Selected Experimental Protocols - Cenozoic: 2018 iGEM at University of California, Davis
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Cenozoic: iGEM at UC Davis Experimental Protocol
Selected Experimental Protocols
Cenozoic: 2018 iGEM at
University of California, Davis
http://2018.igem.org/Team:UC_DavisCenozoic: iGEM at UC Davis Experimental Protocol
Overview of Experiment
1. Make Genetic Constructs
A. Obtain promoter sequences
Synthesize suitable promoters (IDT)
Anneal single stranded DNA
Amplify remaining promoters out of mammalian genomic DNA
Amplify via PCR
B. Clone plasmid
Grow up in DH5a
Cryogenically preserve a sample in glycerol at -80º C for stock solution
miniprep
C. Insert promoters into plasmid backbone
Restriction digest to remove sequence from plasmid and insert promoters
SLIC
D. Clone plasmid constructs in DH5a
Heat shock transformation
Plate on agar
Pick colonies with desired phenotype
Confirm promoter presence by qualitative PCR and gel electrophoresis
Confirm by sequencing
Miniprep
Cryogenically preserve samples in glycerol at -80º C for stock solution
Miniprep
2. Creation and Use of Bioassay
A. Grow up mammalian cells
Cryogenically preserve samples at -80º C for stock solution
Record the passage number of each sample
B. Transfection
Plate cells on 96 well plate
Allow cells to reach 70-90% confluency
Transfect with constructs using Lipofectamine
Expose bioassay to chemicals of concern
Measure fluorescence
Standardize fluorescence measurements
Measure confluenceCenozoic: iGEM at UC Davis Experimental Protocol
Measure cell viability
Part 1: make genetic constructs
Amplify not-yet-synthesized promoters out of the specific cell it comes from (can likely use the
primers that have already been made)
Isolation of Genomic DNA from Mammalian Cells:
https://www.sciencedirect.com/science/article/pii/B9780124186873000136
https://ac.els-cdn.com/B9780124186873000136/1-s2.0-B9780124186873000136-main.pdf?_tid=
a38498be-26c2-4784-9e12-b78c37adc726&acdnat=1533684670_fda7109ee9845406bfec2be452
843585
PROTOCOL
4.1. Preparation
Make the buffered phenol. Add 0.5 g 8-hydroxyquinoline to a glass beaker containing a stir bar.
Add 500 ml phenol and 500 ml 50 mM Tris–HCl, pH 8.0. Cover with aluminium foil to protect
light-sensitive reagents from oxidation. Stir for 10 min at room temperature, allowing the phases
to separate. Decant most of the upper aqueous phase into an appropriate waste container.
Carefully remove the remainder with a 10-ml pipette. Add another 500 ml 50 mM Tris–HCl, pH
8.0. Stir and decant aqueous phase as before. Check the pH of the lower phenol phase with pH
paper. Repeat equilibrations with 50 mM Tris–HCl, pH 8.0 until the pH of the phenol phase
reaches 8.0. Add 250 ml of 50 mM Tris–HCl, pH 8.0. Store at 4 C in either a brown glass bottle
or a clear glass bottle wrapped in aluminium foil to protect from light. Have cells ready to extract
DNA.
5. STEP 1: COLLECTION OF CELLS
5.1. Overview
Cells grown in culture are harvested and washed.
5.2. Duration 20 min
1.1 Collect cells in a 15-ml conical centrifuge tube. If starting with tissue culture cells in
suspension, directly collect suspension in a conical tube. If starting with adherent cells in culture,
trypsinize and collect cells in a conical tube.
1.2 Determine the number of cells collected. Figure 13.1 Flowchart of the complete
protocol, including preparation. Isolation of Genomic DNA from Mammalian Cells 165
1.3 Centrifuge at 500g for 5 min. Discard supernatant.
1.4 Resuspend in 10 ml ice-cold PBS.Cenozoic: iGEM at UC Davis Experimental Protocol
1.5 Centrifuge at 500g for 5 min. Discard supernatant.
1.6 Repeat PBS wash (Steps 1.4 and 1.5).
6. STEP 2: CELL LYSIS
6.1. Overview
A detergent-based buffer is used to disrupt cell membranes and release DNA, protein, and other
cell components. Proteinase K removes protein contaminants (see also Lysis of mammalian and
Sf 9 cells).
