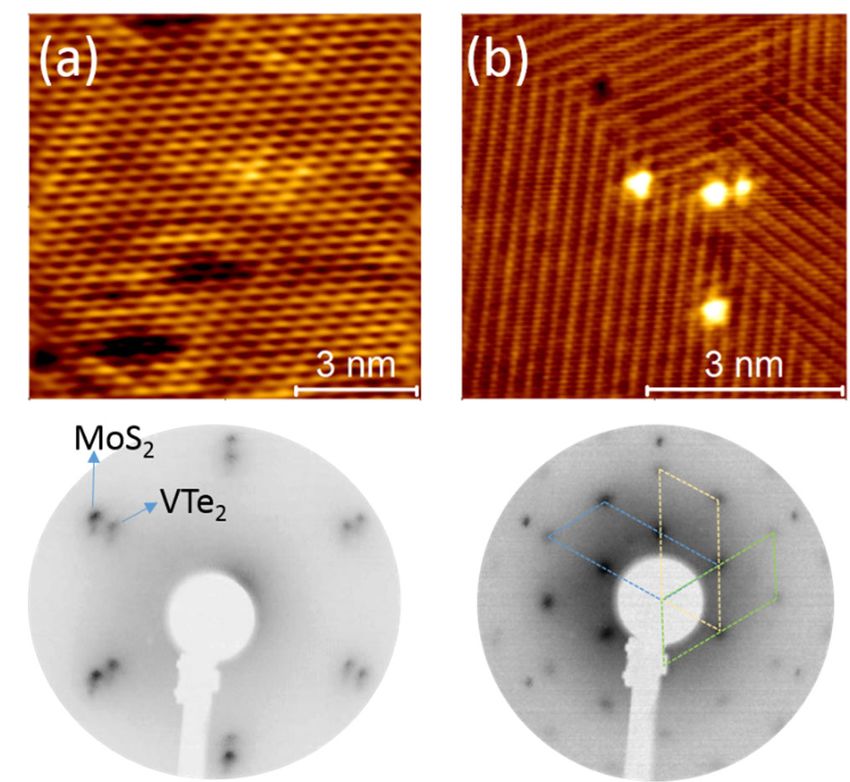
NOTTINGHAM Symmetry Considerations for Interactions on MoS2 Prescott E. Evans,1* and Peter A. Dowben1 *Nottingham Contestant 1 Department of Physics and Astronomy, University of Nebraska-Lincoln, 855 N 16th St., Lincoln, Nebraska 68588-0299, United States. Email: prescott.evans@huskers.unl.edu MoS2 is a well-recognized 2D material and catalyst. From extensive studies of the electronic band structure, it is now understood that, like the other transition metal dichalcogenides [1,2], the electronic structure of monolayer MoS2 differs from bulk MoS2(0001). Furthermore, other electronic band structure studies have found marked differences between the theoretical surface symmetry of the Brillouin zone and an observed retention of high symmetry in MoS2(0001) band structure [3]. The massive role of symmetry in the adsorbate-MoS2 interaction cannot be underscored enough, and is another experimental verification of the retention of local symmetry in the electronic structure. The role of adsorbate frontier orbital symmetry and preservation of MoS2 surface symmetry has been demonstrated by comparing the adsorption of the isomers of diiodobenzene on MoS2 [4]. Experiments indicate that the adsorption of 1,3-diiodobenzene on MoS2(0001) is much more facile compared to other isomers, contrary to negligible differences in calculated adsorption energies and electron affinities. The combination of density functional theory and experiment highlights frontier orbital symmetry as key to diiodobenzene adsorption on MoS2(0001). The surface symmetry of MoS2 also plays a role in defect creation at the MoS2(0001) surface, wherein defects and symmetry have a hand in catalytic activity and application. Surface reactions on MoS2 can be enhanced by defects [5, 6], but we have shown that surface reactions can also create defects, as in the example of methanol to methoxy reactions [7]. The difference in electronic structure and application of the monolayer from bulk is emphasized by the ability to anneal out defects for bulk MoS2(0001) [8], which will persist for the monolayer. From experiment it is clear moving forward theory must include further considerations of symmetry such as frontier orbital symmetry as well as monolayer and bulk considerations such as band bending, and interface charge transfer to gain more accurate insight into reaction mechanisms on MoS2. References: [1] I. Tanabe et al., J. Phys. Conden. Matter 28 (2016) 345503 [2] I. Tanabe, et al., Applied Physics Letters 108 (2016) 252103 [3] T. Komesu, et al., J. Phys.: Condens. Matter 26 (2014) 455501 [4] P. E. Evans, et al., Chem. Comm. (2019) submitted [5] T. B. Rawal, D. Le, T. S. Rahman, T. S., J. Phys. Chem. C 121 (2017) 7282-7293. [6] T. B. Rawal, D. Le, T. S. Rahman, J. Physics: Cond. Matter 29 (2017) 415201. [7] P. E. Evans, et al., J. Phys. Chem. C 122 (2018) 10042-10049 [8] P. E. Evans, H. K. Jeong, P. A. Dowben, MRS Advances (2018); doi: 10.1557/adv.2018.634 N.6
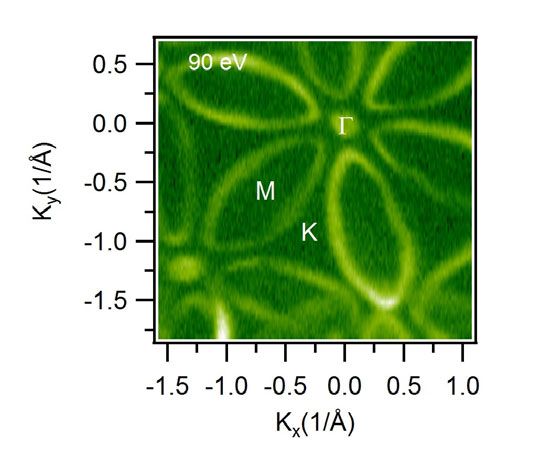
