Quels progrès dans la recherche sur les maladies rénales génétiques en pédiatrie ? - Denis Morin Montpellier
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Quels progrès dans la recherche
sur les maladies rénales
génétiques en pédiatrie ?
Denis Morin
Montpellier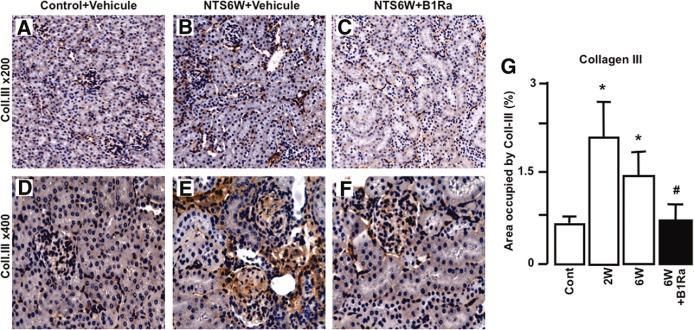
Recherche clinique en pédiatrie
• Faibles nombres de patients parfois
– Études multicentriques +++
• Considérations éthiques
– Recherche qu’on ne peut pas faire chez des
patients adultes
• Importances des protocoles (PHRC, autres,..)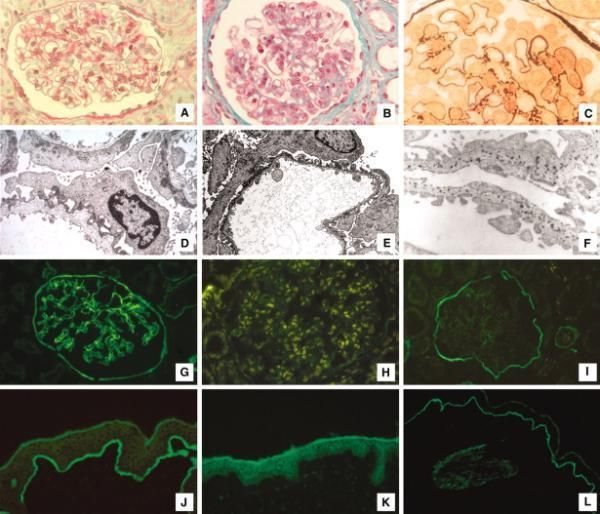
Néphrologie Pédiatrique
Basalopathies SHU atypique
Maladies kystiques
Glomérulopathies Autres…
Tubulopathies
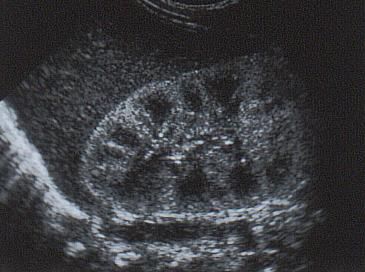
Maladies rénales kystiques • Polykystose dominante • Polykystose récessive • Maladie kystique liée à TCF2
Polykystose dominante
• Diagnostic anténatal possible
– Echographie / IRM
• Questions concernant la prise
en charge
– Surveillance simple de loin
en loin ?
– Possibilités d’attitudes
thérapeutiques préventives
à l’image de ce qui existe
pour les adultes ?
Polykystose récessive
• Maladie rare 1/25 000 naissances
• Transmission autosomique
récessive
• Expression anténatale fréquente
– Mise en évidence de gros reins
hyperéchogènes
– Parfois retentissement fœtal
• Oligoamnios
• Diagnostic génétique possible
– Etude de corrélations
génotype/phénotype
• Pas de résultats probants en
terme de conseil génétique
(CJASN 2010)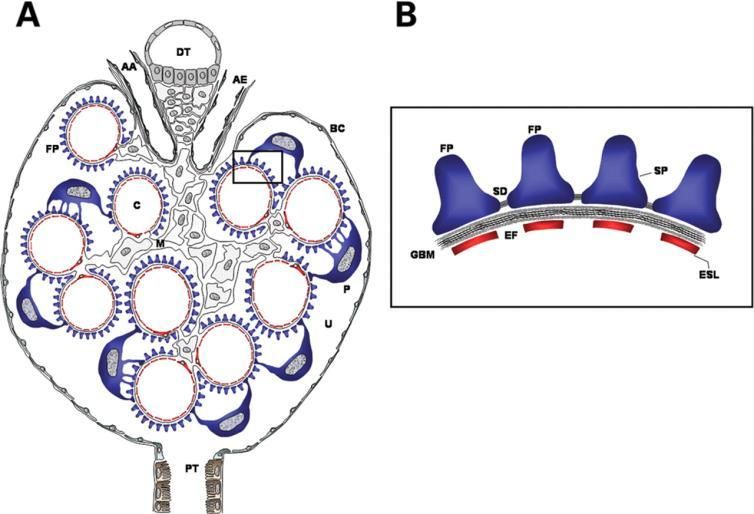
Polykystose récessive • Grande variabilité d’expression clinique – Atteinte rénale – Atteinte hépatique • Prise en charge symptomatique – Traitement anti-HTA – Prise en charge insuffisance rénale chronique • Place d’une approche thérapeutique spécifique visant à limiter le développement des kystes ?
