Agence fédérale des médicaments et des produits de santé Federaal agentschap voor geneesmiddelen en gezondheidsproducten - MD in the retailsector ...
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Agence fédérale des médicaments
et des produits de santé
Federaal agentschap voor geneesmiddelen
en gezondheidsproducten
MD in the retailsector
Valerie Nys - Katrien Martens
04/10/2017
Who are we?
FAMHP - AFMPS – FAGG
http://www.fagg-afmps.be
• Address : Victor Horta Square 40/40
In front of the Midi Station
• More than 400 employees
• Minister of Public Health and Social
Affairs : Maggie de Block
• CEO : Xavier de Cuyper
2
Who are we?
3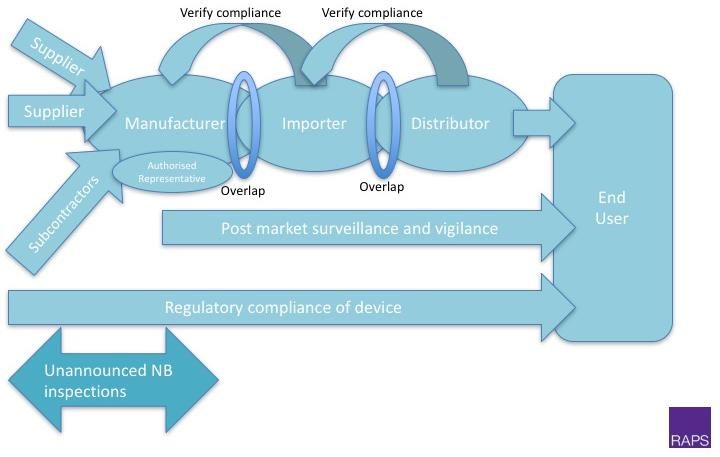
Who are we?
4
Legal basis
5
Legal basis medical devices
Legislation Guidelines
European Directives Meddev guidelines
90/385/EEC AIMDD – RD (Harmonized) standards
15/07/1977 EU consensus statements
93/42/EEC MDD – RD EU interpretative documents
18/03/1999 NBOG best practise guides
98/79/EC IVDD – RD 14/11/2001 Autocontrol guides
European Regulations
2017/745 MDR
2017/746 IVDR
Medicines law 25/03/1964
Medical devices law 15/12/2013
Ministrial degree 18/05/2005
Common specifications
Implementing acts
6
Definitions
7
Medical Device
MDD MDR
‘medical device’ means any instrument, ‘medical device’ means any instrument,
apparatus, appliance, software, material or other apparatus, appliance, software, implant, reagent,
article, whether used alone or in combination, material or other article intended by the
including the software intended by its manufacturer to be used, alone or in combination,
manufacturer to be used specifically for diagnostic for human beings for one or more of the following
and/or therapeutic purposes and necessary for its specific medical purposes:
proper application, intended by the manufacturer — diagnosis, prevention, monitoring, prediction,
to be used for human beings for the purpose of: prognosis, treatment or alleviation of disease,
— diagnosis, prevention, monitoring, treatment or — diagnosis, monitoring, treatment, alleviation of,
alleviation of disease, or compensation for, an injury or disability,
— diagnosis, monitoring, treatment, alleviation of — investigation, replacement or modification of
or compensation for an injury or handicap, the anatomy or of a physiological or pathological
— investigation, replacement or modification of process or state,
the anatomy or of a physiological process, — providing information by means of in vitro
— control of conception, and which does not examination of specimens derived from the
achieve its principal intended action in or on human body, including organ, blood and tissue
the human body by pharmacological, donations, and which does not achieve its
immunological or metabolic means, but which principal intended action by pharmacological,
may be assisted in its function by such means immunological or metabolic means, in or on the
human body, but which may be assisted in its
function by such means.
The following products shall also be deemed to be
medical devices:
— devices for the control or support of
conception;
— products specifically intended for the cleaning,
disinfection or sterilisation of devices as referred
to in Article 1(4) and of those referred to in the
first paragraph of this point.
