Non-model organism journal club 3 September 2021 Verena Kutschera - WABI wiki
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Non-model organism journal club
3 September 2021
Verena Kutschera

What can we learn from this data? • We can learn a lot from comparative genomics, e.g. • Which variants lead to changes in phenotypes (incl. disease)? • Which genomic variation is shared and which variation is lineage-specific? • How are species related to each other? • This requires multiple genome alignments
How are we supposed to analyze this data?
• Genome alignment is complex
• Different aligners, different results
• Solution: reduce complexity to simplify the problem?
• Common limitations of alignment software
• Reference bias (aligning only regions from reference genome)
• Restriction to a single alignment in any column in any given genome (missing
multiple-orthology relationships from duplications)
• More genomes à increased computational requirements
Armstrong et al. 2020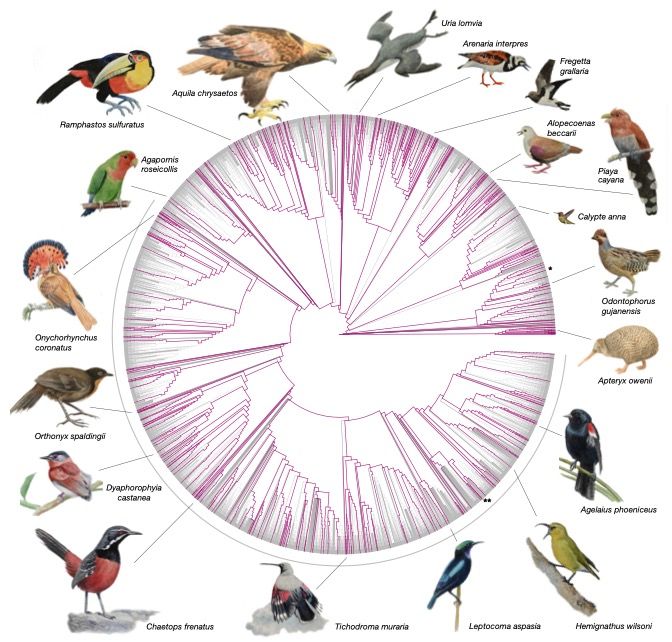
Progressive cactus
• Reference-free multiple
alignment
• Allows the detection of multiple-
orthology relationships
• Runtime scales linearly with the
number of genomes (unlike
“Star”, equivalent to previous
version Cactus from 2012)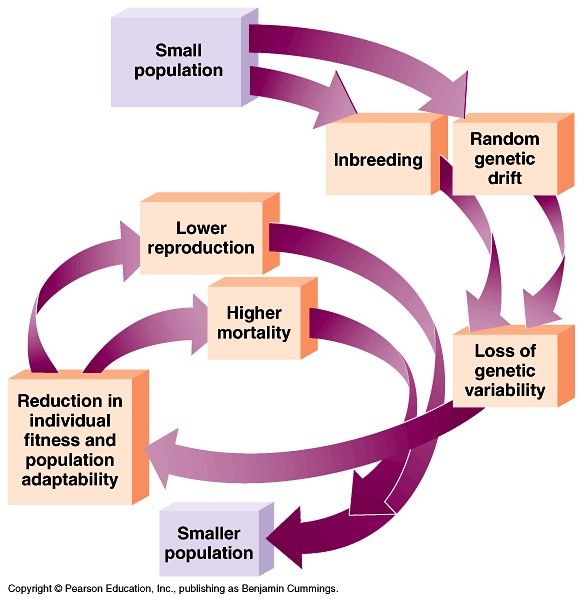
• A phylogenetic tree of the species is used as guide tree (from another source, e.g. timetree.org) • The guide tree is split into subproblems • An ancestral genome is constructed at each internal node of the guide tree • Each ingroup genome is aligned to its ancestral genome • The outgroup genome is used to find structural rearrangements (e.g. copy number variations) • The ancestral genome is used as input for subproblems further up the tree • Parent-child alignments are later combined to form the the full alignment
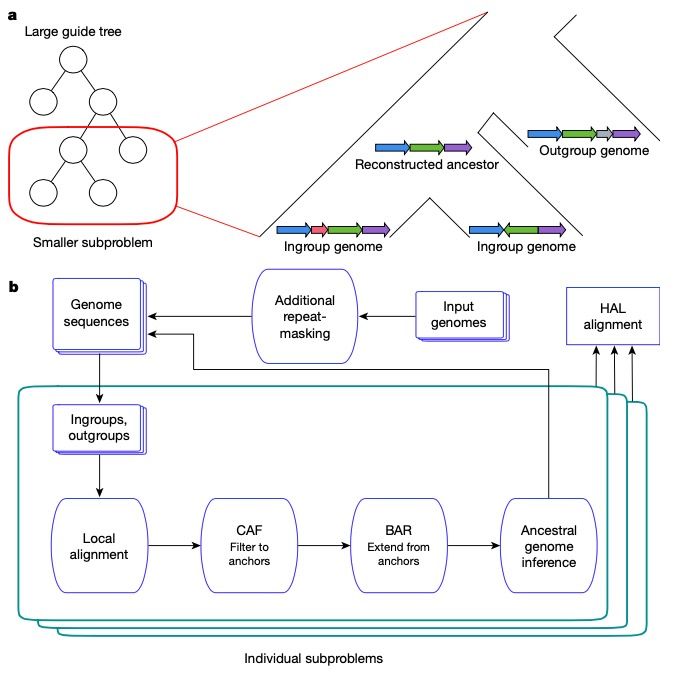
(full alignment) (LASTZ) (cactus graph)
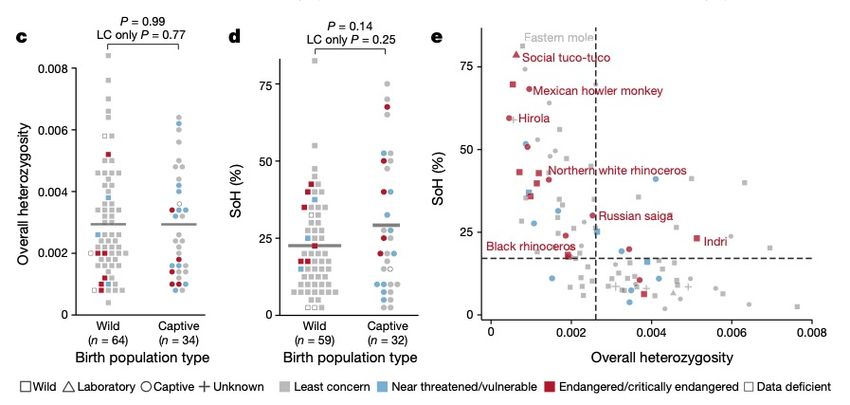
Implementation
• Runs on Toil, breaking the
problem into smaller pieces
• Supports container execution