Pathologie kystique liée à des mutations du gène TCF2/HNF1b
Syndrome d’Alport
Syndrome d’Alport • Basalopathie héréditaire - Forme liée à l’X - Forme autosomique récessive - Forme autosomique dominante - Anomalie chaines a du collagène • Evolution ⁻ Protéinurie ⁻ HTA ⁻ Insuffisance rénale
Syndrome d’Alport
Heidet L , Gubler M JASN 2009;20:1210-1215
©2009 by American Society of NephrologyFigure 2. Schematic algorithm in case of Alport syndrome suspicion because of hematuria {+/-} proteinuria
Heidet, L. et al. J Am Soc Nephrol 2009;20:1210-1215
Copyright ©2009 American Society of NephrologySyndrome d’Alport Cyclosporine A treatment in patients with Alport syndrome: a single-center experience. Massela et al Ped Nephrology 2010 “Our data do not support the use of CsA therapy for proteinuric patients with AS, particularly if they have chronic renal failure”
Syndrome d’Alport
• Efficacy and safety of losartan in children with Alport
syndrome results from a subgroup analysis of a prospective,
randomized, placebo- or amlodipine-controlled trial.
Webb J and col. Nephrol Dial Transplant. 2011 Aug;26(8):2521-6
• Inhibition du système rénine angiotensine
– Effet anti-protéinurique
– Anti-cytokine
– Inhibition de la production de collagène
– Inhibition de la fibrose tubulo-interstitielleSyndrome d’Alport • Nephroprotective effect of the HMG-CoA- reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome • Koepke ML et al . NDT 2007 – Action antio-fibrotique – Moindre infiltrat de cellules inflammatoires – Intérêt d’un traitement précoce en pédiatrie ?
Syndrome d’Alport
• Modèle animal de souris COL4A3 -/-
• Intérêt de la transplantation médullaire ?
– Recrutement de cellules aptes à devenir des
podocytes et des cellules mésangiales
– A confirmerSyndrome hémolytique et
urémiqueSyndrome hémolytique et urémique • Forme post-diarrhéique : typique – Nourrisson avant 2 ans – Infection à E.Coli entéropathogène – Anémie, thrombopénie, IRA – Dialyse dans 50% des cas – Pronostic dominé par atteinte neurologique – Présence schizocytes
Syndrome hémolytique et
urémique
• Forme atypique
– Age de début variable
• Formes néonatales
– Pas de contexte infectieux
• SHU D-
– Début plus insidieux
• HTA plus fréquente
– Risque de rechuteSHU atypique • Déficit en ADAMTS 13 • Anomalie du métabolisme Vitamine B12 • Anomalie de la régulation de protéines du système du complément
Système du complément
Système du complément
SHU atypique et anomalies du complément • Différents facteurs en cause – MCP , – C3, H, I, B • Etude de l’expression de MCP • Dosages du C3 et des facteurs H, I, B • Anticorps anti-H ? • Génotypage même si taux plasmatiques normaux – anomalies qualitatives ?
SHU atypique et anomalies du complément
Prise en charge
• Traitement symptomatique : HTA, IRA, Anémie,…
• Traitement « spécifique » récent
– Plasmathérapie débutée dès que possible
• Perfusion de plasma
• Echanges plasmatiques +++
• Questions :
– Combien de temps ?
– Problèmes abords vasculairesSHU atypique et anomalies du complément
Prise en charge
• A long terme ?
– Echanges plasmatiques
– Anticorps monoclonal anti-C5
– Concentré de facteur H
• Si insuffisance rénale terminale
– Transplantation rénale + EP
– Transplantation combinée foie – rein
– Concentré de facteur H
– Anticorps monoclonal anti-C5SHU atypique et anomalies du complément
Prise en charge
• Actuellement
• Protocole de prise en charge des SHU
atypique de l’enfant par EculizumabSHU typique et activation du
système du complément
• « Epidémie » de SHU post-diarrhéique en
2011 (Allemagne, Bordeaux, Lille)
• Formes avec atteinte neurologique
– Intérêt du traitement par Eculizumab dans les
formes sévère de SHU typiqueCystinose
PHYSIOPATHOLOGIE
• Défaut du transporteur
lysosomal de cystine:
CYSTINOSINE
• Gène CTNS (23 kb)
• Transmission autosomique
récessive: 1/200 000
naissances vivantes
• Accumulation et cristallisation
intralysosomiale de cystine =
Maladie systémique
W Gahl, 2002Cystinose - Traitement • Cystagon = chlorydrate de cystéamine – 10-50 mg/Kg/jour sans dépasser 1300 mg/m2 – En 4 prises dont une nocturne +++ – Cible thérapeutique – Cystine intraleucocytaire < 1 nmole/mg protéine – Collyre cystéamine • Traitement symptomatique du syndrome de Fanconi
Cystinose - Evolution
Age distribution of patients starting renal replacement therapy (RRT) in different eras.