8
Medical Device
9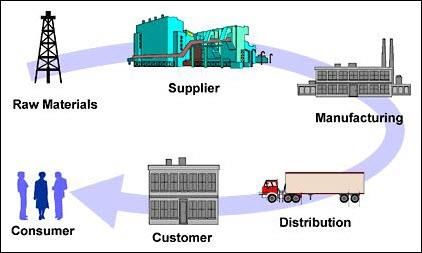
Medical Device
10MDR Annex XVI – products without
intented medical purpose – 26/05/2020
1. Contact lenses or other items intended to be introduced into or
onto the eye.
2. Products intended to be totally or partially introduced into the
human body through surgically invasive means for the purpose of
modifying the anatomy or fixation of body parts with the exception
of tattooing products and piercings.
3. Substances, combinations of substances, or items intended to be
used for facial or other dermal or mucous membrane filling by
subcutaneous, submucous or intradermal injection or other
introduction, excluding those for tattooing.
4. Equipment intended to be used to reduce, remove or destroy
adipose tissue, such as equipment for liposuction, lipolysis or
lipoplasty.
5. High intensity electromagnetic radiation (e.g. infra-red, visible
light and ultra-violet) emitting equipment intended for use on the
human body, including coherent and non-coherent sources,
monochromatic and broad spectrum, such as lasers and intense
pulsed light equipment, for skin resurfacing, tattoo or hair removal
or other skin treatment.
6. Equipment intended for brain stimulation that apply electrical
currents or magnetic or electromagnetic fields that penetrate the
cranium to modify neuronal activity in the brain.
11Medical Device accessory
MDD MDR
‘Accessory’ means an article which whilst not ‘Accessory for a medical device’ means an article
being a device is intended specifically by its which, whilst not being itself a medical device, is
manufacturer to be used together with intended by its manufacturer to be used together
a device to enable it to be used in accordance with one or several particular medical device(s) to
with the use of the device intended by the specifically enable the medical device(s) to be
manufacturer of the device; used in accordance with its/their intended
purpose(s) or to specifically and directly assist the
medical functionality of the medical device(s) in
terms of its/their intended purpose(s);
12In vitro diagnostic medical device
IVDD IVDR
‘in vitro diagnostic medical device’ means any ‘in vitro diagnostic medical device’ means any
medical device which is a reagent, reagent medical device which is a reagent, reagent
product, calibrator, control material, kit, product, calibrator, control material, kit,
instrument, apparatus, equipment or system, instrument, apparatus, piece of equipment,
whether used alone or in combination, intended software or system, whether used alone or in
by the manufacturer to be used in vitro for the combination, intended by the manufacturer to be
examination of specimens, including blood and used in vitro for the examination of specimens,
tissue donations, derived from the human body, including blood and tissue donations, derived
solely or principally for the purpose of providing from the human body, solely or principally for the
information: purpose of providing information on one or more
— concerning a physiological or pathological of the following:
state, or (a) concerning a physiological or pathological
— concerning a congenital abnormality, or process or state;
— to determine the safety and compatibility with (b) concerning congenital physical or mental
potential recipients,or impairments;
— to monitor therapeutic measures. (c) concerning the predisposition to a medical
Specimen receptacles are considered to be in vitro condition or a disease;
diagnostic medical devices. (d) to determine the safety and compatibility with
potential recipients;
‘Specimen receptacles’ are those devices, (e) to predict treatment response or reactions;
whether vacuum-type or not, specifically intended (f) to define or monitoring therapeutic measures.