(Docker & Singularity)
• Supports adding and removing
genomes without re-computing
the entire alignment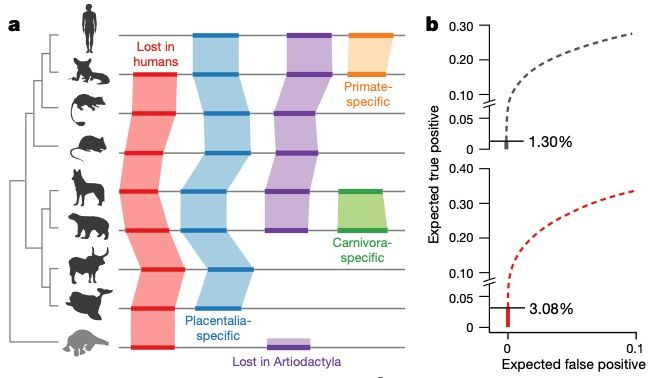
Alignment quality
• 20 simulated 30-Mb genomes
(Evolver) along a tree of
catarrhines (primates)
• More accurate than previous
version
• Maintains accuracy as the number
of species increases
• Alignathon data
• Higher accuracy than any aligner
that participated in the Alignathon
(2014)Confounding effects
• Guide tree
• Subset of 48 bird genomes
• 4 different guide trees, incl. a randomized tree
• On average 98.5% of aligned pairs were identical between any two alignments
• Assembly quality
• 11 mammals, 7 with one short-read and one high quality (often long-read)
assembly
• 2 alignments (low vs. high assembly quality) differed, but were more similar
than alignments from the same data and different alignment strategiesBird 10,000 genomes (B10K) project
üPhase I (2014): 48 species
üPhase II: 363 species in 92.4% of
avian families across all
continents (267 newly
sequenced)
• Short-read assemblies (incl. data
from museum samples)
• Sequenced at BGI, assembly with
SOAPdenovo & Allpaths-LGBird 10,000 genomes (B10K) project
• 267 new genome assemblies
• Comparable quality to previously published bird genomes
• Variation in contiguity (avg scaffold N50 = 1.42 Mb, contig N50 = 42.57 kb)
• Coverage ranged from 35X to 368X
• Analysis of all 363 available genome assemblies
• Annotation via homology-based approach à avg 15,464 protein-coding genes
per species
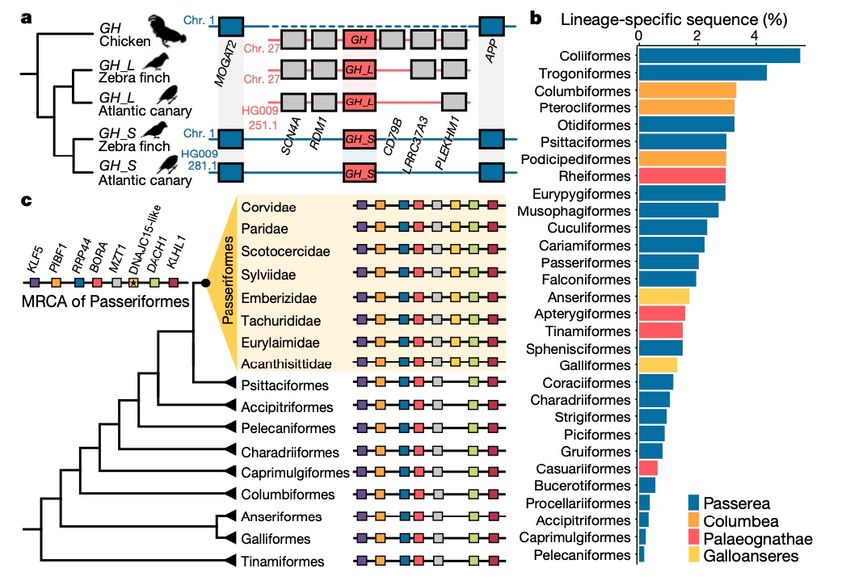
• 96.1% of all genomes with transposable element contentWhat have we learned from the additional bird genome sequences? • Reference-free alignment of 363 genomes increased the proportion of orthologous sequences (compared to a 48 genome alignment relative to chicken and zebra finch): • 981 Mb across the whole genome (149% increase) • 24 Mb of orthologous coding sequence (84.4% increase) • 141 Mb of orthologous introns (631% increase)
What have we learned from the additional
bird genome sequences?