Van Stralen K J et al. CJASN 2011;6:2485-2491
©2011 by American Society of NephrologyCystinose - Traitement
• Etude « Raptor »
– Vise à étudier la tolérance et l’efficacité d’une
forme galénique nouvelle de cystéamine qui
permettra une prise toutes les 12 heures
– Etude qui s’est déroulé aux USA et en Europe
– Doit permettre une diminution des prises de
médicaments, source d’une meilleure observance
• Equivalence entre Raptor et Cystagon
– Démarche en vue d’obtenir l’AMM en coursSyndromes néphrotiques
Syndromes néphrotiques
Machuca E et al. Hum. Mol. Genet. 2009;18:R185-R194
© The Author 2009. Published by Oxford University Press. All rights reserved. For Permissions,
please email: journals.permissions@oxfordjournals.orgSyndromes néphrotiques • SN génétiquement déterminés • Néphrose lipoïdique
Syndromes néphrotiques • SN génétiquement déterminés • Néphrose lipoïdique
Syndromes néphrotiques • Début précoce, parfois néonatal, peut être tardif • ATCD familiaux • Transmission AD ou AR • Corticorésistance • Pas de récidive après transplantation rénale • Etude génétique nécessaire pour affirmer le diagnostic
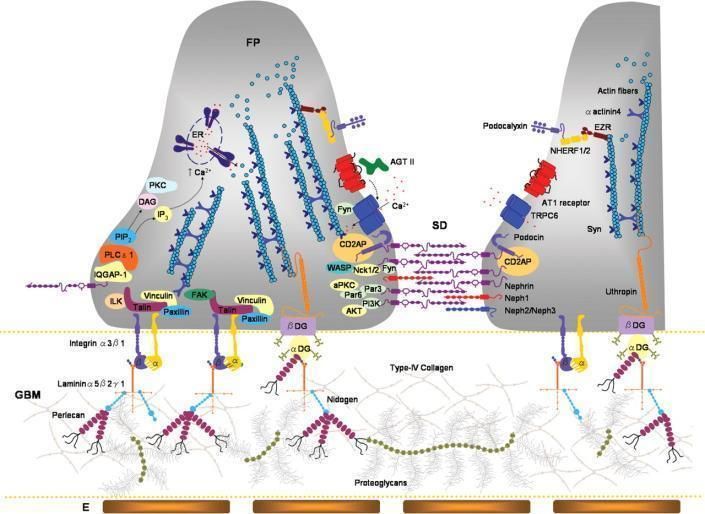
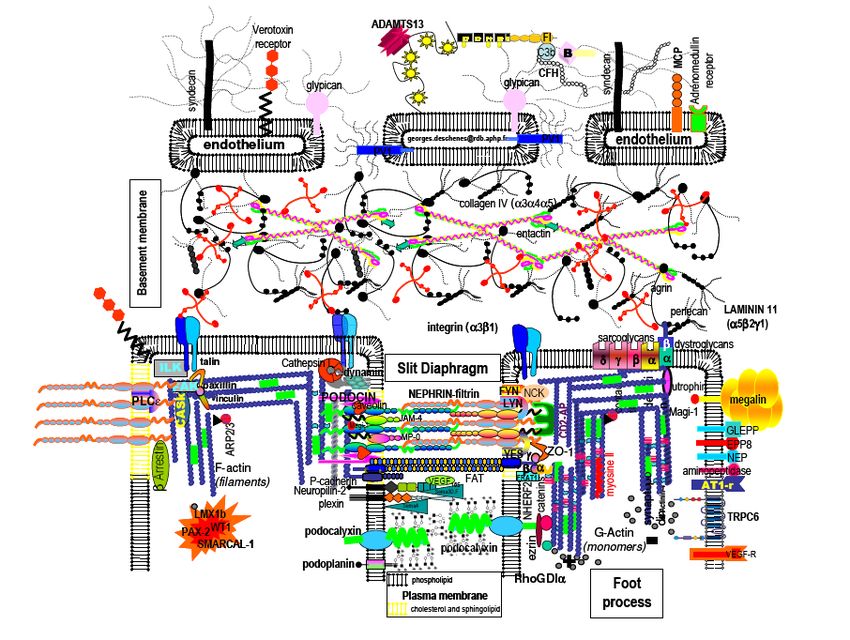
Diaphragme de fente / Podocytes
Machuca E et al. Hum. Mol. Genet. 2009;18:R185-R194
© The Author 2009. Published by Oxford University Press. All rights reserved. For Permissions,
please email: journals.permissions@oxfordjournals.orgAge de début du SN selon le type d’anomalie génétique
Machuca E et al. Hum. Mol. Genet. 2009;18:R185-R194
© The Author 2009. Published by Oxford University Press. All rights reserved. For Permissions,
please email: journals.permissions@oxfordjournals.orgSyndromes néphrotiques • SN génétiquement déterminés • Néphrose lipoïdique
Néphrose lipoïdique
• 1 an à 12 ans
• Protéinurie / rechutes
• Lésion glomérulaires minimes
• Pronostic dominé par évolution
– Corticosensibilité ?