6.2. Duration 12–18 h
2.1 Resuspend cells in a suitable amount of Lysis Buffer. In general, use 1 ml of buffer
per 108 cells.
2.2 Transfer to a 1.5-ml microcentrifuge tube. Vortex the samples.
2.3 Incubate at 50 C for 12–18 h.
7. STEP 3: ORGANIC EXTRACTION
7.1. Overview
Organic extraction is used to separate DNA from other cellular contaminants.
7.2. Duration 10 min
3.1 Add an equal volume of phenol extraction buffer to the lysed cell suspension.
3.2 Vortex for 10 s.
3.3 Centrifuge at 2000g for 5 min in a microcentrifuge at room temperature.
3.4 Transfer the upper aqueous phase, which contains DNA, to a new microcentrifuge
tube. Determine the volume of the aqueous phase obtained.
3.5 Optional: Add RNase A to a final concentration of 20 mg ml1 . Incubate sample at 37
C for 20 min
8. STEP 4: ETHANOL PRECIPITATION
8.1. Overview
Ethanol is used to precipitate DNA from solution.
8.2. Duration 40 min
4.1 Add 0.5 volumes of 7.5 M ammonium acetate and 2 volumes of 100% ethanol.
Vortex.
4.2 Centrifuge at 2000g for 10 min in a microcentrifuge at room temperature.
4.3 Remove supernatant carefully, without disrupting DNA pellet.
4.4 Add 1 ml of 70% ethanol and invert tube several times to wash the DNA pellet.
4.5 Centrifuge at 1700g for 5 min in a microcentrifuge at room temperature.
4.6 Remove supernatant. Uncap the tube and allow the pellet to air-dry for 10–15 min.
4.7 Resuspend DNA in TE buffer or sterile water.Cenozoic: iGEM at UC Davis Experimental Protocol
8.3. Tip
Resuspension in TE buffer stabilizes the DNA for long-term storage. However, the EDTA in the
TE buffer may interfere with certain downstream applications, in which case, resuspension in
water is preferred.
Mini/Midiprep pcDNA-EGFP plasmid - Follow instructions from kit
http://omegabiotek.com/store/wp-content/uploads/2013/05/D6904.D6922-Plasmid-DNA-Midi.M
axi-Kit-Combo-Online.pdf
1. Transfer 20-50 mL overnight culture to a 50 mL centrifuge tube (not provided). Note:
The optimal volume to use depends on the culture density and plasmid copy number. The
optimal cell mass (OD600 x mL culture) for the HiBind® DNA Midi Column is 80-100. For
example, if the OD600 of a culture is 4.0, the optimal culture volume should be 20-25 mL. If
excess culture cell mass is used, alkaline lysis will be inefficient, the HiBind® membrane will be
overloaded, and the performance of the system will be decreased. The increase in lysate viscosity
will require vigorous mixing which may result in shearing of genomic DNA and contamination
the plasmid DNA.
2. Centrifuge at 4,000 x g for 10 minutes at room temperature.
3. Decant or aspirate and discard the culture media. Note: To ensure that all traces of the medium
are removed, use a clean paper towel to blot excess liquid from the wall of the tube.
4. Add 2.25 mL Solution I/RNase A. Vortex or pipet up and down to completely resuspend the
cells.
5. Transfer the cell suspension to a 30 mL or 50 mL centrifuge tubes capable of withstanding
15,000 x g (not provided).
6. Add 2.25 mL Solution II. Invert and rotate the tube gently 8-10 times to obtain a cleared
lysate. This may require a 2-3 minute incubation at room temperature with occasional mixing.
Note: Avoid vigorous mixing as this will shear chromosomal DNA and lower plasmid purity. Do
not allow the lysis reaction to proceed more than 5 minutes. Store Solution II tightly capped
when not in use to avoid acidification from CO2 in the air.
7. Add 3.2 mL Solution III. Invert and rotate the tube gently until flocculent white precipitates
form. This may require a 2-3 minute incubation at room temperature with occasional mixing.