• Orthologue assignment pipeline
(based on Progressive Cactus
alignment) confirms gene
duplication in Passeriformes
(songbirds, >50% of all bird
species)
• Putative lineage-specific gene
(DNAJC15L) found in 131 of 173
sequenced Passeriformes and
their ancestral genome, but not
in non-PasseriformesWhat have we learned from the additional
bird genome sequences?
• 498 genes lost from all studied genomes
• Consistent with previous suggestion of gene loss in the ancestor of birds
• Not discussed: GC content varies across chromosome classes in birds and has
an effect on assembly quality from short-read data. Maybe these genes are
present but located in difficult regions?
• Passeriformes had higher GC-content in coding regions than other
birds, but not in non-coding regions
• Bias in synonymous codon usageWhat have we learned from the additional
bird genome sequences?
• Denser sampling of species
increased the power to detect
conserved sites (under selective
constraint), calculated for the
chicken genomeZoonomia project
üMammals project (2011): 29
species
üZoonomia project: 240 species
(131 newly sequenced), from >80%
of all mammalian families
• 131 genomes: short-read
sequencing, assembly with
DISCOVAR
• 9 of these (+ 1 pre-existing):
scaffolding with proximity ligation
(Dovetail Chicago, HiRise)What have we learned from the additional mammal genome sequences? • Empirical support for speciation via allopatry and positive selection on postzygotic isolation mechanisms from the genomes of the endangered Mexican howler monkey and the Guatemalan black howler monkey • Positive selection on anti-cancer pathways in the capybara, consistent with Peto’s paradox (cancer is rarer in large mammals)
What have we learned from the additional mammal genome sequences? • Molecular convergence of the KLK1 gene that is responsible for venom production in solenodons and shrews • Comparative analysis of ACE2 (receptor for SARS-CoV-2) identified 47 mammals with a (very) high likelihood of being virus reservoirs, intermediate hosts or good model organisms for studying COVID-19
What have we learned from the additional mammal genome sequences? • Genetic diversity and extinction risk
What have we learned from the additional
mammal genome sequences?
• Genetic diversity and extinction risk
• Heterozygosity and segments of homozygosity
(SoH; proportion of the genome that resides
in an extended region without any variation)
• Calculated for 126 of the 131 newly
sequenced genomes
• Overall heterozygosity is correlated with
contig N50 values, but not SoH
• Only a small fraction of threatened mammals
were included (2.6%) but the results show
that useful information can be obtained from
only one individual per speciesWhat have we learned from the additional
mammal genome sequences?
Heterozygosity decreases & segments of homozygosity (SoH) increase with increasing
levels of conservation concernWhat have we learned from the additional
mammal genome sequences?
No difference between wild and captive Critically endangered species have higher SoH
individuals values than the median of least concernWhat have we learned from the additional
mammal genome sequences?
• 240 mammal genome alignment
with Progressive Cactus
• More conserved sites found in
human genome than with 100
vertebrate alignment
• Largest improvement in non-
coding regionsAlignment of 605 genomes from B10K +
Zoonomia
Armstrong et al. 2020Alignment of 605 genomes from B10K +
Zoonomia
• Coverage tracks phylogenetic
distance and genome size
• Ancestral reconstructions highly
complete for functional
sequence
• 86% of human coding bases in
ancestor of all placental mammals
• 95% of chicken coding bases in
ancestor of all birds
Armstrong et al. 2020You can also read

















