– Corticodépendance ?
– Corticorésistance ?
• Probablement « polygénique »Néphrose
Néphrose
Néphrose
Traitement
Etude Néphromycy (V Baudoin)
• Vise à comparer l’efficacité et la tolérance de deux
thérapeutiques utilisées dans la néphrose cortico-
dépendante
– Cyclophosphamide
– Mycophénolate mophétil
• Débutée début 2011Néphrose
Traitement
• Etude NEPHRUTIX
– Vise à mesure l’efficacité et la tolérance du
Rituximab dans certains cas de néphrose cortico-
dépendante et antiocalcineurine dépendante
– Rituximab :
• antiCD20
• Action sur les B lymphocytes
• Efficacité / risquesConclusion • Beaucoup d’autres sujets – Tubulopathies – Oxalose – Cystinurie –…
Diabète Insipide Néphrogénique
congénitalDINc
• Pathologie rare ++
• Expression néonatale précoce
– Polyurie – perte de poids – déshydratation
– Hypernatrémie – trouble de concentration des
urines
– Résistance du tubule rénal à l’action de la
vasopressineDINc
DINc - Génétique Récépteur V2 de la vasopressine Aquaporine 2 Sexe masculin Expression dans les deux sexes Femme transmettrice
Diabète insipide néphrogénique
congénital
• Traitement symptomatique
– Hydratation : NEDC parfois
– AINS
– Hydrochlorothiazide
• Traitement spécifique en cas de mutation du
RV2 : molécules chaperonnes ?Molécule Chaperonne
Bouvier et al 2006Effets chez les patients ?
Mais….
Efficacité fonction de la nature des mutations en cause
Bouvier et al 2006Recherche de pharmacochaperones agonistes
Expression à la surface cellulaire-Microscopie confocale
Contrôle MCF57 18H
V2
A294P
L44P
R337X
JASN 2009Effet agoniste des MCF sur les récepteurs DNIc MCF = Pharmacochaperones Agonistes pour A294P et L44P Pas d’effet sur le mutant R337X malgré une restauration à la membrane JASN 2009
Autre pathologie lié à une
mutation du Récepteur V2 de la
vasopressine :
Syndrome d’antidiurèse d’origine
néphrogéniqueNSIAD décrit en 2005
L
e2 C
R
A V K
TM3 Y L Q M
V G M 120
125 A S S Y
M Y
I L A
M T
L
D 135
C i2
NSIAD R H DINc
L H
R
A INSIAD • Signes cliniques: variabilité phénotypique – Crises convulsives ++, âge d’apparition variable – Mais aussi patients asymptomatiques • Signes biologiques: – Hyponatrémie – Baisse de l’osmolalité plasmatique – Osmolalité urinaire inadaptée anormalement élevée – Absence ou faible sécrétion de vasopressine – Réponse anormale à une épreuve de charge hydrique • Principal diagnostic différentiel: SIADH
NSIAD
• Possibilité d’évolution neurologique péjorative si
absence de diagnostic et récidive des convulsions
• Circonstance déclenchant la symptomatologie:
ingestion excessive d’eau (chaleur, relais de
l’allaitement maternel)
• Traitement:
– Restriction hydrique +/- urée
– Inefficacité des antagonistes non peptidiques du récepteur
V2
• Femmes transmettrices:
– Souvent antidiurèse inappropriée
– Parfois signes cliniques ou biologiquesNSIAD • Pathologie de description récente • Variabilité phénotypique patients et femmes transmettrices • Fréquence probablement sous-estimée • Nécessité d’une information médicale ciblée pour permettre le diagnostic • Avancées dans la compréhension des mécanismes physiopathologiques • Meilleure appréhension de la structure du récepteur V2
Protein
Implicated Principal
OMIM Gene/locus Genetic expression in Other features
molecule featuresa
renal tubule
Polyhydramnio
s, prematurity,
Antenatal
SLC12A1/15q2 Na-K-2Cl nephrocalcinos
Bartter 601678 Type I TAL
1.1 cotransporter is, polyuria,
syndrome
failure to
thrive
Transitory
Kir 1.1 hyperkalaemia
241200 KCNJ1/11q24 Type II potassium TAL and CCD in neonatal
channel period in most
patients
ClC-Kb Nephrocalcino
602522 CLCNKB/1p36 Type III chloride TAL and DCT sis in some
channel patients
Antenatal Barttin ClC-Ka
BSND/1p31 or TAL and DCT CRI with some
Bartter and ClC-Kb
607364 CLCNKA– Type IV tAL, TAL, DCT Barttin
syndrome with chloride
CLCNKB/1p36 and CCD mutations
SNHL channels
Severe to mild
ClC-Kb salt wasting
CLCNKB/1p36 Sometimes
Classic Bartter chloride TAL and DCT with or
602522 SLC12A3/16q1 Type III hypomagnesae
syndrome channel Na-Cl DCT without
3 mia
cotransporter nephrocalcinos
is
Sometimes
Na-Cl polyuria,
SLC12A3/16q1 cotransporter Hypomagnesa failure to
Gitelman DCT TAL and
263800 3 ClC-Kb emia, thrive or
Syndrome DCT
CLCNKB/1p36 chloride hypocalciuria growth
channel hormone
deficiencyPlasma potassium, chloride and bicarbonate concentration at diagnosis in patients with
antenatal and neonatal Bartter syndrome according to the gene involved.
Brochard K et al. Nephrol. Dial. Transplant. 2009;24:1455-
1464
© The Author [2008]. Published by Oxford University Press on behalf of ERA-EDTA. All rights