NOTTINGHAM Distinct multiple fermionic states in a single topological metal M. Mofazzel Hosen1, and M. Neupane1 1 Department of Physics, University of Central Florida, Orlando, FL 32816, USA Email: dolar.kabir@knights.ucf.edu Among the quantum materials that have recently gained interest are the topological insulators, wherein symmetry-protected surface states cross in reciprocal space, and the Dirac nodal-line semimetals, where bulk bands touch along a line in k-space. However, the existence of multiple fermion phases in a single material has not been verified yet. Using angle-resolved photoemission spectroscopy (ARPES) and first-principles electronic structure calculations, we systematically study the metallic material Hf2Te2P and discover properties, which are unique in a single topological quantum material. We experimentally observe weak topological insulator surface states and our calculations suggest additional strong topological insulator surface states. Our first-principles calculations reveal a one- dimensional Dirac crossing-the surface Dirac-node arc-along a high- Fig. 1. ARPES measured symmetry direction which is confirmed by our ARPES measurements. Fermi surface at a photon This novel state originates from the surface bands of a weak topological energy of 90 eV. High insulator and is therefore distinct from the well-known Fermi arcs in symmetry points are noted in semimetals. Our discovery opens a unique platform to study the interplay the plots. Here, Gamma is the between various novel topological states. center of the Brillouin zone and K is the corner and M is the mid-point between Ref. adjacent corners [1]. [1] M. M. Hosen, et al., Nat. Commun. 9, 3002 (2018). N.7
NOTTINGHAM Oxidation of Semiconductors and Semimetals by Supersonic Beams of O2 with Scanning Tunneling Microscopy Visualization Ross Edel* and Steven J. Sibener *Nottingham Contestant Department of Chemistry, University of Chicago, 5741 S Drexel Ave, Chicago, IL, 60637, USA Email: redel@uchicago.edu Our research examines the oxidation of semiconductor and semimetal surfaces using a novel, one- of-a-kind instrument that combines a supersonic molecular beam with an in-line scanning tunneling microscope (STM) in ultra-high vacuum. This new approach to surface reaction dynamics provides spatiotemporal information on surface oxidation over nanoscopic and mesoscopic length scales. We have uncovered the kinetic and morphological effects of oxidation conditions on three technologically relevant surfaces: Si(111)-7×7, highly oriented pyrolytic graphite (HOPG), and GaAs(110). A complete understanding of the oxidation mechanism of these surfaces is critical due to their technological applications and roles as model systems. Samples were exposed to O2 with kinetic energies from 0.4-1.2 eV and impingement angles 0-45° from normal, with STM characterization between exposures. In some cases, we were able to monitor the evolution of specific features by revisiting the same nanoscopic locations. Our study of Si(111)-7×7 revealed two oxidation channels, leading to the formation of dark and bright reacted sites. The dark sites dominated the surface and exhibited almost no site selectivity while the bright sites preferred the corner sites of the 7×7 unit cell. Our observations suggest that two adsorption pathways, trapping-mediated and direct chemisorption, occur simultaneously. On HOPG, we found that different oxygen energies, incident angles, and surface temperatures produce morphologically distinct etching features: Anisotropic channels, circular pits, and hexagonal pits. Reaction