Note: It is vital that the solution is mixed thoroughly and immediately after the addition of
Solution III to avoid localized precipitation.
8. Centrifuge at 15,000 x g for 10 minutes at room temperature (preferably at 4°C). A compact
white pellet will form. Promptly proceed to the next step. Note: Steps 9-26 should be performedCenozoic: iGEM at UC Davis Experimental Protocol in a swing bucket rotor for maximum plasmid DNA yield. All of centrifugation steps should be carried out at room temperature. 9. Insert a HiBind® DNA Midi Column into a 15 mL Collection Tube (supplied). Optional Protocol for Column Equilibration: 1. Add 1 mL 3M NaOH to the HiBind® DNA Midi Column. 2. Let sit at room temperature for 4 minutes. 3. Centrifuge at 4,000 x g for 3 minutes. 4. Discard the filtrate and reuse the collection tube. 10. Transfer 3.5 mL cleared supernatant from Step 8 by CAREFULLY aspirating it into the HiBind® DNA Midi Column. Be careful not to disturb the pellet and that no cellular debris is transferred to the HiBind® DNA Midi Column. 11. Centrifuge at 4,000 x g for 3 minutes 12. Discard the filtrate and reuse the collection tube. 13. Repeat Steps 10-12 until all of the cleared supernatant has been transferred to the HiBind® DNA Midi Column. 14. Add 3 mL HBC Buffer. Note: HBC Buffer must be diluted with 100% isopropanol prior to use. Please see the instructions in the “Preparing Reagents” section on Page 6. 15. Centrifuge at 4,000 x g for 3 minutes. 16. Discard the filtrate and reuse the collection tube. 17. Add 3.5 mL DNA Wash Buffer. Note: DNA Wash Buffer must be diluted with 100% ethanol prior to use. Please see the instructions in the “Preparing Reagents” section on Page 6. 18. Centrifuge at 4,000 x g for 3 minutes. 19. Discard the filtrate and reuse the collection tube. 20. Repeat Steps 17-19 for a second DNA Wash Buffer wash step. 21. Centrifuge the empty HiBind® DNA Midi Column at 4,000 x g for 10 minutes to dry the column matrix. Note: It is important to dry the HiBind® DNA Midi Column matrix before elution. Residual ethanol may interfere with downstream applications. 22. Transfer the HiBind® DNA Midi Column to a nuclease-free 15 mL centrifuge tube (not supplied). 23. Add 0.5-1 mL Elution Buffer or sterile deionized water directly to the center of the column matrix. 24. Let it sit at room temperature for 3 minutes 25. Centrifuge at 4,000 x g for 5 minutes. Note: This represents approximately 65-80% of bound DNA. An optional second elution will yield any residual DNA, though at a lower concentration. Alternatively, a second elution may be performed using the first eluate to maintain a high DNA concentration. 26. Store DNA at -20°C. 27. Nanodrop the resulting DNA solution. Nanodrop General Use Protocol
Cenozoic: iGEM at UC Davis Experimental Protocol
Rinse with a kim wipe and small amount of water
● 2 ul molecular water- initialize
● 2 ul of chosen buffer (usually water) - blank
● 2 ul of actual sample. MAKE SURE TO LABEL THESE
For Gel Extraction:
● 1 uL of loading dye per 5uL of DNA sample
○ So, a 50 uL DNA sample would have 10ul of loading dye
○ For a 50 uL sample, tape together three teeth of the gel comb to make one big
tooth, and load entire sample in
---------------------------------------------------------------------------------------------------------------------
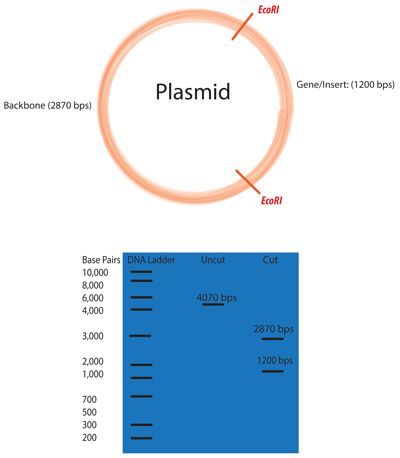
For Promoters that are Inserted Into the Plasmid Using Restriction Digest
1. Digest Plasmid & Promoter w/ proper restriction enzymes
https://www.addgene.org/protocols/restriction-digest/
Select restriction enzymes to digest your plasmid.