reserved. For Permissions, please e-mail: journals.permissions@oxfordjournals.orgGrowth under treatment (water and electrolyte + indomethacin) according to the gene
involved.
Brochard K et al. Nephrol. Dial. Transplant. 2009;24:1455-
1464
© The Author [2008]. Published by Oxford University Press on behalf of ERA-EDTA. All rights
reserved. For Permissions, please e-mail: journals.permissions@oxfordjournals.orgThe kidney diseases discussed above can be life-threatening and most have limited, often unsuccessful, treatment options. Many patients with MPGN and aHUS experience recurrent episodes that eventually lead to end-stage renal failure.40,57,84 Even when kidney transplants are successful, diseases that are caused by systemic factors such as mutated fH, C3 and fB can present again and the outcome is often fatal.72,103 In such situations, combined kidney and liver transplantation may be the only way to correct the underlying defects, and success with such an approach has been described in the literature but the high risk for adverse events in such procedures makes this a less desirable option.104,105 By the same principle, kidney transplantation may be an acceptable option for end-stage aHUS patients whose diseases are attributable to mutations in the membrane regulator MCP.91,106 Given the well-established role of complement in the pathogenesis of these kidney diseases, it is envisioned that systemic or targeted local complement inhibition may represent a promising therapeutic strategy. In this context, the recent approval and successful clinical application of a first-in-class complement inhibitor Eculizumab, a humanized anti-C5 monoclonal antibody,107 for treatment of the complement-mediated disease paroxysmal nocturnal haemaglobinuria108–110 is particularly encouraging. Based on a number of animal studies in which C5 deficiency or C5-blocking antibodies reduced renal injury,59,69,111 it may be anticipated that Eculizumab will prove to be efficacious for some, if not all, complement-mediated kidney disorders as well. Indeed, two case reports on the successful treatments of paediatric aHUS patients with Eculizumab have already appeared in the literature112,113 and clinical trials on the use of Eculizumab in aHUS are currently underway.114 Other complement-based therapeutic strategies include chemical and biological agents that target additional complement components. A chemical inhibitor for C3aR and two antagonists for C5aR, a cyclic hexapeptide and a recombinant C5a analogue, have been developed and shown to effectively block anaphylatoxin-mediated inflammatory injury in a variety in vitro and in vivo studies including models of renal IRI and transplantation.115–118 A synthetic peptide, named Compstatin, with potent human C3-inhibiting activity has also been developed by phage display and shown to effectively shut down human complement activation in several experiments including an ex vivo model of hyperacute rejection of kidney xenotransplantation model.119–121 Compstatin is currently being evaluated in clinical trials for the treatment of AMD, a disease that also implicates abnormal AP complement activation.122 One of the concerns of targeting C3 with agents like Compstatin is that they obliterate the complement system completely, potentially compromising host defence and leaving the patients susceptible to infection. Because the AP complement is principally involved in many of the complement-mediated diseases, efforts have also been made to develop inhibitors that target the AP only. For example, two anti-C3b mAbs that specifically inhibit the AP C3 convertase with no activity on classical and lectin pathway complement activation have been described recently.123,124 A third area of promising research for treating complement-mediated kidney injury is the creation of soluble recombinant forms of complement regulatory proteins. Several studies have shown that administering a soluble form of CR1 or Crry can reduce renal injury125,126 and such proteins have an extended half-life when fused to an Ig Fc domain.127 More recently, strategies have been developed to target the recombinant protein to sites of injury. He et al. targeted recombinant regulatory proteins to the kidney using an Ag-specific single chain Ab fragment.128 In other efforts, the inhibitors were directed to sites of complement activation with the design of a fusion protein consisting the C3d-binding domain of CR2 and a regulatory protein partner, either Crry (CR2-Crry) or the SCR1-5 region of fH (CR2-fH).129 In one study of MRL/lpr mice, which are prone to autoimmune glomerulonephritis and vasculitis, CR2-Crry ameliorated disease symptoms compared with untreated mice.130 Studies with these recombinant proteins have also been performed