probability increased with beam energy and demonstrated non-Arrhenius behavior with respect to surface temperature, peaking at around 1375 K. Finally, oxidation of GaAs(110) was found to proceed by two morphologically distinct, competing mechanisms: a homogeneous process leading to layer-by-layer oxide growth, and a heterogeneous process with oxide islands nucleating from surface defects. The rates of both mechanisms change with O2 kinetic energy, with homogeneous oxidation dominating at lower energies (
NOTTINGHAM Growth and Characterization of Large-Area Ultrathin InSe Hadallia Bergeron1* and Mark C. Hersam2 *Nottingham Contestant 1 Department of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA 2 Department of Materials Science and Engineering, Department of Chemistry, and Department of Electrical Engineering and Computer Science, Northwestern University, Evanston, Illinois 60208, USA Email: hadalliabergeron2014@u.northwestern.edu The lack of large-area synthesis methods for emerging 2D materials presents a significant challenge for nanoelectronic devices and systems. In particular, InSe is a semiconducting van der Waals (vdW) material that possesses exceptional band-gap tunability as a function of thickness in the ultrathin limit. Despite its desirable electronic properties, InSe is a relatively under-investigated 2D material, primarily due to the fact that high-quality samples have only been achieved via mechanical exfoliation as opposed to large-area thin-film growth. While many of the extensively studied and synthesized vdW 2D material systems have relatively simple phase diagrams (e.g., MoS2), the complex phase diagram for InSe has hindered the development of large-area thin films, thus motivating more fundamental studies aimed at characterizing and understanding growth mechanisms. Towards that end, we use in operando X-ray diffraction (XRD) and diverse surface characterization techniques (e.g., X-ray photoelectron spectroscopy, Raman spectroscopy, atomic force microscopy, transmission electron microscopy, and selected area electron diffraction) to study the structural and compositional evolution of ultrathin InSe films grown by pulsed laser deposition and subsequent vacuum thermal annealing. The complexity of the indium– selenium phase diagram manifests itself during annealing, with the film initially passing through a partial In4Se3 crystallization at low temperatures and ultimately degrading into an In2Se3 matrix at higher temperatures. However, by monitoring all temperatures in operando, this methodology Fig. 1. in operando XRD of a PLD InSe efficiently determined that InSe films with no detectable thin-films on 300 nm SiO2 as a function impurity phases can be achieved at post-deposition annealing of annealing temperature, with the as- temperatures between 350 C and 450 C (Fig. 1). The deposited amorphous film in black. Peaks resulting InSe films possess high uniformity in stoichiometry indexed as InSe are denoted by the black and crystallinity over large areas with controlled thickness, dashed lines. thus providing an ultrathin InSe synthesis technique that is generalizable to a wide range of substrates suitable for nanoelectronic applications. N.9