2. *Pro-Tip* To determine which restriction enzymes will cut your DNA sequence (and
where they will cut), use a sequence analysis program such as Addgene's Sequence
Analyzer.
3. Determine an appropriate reaction buffer by reading the instructions for your enzyme.
4. *Pro-Tip* If you are conducting a double digest (digesting with two enzymes at the
same time), you will need to determine the best buffer that works for both of your
enzymes. Most companies will have a compatibility chart, such as the double digest
finder tool from NEB.
5. In a 1.5mL tube combine the following:
a. DNA
b. Restriction Enzyme(s)
c. Buffer
d. BSA (if recommended by manufacturer)
e. dH2O up to total volume
6. *Pro-Tip* The amount of DNA that you cut depends on your application. Diagnostic
digests typically involve ∼500 ng of DNA, while molecular cloning often requires 1-3 µg ofCenozoic: iGEM at UC Davis Experimental Protocol
DNA. The total reaction volume usually varies from 10-50 µL depending on application
and is largely determined by the volume of DNA to be cut.
7. *Pro-Tip* A typical restriction digestion reaction could look like this:
a. 1 µg DNA
b. 1 µL of each Restriction Enzyme
c. 3 µL 10x Buffer
d. 3 µL 10x BSA (if recommended)
e. x µL dH2O (to bring total volume to 30µL)
8.
9. *Pro-Tip* The amount of restriction enzyme you use for a given digestion will depend on
the amount of DNA you want to cut. By definition: one unit of enzyme will cut 1 µg of
DNA in a 50 µL reaction in 1 hour. Using this ratio, you can calculate the minimal amount
of enzyme for your reaction. However, keep in mind that restriction enzyme activity is
determined under ideal conditions with very clean DNA, so using a little more enzyme is
advisable. Reactions are often performed with 0.2-0.5 µL of enzyme because it is difficult
to pipette less volume than this; 0.2-0.5 µL will likely be more enzyme than you will need,
but that's okay because a little more enzyme is usually better.Cenozoic: iGEM at UC Davis Experimental Protocol
10. Mix gently by pipetting.
11. Incubate tube at appropriate temperature (usually 37 °C) for 1 hour. Always follow the
manufacturer’s instructions.
12. *Pro-Tip* Depending on the application and the amount of DNA in the reaction,
incubation time can range from 45 mins to overnight. For diagnostic digests, 1-2 hours is
often sufficient. For digests with >1 µg of DNA used for cloning, it is recommended that
you digest for at least 4 hours.
13. *Pro-Tip* If you will be using the digested DNA for another application (such as a
digestion with another enzyme in a different buffer), but will not be gel purifying it, you
may need to inactivate the enzyme(s) following the digestion reaction. This may involve
incubating the reaction at 70 °C for 15 mins, or purifying the DNA via a purification kit,
such as a QIAGEN DNA cleanup kit. See the enzyme manufacturer's instructions for more
details.
14. To visualize the results of your digest, conduct gel electrophoresis.
15. Ligate promoter into plasmid
---------------------------------------------------------------------------------------------------------------------
For Promoters that are Inserted Using SLIC(the three small MT2 constructs)
Digest plasmid with restriction enzymes
https://www.addgene.org/protocols/restriction-digest/
Alternative Digest protocol (use when getting too low of yields of DNA):
44uL plasmid (around 100-200 ng/uL)
1uL of restriction enzyme
5uL NEBuffer
Silica column clean up, elute with 40uL water
Repeat with other Enzyme
For Gel Extraction:
● 1 uL of loading dye per 5uL of DNA sampleCenozoic: iGEM at UC Davis Experimental Protocol
○ So, a 50 uL DNA sample would have 10ul of loading dye
○ For a 50 uL sample, tape together three teeth of the gel comb to make one big
tooth, and load entire sample in
Ligation Protocol with T4 DNA Ligase (M0202)
Protocols.io also provides an interactive version of this protocol where you can discover
and share optimizations with the research community.