The basic defect in Alport syndrome is either the lack, in the mature GBM, of the 3- 4- 5(IV) network and its failure to replace the 1- 2 network, which is known to be less resistant to proteolysis, or the presence of a defective 3- 4- 5(IV) network. There are several animal models for AS, in dogs and mice that faithfully recapitulate autosomal and X-linked forms of the disease. They have brought novel data to the understanding of the mechanisms responsible for the progression of AS nephropathy and in the elaboration of future therapies. The re-expression of the 3(IV) chain in Col4a3-/- mice, for example, was shown to restore the expression of 4 and 5 (IV), thus demonstrating that the expression of all three 3- 4- 5(IV) chains is required for network assembly.12 The downstream mechanisms responsible for progressive alteration of the GBM and renal failure are not fully understood. In young Alport mice, the ultrastructurally normal GBM is known to already be abnormally permeable.13 The concomitant accumulation of mRNAs encoding TGFβ1 and extracellular matrix components in human and mouse Alport podocytes are thought to reflect key events in renal disease progression.14 Blocking the TGFβ1 pathway prevents GBM thickening in Alport mice.15 The role of metalloproteinases in Alport disease has been underlined by recent studies. Increased expression of MMP2, MMP3, and MMP9 has been described, both at the transcriptional and the protein level, in AS kidneys in humans, mice, and dogs.16,17 Such MMP up-regulation is not unique to Alport nephropathy. However, AS kidney basement membranes were shown to be more readily degradable in vitro by collagenase, elastase, and cathepsins, compared with normal kidney basement membranes,18 and this is thought to be due to the lack of the highly cross linked 3- 4- 5(IV) network. Blocking simultaneously at least MMP2, MMP3, and MMP9 in Col4a3-/- mice delays the progression of the disease if treatment is given before development of GBM injury and occurrence of proteinuria in a C57BL6 genetic background.16 In addition, a recent study found an increase of MMP12 expression in podocytes of humans, mice, or dogs affected with AS, possibly linked to MCP1-mediated activation of the podocyte CCR2 receptor.19 Either MMP12 inhibitor or CCR2 receptor antagonist attenuates the GBM thickening in Col4a3-/- mice.19 Pharmacologic therapeutic approaches have been tested in animal models and in humans. Cyclosporine A was found to delay progression of renal failure in humans and dogs in initial studies.20,21 However, cyclosporine is also found to be rapidly associated with nephrotoxicity, thereby precluding its long-term use.22 Angiotensin-converting enzyme inhibitors and/or angiotensin 2 type 1 receptor antagonists reduce urinary protein excretion and preserve glomerular filtration in dogs affected with X-linked AS, in Col4a3-/- mice,23 and in a few pediatric patients.24 Larger controlled studies are necessary in humans to clarify the long-term benefit of the treatment and the nature and doses of drugs that are effective. Also, criteria for micro- or macroalbuminuria for starting renoprotective treatment by blockade of the renin-angiotensin system remain to be precisely determined. In Alport mice, chemokine receptor-1 blockade as well as statin treatment improves survival and renal lesions.25 Finally, bone marrow transplantation of Col4a3-/- mice shows recruitment of bone marrow cells as future podocytes and mesangial cells, partial restoration of the expression of the 3- 4- 5(IV) network, and clinical and histologic improvement.26–28 However, a recent study suggested that irradiation, which preceded bone marrow transplantation, may improve the survival of Col4a3-/- mice by itself, through as yet unidentified mechanisms.29 Overt anti-GBM nephritis occurs in only 3 to 5% of transplanted Alport patients.30 The risk of graft loss is very high, and treatment with plasmapheresis and cyclophosphamide has shown limited benefit. The risk of recurrence on subsequent transplantation is very high. This complication is more likely to occur in patients with deletions or frameshift mutations, who do not express the 3 4 5(IV) GBM network. However, many patients with COL4A5 deletion have been successfully transplanted, without developing anti GBM nephritis, and predictive factors for developing the disease are currently unknown.