NOTTINGHAM Borophene and its Functionalization Xiaolong Liu1* and Mark C. Hersam1,2 *Nottingham Contestant (current affiliation: Physics, Cornell University) 1 Applied Physics Graduate Program, Northwestern University, 2220 Campus Drive, Evanston, Illinois 60208-3108, USA 2 Department of Materials Science and Engineering, Department of Chemistry, Department of Medicine, Department of Electrical Engineering and Computer Science, Northwestern University, 2220 Campus Drive, Evanston, Illinois 60208-3108, USA Email: xiaolongliu2013@u.northwestern.edu Following theoretical predictions, 2D boron (i.e., borophene) was experimentally realized in late 2015, showing intriguing properties such as high in-plane anisotropy. As a synthetic 2D material on Ag(111) substrates, its structural properties such as intrinsic defects and polymorphs cannot be deduced from bulk boron. In particular, the complex lattice structures of borophene polymorphs remain to be experimentally verified. Given the dominant role of structural defects in 2D systems and the knowledge gap in borophene surface chemistry, we perform atomically-resolved study of borophene structures and chemical functionalization using ultra-high vacuum scanning tunneling microscopy (STM) and non-contact atomic force microscopy (nc-AFM). Borophene phases corresponding to the v1/6 and v1/5 models are found to coexist and intermix periodically in the form of line defects, giving rise to new borophene phases. To overcome the imaging ambiguity of conventional STM, we use CO-functionalized AFM to geometrically reveal features corresponding to B-B covalent bonds and assemble a growth phase diagram of borophene, confirming the v1/5 and v1/6 borophene models as the unifying structures for all observed borophene polymorphs even in newly discovered incommensurate phases. Towards the realization of borophene-based nanoelectronics, we chemically functionalize borophene with self-assembled organic molecules, resulting in the spontaneous formation of electronically abrupt lateral heterostructures that is further explored as a platform for covalent chemistry. Overall, our work informs both fundamental studies of borophene material properties and emerging efforts in realizing borophene-based nanoelectronics. N.10
ORAL Sulfur-Metal Interactions on Ag(100) and Ag(110) at Low Coverages Peter M. Spurgeon,1 Da-Jiang Liu,2 Junepyo Oh,3 Yousoo Kim,3 and Patricia A. Thiel1,2,4 1 Department of Chemistry, Iowa State University, Ames, IA, 50010, USA 2 Ames Laboratory of the USDOE, Ames, IA, 20010, USA 3 RIKEN Surface and Interface Science Laboratory, Wako, Saitama 351-0198, Japan 4 Department of Materials Science and Engineering, Iowa State University, Ames, IA, 50010, USA Email: peterms@iastate.edu Coinage metals (Cu, Ag, Au) are of interest due to their catalytic and plasmonic properties, especially when they are in the form of nanoparticles. Sulfur is an important adsorbate on these metals because sulfur is known to enhance mass transport of coinage metal nanoparticles. Recent studies of chemisorbed sulfur on the coinage metals have shown that even at extremely low sulfur coverages, well below 0.10 monolayers, indigenous metal atoms can react to from complexes on some surfaces. (A complex is an isolated species consisting of metal and sulfur atoms, and with well-defined shape, size, and stoichiometry.) In other systems, isolated adatoms or extended