Protocol
1. Set up the following reaction in a microcentrifuge tube on ice.
2. (T4 DNA Ligase should be added last. Note that the table shows a ligation using a molar
ratio of 1:3 vector to insert for the indicated DNA sizes.) Use NEBioCalculator to calculate
molar ratios.
3. COMPONENT 4. 20 μl REACTION
5. T4 DNA Ligase Buffer (10X)* 6. 2 μl
7. Vector DNA (4 kb) 8. 50 ng (0.020 pmol)
9. Insert DNA (1 kb) 10. 37.5 ng (0.060 pmol)
11. Nuclease-free water 12. to 20 μl
13. T4 DNA Ligase 14. 1 μl
15. * The T4 DNA Ligase Buffer should be thawed and resuspended at room temperature.
16. Gently mix the reaction by pipetting up and down and microfuge briefly.
17. For cohesive (sticky) ends, incubate at 16°C overnight or room temperature for 10
minutes.
18. For blunt ends or single base overhangs, incubate at 16°C overnight or room
temperature for 2 hours (alternatively, high concentration T4 DNA Ligase can be used
in a 10 minute ligation).
19. Heat inactivate at 65°C for 10 minutes.
20. Chill on ice and transform 1-5 μl of the reaction into 50 μl competent cells.
https://www.neb.com/protocols/1/01/01/dna-ligation-with-t4-dna-ligase-m0202
SLIC Protocol:Cenozoic: iGEM at UC Davis Experimental Protocol
1. Prepare the following reactions for both the backbone (pcDNA-EGFP) and promoter in 1.5
mL microcentrifuge tubes.
2. Add 2 uL of 10X NEB Buffer 2, 2uL of 10X BSA, and 15 uL total of equimolar DNA insert
(promoter) and backbone (digested pcDNA-EGFP).
1. Lastly, add 1uL of T4 DNA Polymerase (diluted to 0.5 U/uL).
3. Incubate this reaction at room temperature for 2.5 minutes.
4. Add 2 µL of 10 mM dCTP to stop the reaction. Mix. Place tubes on ice. ☐
-----
● Colony PCR protocol:
○ Fill a PCR tube with 10 uL of molecular water
○ Take 10uL pipette, touch colony from whatever you’re using, touch the tip to
your replicate plate (label this), touch to LB media if desired, then pipet up and
down the molecular water in the PCR tube.
○ After all samples have been processed in the previous manner, lyse the bacterial
cells in the PCR tubes in a thermocycler. Lyse at 98º C for ten minutes.
○ Use the lysate as DNA template for a 20 uL Taq. polymerase PCR reaction.
Follow specific protocol from your supplier of enzyme. An example Protocol is
available here.
○ Run a gel (use 1 gram of agarose and 50 mL of TAE buffer at 120 volts for 27
minutes)
● Making Freezer Stock of E. Coli:
○ Equal volume of cells w/media to pure 80% glycerol (the cryo tubes are 2mL, so
add 800uL of each)
○ Store at -80º C
Part 2. Creation and Use of Bioassay
Overview
1. Grow up mammalian cells
a. Cryogenically preserve samples at -80º C for stock solution
b. Record the passage number of each sample
2. TransfectionCenozoic: iGEM at UC Davis Experimental Protocol
a. Plate cells on 96 well plate
b. Allow cells to reach 70-90% confluency
c. Transfect with constructs using Lipofectamine
d. Select for transfected cells using geneticin (G418)
e. Expose bioassay to chemicals of concern
f. Measure fluorescence
g. Standardize fluorescence measurements
h. Measure confluence
i. Measure cell viability
Use Lipofectamine for Transfection
https://tools.thermofisher.com/content/sfs/manuals/lipofectamine3000_protocol.pdf
Concentrations at which chemicals of concern will be exposed to bioassay
Chemical name Concentration [1] [2] [3] [4] [5]
Hydrogen 0 μM 1 μM 10 μM 60 μM 100 μM
Peroxide
Copper sulfate 0 μM 1 μM 10 μM 50 μM 100 μMYou can also read

















