Cemara
1. Determination of C3, CFH, CFI and CFB levels, expression of MCP and screening
for anti-CFH antibodies is indicated for all patients with aHUS. Normal C3 level
does not eliminate the presence of CFH or CFI mutation or of anti-CFH
antibodies.
2. Genotyping of CFH, CFI and MCP, and if possible CFB and C3, is indicated for all
patients with aHUS, even if plasma levels are normal.
3. The identified mutation has to be regarded as a risk factor for HUS, not as the
direct cause. The association of mutations in several genes is not exceptional.
Penetrance of the disease is 50% in patients with a mutation in complement.
Therefore, the risk of developing HUS is difficult to predict in family members
with the mutation. Intrafamilial genetic heterogeneity exists, suggesting that
unknown genetic factors are present.
4. A post-diarrheal onset of HUS can be observed in all groups. Therefore,
genotyping must be performed for patients with uncertain diagnosis of D +
/STEC + HUS, especially before transplantation. The worst prognosis is in
patients with CFH mutation, who are at high risk of ESRD as soon as at first flare
or within the year of onset.
5. Plasmatherapy (PE with FFP) should be started as early as possible. Although
evidence is lacking, benefit is expected mainly in CFH-mutated patients and in
patients with anti-CFH antibodies. Benefit is likely in all other subgroups of
aHUS, except the MCP subgroup, where spontaneous remission generally
occurs.
6. The risk of graft loss due to HUS recurrence or graft thrombosis is high in
patients with CFH and CFI mutations, while it is very low in patients with MCP
mutations. Family living donor transplantation is contraindicated, because of
the risk of graft loss due to recurrence and the risk that donors themselves
might have HUS after donation, due to unknown genetic factors shared with
the recipient.
Kidney transplantation under pre-, intra- and post-operative intensive
plasmatherapy may be successful in some patients.
Combined liver and kidney transplantation under pre- and intra-operative
plasmatherapy, and post-operative anticoagulation, has been successful in a
few patients with CFH mutation. This option will now have to be considered on
an individual basis for patients with mutations in other factors synthesized in
the liver.
7. Hope for the future relies on therapies which could prevent ESRD, such as CFH
concentrate or anti-C5 monoclonal antibodies.Figure 1. Clinical course and laboratory findings
Mache, C. J. et al. Clin J Am Soc Nephrol 2009;4:1312-1316
Copyright ©2009 American Society of NephrologyInfiltration of CD3-positive T-cells (A–D), F4/80-positive macrophages (E–F) and α-SMA-
positive activated fibroblasts (H–J).
Koepke M et al. Nephrol. Dial. Transplant. 2007;22:1062-
1069
© The Author [2007]. Published by Oxford University Press on behalf of ERA-EDTA. All rights
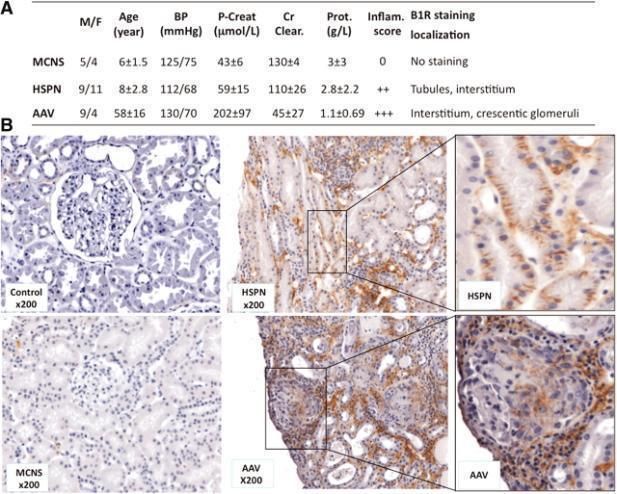
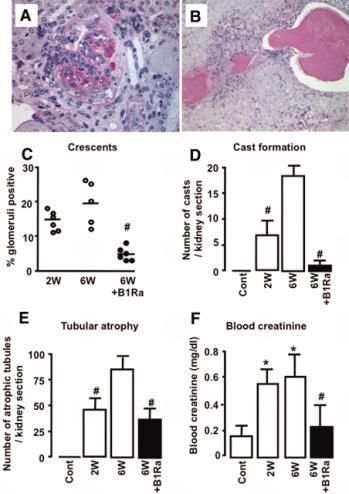
reserved. For Permissions, please email: journals.permissions@oxfordjournals.orgFigure 2. Representative human B1R expression in normal and pathologic human kidney biopsies
Klein, J. et al. J Am Soc Nephrol 2010;21:1157-1164
Copyright ©2010 American Society of NephrologyFigure 4. Delayed B1Ra treatment reduces renal lesions and improves renal function
Klein, J. et al. J Am Soc Nephrol 2010;21:1157-1164
Copyright ©2010 American Society of NephrologyFigure 5. B1R blockade inhibits the development of renal fibrosis
Klein, J. et al. J Am Soc Nephrol 2010;21:1157-1164
Copyright ©2010 American Society of NephrologyRecherche plus fondamentale
Phenotype or
Gene Locus Inheritance Protein Functiona
Syndrome
Slit-Diaphragm protein complex
Main component of the
SD. Anchors the SD to the
CNS of the Finnish type.