reconstructions are observed. For example, sulfur and copper form a heart-shaped Cu2S3 complex on Cu(111)1, whereas sulfur forms only isolated adatoms on Au(111)2. Here, we report a study of sulfur on Ag(110) and Ag(100) at multiple coverages. The main experimental technique is low-temperature scanning tunneling microscopy (LT-STM). At low coverages (~0.02 monolayer) we observe that sulfur forms isolated adatoms on Ag(110), but at higher coverages (~0.25 monolayer) there is evidence of AgS2 complexes coexisting with S adatoms. For sulfur on Ag(100), we observe that sulfur forms isolated adatoms within our entire experimental coverage range (0.01 - 0.1 monolayer). References: [1] H. Walen, D. -J. Liu, J. Oh, H. Lim, J. W. Evans, C. M. Aikens, Y. Kim, and P. A. Thiel. "Cu2S3complex on Cu(111) as a candidate for mass transport enhancement". Phys. Rev. B 91, 045426 (2015) [2] H. Walen, D. -J. Liu, J. Oh, H. Lim, J. W. Evans, Y. Kim, and P. A. Thiel. "Self-organization of S adatoms on Au(111): Rt.3R30 rows at low coverage" J. Chem. Phys. 143 014704 (2015). Fig. 1. STM image of AgS2 complexes and S adatoms at 0.25 ML. Image is 10 x10 nm2. O.1
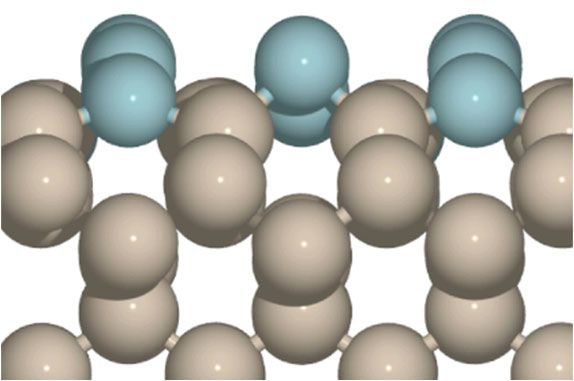
ORAL Defect-laden Hexagonal Boron Nitride for the Reduction of Carbon Dioxide Richard G. Blair1, Katerina L. Chagoya2, 1, Talat S. Rahman3 1 Florida Space Institute, University of Central Florida, 12354 Research Parkway, Suite 214 Orlando, FL 32826-2933 2 Mechanical Engineering, University of Central Florida, 12760 Pegasus Drive Orlando, FL 32828 3 Physics, University of Central Florida, 4000 Central Florida Blvd. - PSB 430 Orlando, FL 32816-2385 Email: richard.blair@ucf.edu Heterogeneous catalysis is often most rapid on surface discontinuities such as grain boundaries or steps. Much work has gone into growing non-equilibrium structures such as nanostructures with specific shapes to control surface discontinuities and enhances catalytic pathways. Improved catalysis is realized by controlling the localized molecular orbital structure, which can vary significantly from the bulk material. Catalytic activity can be introduced into surfaces previously thought of as inert through the introduction of discontinuities in the form of defects. Defects can be introduced through mechanical processing. Catalyst surfaces undergo plastic deformation when processed with forces Fig. 1. A nitrogen vacancy beyond the ultimate tensile strength of the bulk material producing and (VN) in the hexagonal boron mobilizing non-equilibrium defect structures. In highly defect-laded nitride sheet has a localized boron nitride surfaces (dh-BN), these defect structures can be exploited electronic structure that for the hydrogenation of olefins[1] and the reduction of carbon dioxide. facilitates catalysis. Defect- Controlling the reaction temperature allows realization of formic acid or free h-BN surfaces are not methanol. Application of a low energy, heterogeneous route to CO2 active. reduction that can be intensified using traditional chemical engineering principles will reduce emissions and produce a profitable product. References: [1] D. J. Nash, D. T. Restrepo, N. S. Parra, K. E. Giesler, R. A. Penabade, M. Aminpour, et al., ACS Omega, 1,(2016). O.2
You can also read

















