actin cytoskeleton.
Early-onset SRNS in cases
NPHS1 19q13.1 AR Nephrin Modulate signalling
carrying at least one mild
events related with actin
mutation
cytoskeleton dynamics,
cell polarity and survival
CNS. Early and late onset
Scaffold protein linking AR SRNS. Juvenile and
plasma membrane to the adult SRNS in cases
NPHS2 1q25–31 AR Podocin actin cytoskeleton. bearing the R229Q
Modulates variant in compound
mechanosensation heterozygous state with a
pathogenic mutation
Involved in cell junction
Early-onset SRNS with
PLCE1 10q23 AR Phospholipase Cε1 signalling and glomerular
DMS and FSGS
development
Not precisely defined in
humans, may cause early-
Adapter protein, may onset SRNS and FSGS.
CD2AP 6p12.3 AR (?) CD2 associated protein anchor the SD to the Mice model exhibits a
actin cytoskeleton severe phenotype
resembling CNS in
humans
Receptor-activated non-
selective calcium
Adult-onset SRNS with
TRPC6 11q21–22 AD TRPC6 permeant cation channel.
FSGS
Involved in
mechanosensationActin cytoskeleton components
ACTN4 19q13 AD α-actinin-4 F-actin cross- Late-onset SRNS
linking protein with incomplete
penetrance and
slow progression
to ESRD
MYH9 22q12.3 complex NMMHC-A Cellular myosin High risk
that appears to haplotypes
play a role in associated with
cytokinesis and increased risk of
cell shape FSGS and ESKD in
African-Americans
Nuclear proteins
LMX1B 9q34.1 AD LIM/homeobox Podocyte and Nail-patella
protein LMX1B GBM development syndrome. NS in
and maintenance 40% of cases
SMARCAL1 2q35 AR hHARP ATP-dependent Schimke immuno-
annealing helicase osseus dysplasia
that rewind stably
unwound DNA
WT1 11p13 AD Wilms’ tumour 1 Zinc finger Denys–Drash
transcription syndrome, Frasier
factor that syndrome, WAGR
functions both as a syndrome, isolated
tumour suppressor FSGS and DMS
and as a critical
regulator of kidney
and gonadal
developmentGlomerular basement membrane proteins
LAMB2 3p21 AR Laminin-β2 GBM component, Pierson syndrome
scaffold for type IV
collagen assembly.
Interactions with
integrin α3β1 links the
GBM to the actin
cytoskeleton
ITGB4 17q25.1 AR Integrin-β4 Cell-matrix adhesion, Epidermolysis bullosa.
critical structural role Anecdotic cases
in the presenting with NS
hemidesmosome of and FSGS
epithelial cells
Mitochondrial proteins
COQ2 4q21–q22 AR Polyprenyltransferase CoQ10 biosynthesis, COQ10 deficiency,
which transfers early-onset SRNS, with
electrons from the or without
mitochondrial encephalomyopathy
respiratory chain
PDSS2 6q21 AR Decaprenyl CoQ10 biosynthesis, COQ10 deficiency,
diphosphate synthase- which transfers Leigh syndrome and
2 electrons from the SRNS
mitochondrial
respiratory chain
MTTL1 mtDNA tRNA-LEU Mitochondrial tRNA for MELAS syndrome.
leucine Mitochondrial
diabetes, deafness and
FSGS, with or without
nephrotic syndrome
Lysosomal proteins
SCARB2 4q13–21 AR LIMP II May act as a lysosomal Action myoclonus
receptor renal failureLight and electron microscopy.
Koepke M et al. Nephrol. Dial. Transplant. 2007;22:1062-
1069
© The Author [2007]. Published by Oxford University Press on behalf of ERA-EDTA. All rights
reserved. For Permissions, please email: journals.permissions@oxfordjournals.orgYou can also read

















