by their manufacturers for the primary Specimen receptacles shall also be deemed to be
containment and preservation of specimens in vitro diagnostic medical devices;
derived from the human body for the purpose of
in vitro diagnostic examination. ‘specimen receptacle’ means a device, whether of
Products for general laboratory use are not in a vacuum-type or not, specifically intended by its
vitro diagnostic medical devices unless such manufacturer for the primary containment and
products, in view of their characteristics, preservation of specimens derived from the
are specifically intended by their manufacturer to human body for the purpose of in vitro diagnostic
be used for in vitro diagnostic examination; examination; (4) ‘accessory for an in vitro
diagnostic medical device’ means an article which,
whilst not being itself 13In vitro diagnostic medical device
14Placing on the market of MD
1. Define MD/IVD + intended use
2. Meet the essential requirements
3. Draw up the technical documentation
4. Determine the risk class
5. Follow the required conformity assessment route (with or without
intervention of a NB
6. Draw up the declaration of conformity and attach the CE marking
7. Perform a post market surveillance
15Placing on the market of MD -
Packaging
What we get What we expect
Best Medical Device Ever
BDE MD Road 45
NL-1072 Amsterdam Texas
USA
Best EU-rep Ever
Hulpmiddelenstraat 45
1000 Brussel
België
Distributor Best Distributor Ever
Verdelerslaan 45
1072 Amsterdam
Nederland
XXXX
16Pay attention to China Export
Placing on the market of MD – DoC and
CE certificate
What we expect
Declaration of Conformity CE certificate
18Placing on the market of MD – DoC and
CE certificate
What we get
Declaration of Conformity CE certificate
19Economic Operators
20Economic operators - Definitions
‘manufacturer’ a natural or legal person who manufactures or fully refurbishes a
device or has a device designed, manufactured or fully refurbished, and markets
that device under its name or trademark;
‘authorised representative’ any natural or legal person established within the
Union who has received and accepted a written mandate from a manufacturer,
located outside the Union, to act on the manufacturer's behalf in relation to
specified tasks with regard to the latter's obligations under this Regulation;
‘importer’ any natural or legal person established within the Union that places a
device from a third country on the Union market;
‘distributor’ any natural or legal person in the supply chain, other than the
manufacturer or the importer, that makes a device available on the market, up until
the point of putting into service;
21Registration of economic operators
22Registration of economic operators –
current situation
RD 18/03/1999 article 10bis
• All distributors (old definition => excluding retail) should notify their activities at the FAMHP
RD ../10(11)/2017
• All distributors (new definition => including retail, excluding manufacturers and importers) should register
themself, their activities and risk class of the distributed devices using the portal of the FAMHP
• All distributors established in BE should fill in the questionnaire allowing the FAMHP to establish a risk based
inspection planning
• One of the main questions will be whether the distributor commits himself to implement the autocontrol guide
23Registration of economic operators – MDR
Article 30 – Electronic system for registration of economic operators
1. The Commission, after consulting the MDCG, shall set up and manage an electronic system to create the
single registration number referred to in Article 31(2) and to collate and process information that is
necessary and proportionate to identify the manufacturer and, where applicable, the authorised
representative and the importer. The details regarding the information to be provided to that electronic
system by the economic operators are laid down in Section 1 of Part A of Annex VI.
2. Member States may maintain or introduce national provisions on registration of distributors of
devices which have been made available on their territory.
RD ../10(11)/2017
• All distributors (new definition => including retail) should register themself, their activities and risk class of
the distributed devices using the portal of the FAMHP
• All distributors established in BE should fill in the questionnaire allowing the FAMHP to establish a risk
based inspection planning
• One of the main questions will be whether the distributor commits himself to implement the autocontrol
guide
24Supplychain
25Current Supply Chain in Belgium
A 1.1 till 1.5 and 1.7
B 1.8 till 1.15
Manufacturer
C 1.6 and/or distributor
A B
D AIMD D Distributor +
Exporter
A B
B D
C A
AB
Health care institution
Dentist
Hospital pharmacy
Other services
B
+ MD 18/05/2005
Doctor Public-pharmacist B
Dentist B
Veterinarian
Nurse B A
B
A Pedicurist,
B physiotherapist, …
+ MD 10/08/2009
Patiënt, representative of a patiënt,
Patiënt in caretraject diabetici
keeper of animals
B RETAIL
26Cat. 1.8 till 1.15 (RD 18/03/1999 Annex XIII) + IVD Distributiecircuit MD / 13/02/2017 afmps-fagg/DGI/Industry/Meddev 27
New Supply Chain in Belgium
28Traceability of medical devices
29Traceability – current situation
Blue Guide
• The traceability requirements allow tracing the history of the product and support market surveillance. It
allows market surveillance authorities to find the liable economic operators and obtain evidence of the
product compliance.
• The traceability requirements include labelling the product and identifying the economic operators in the
distribution chain.
National legislation
• MD 18/05/2005 traceability based upon lot/serienumber for distribution of spec MD to specific healthcare
professionals
Autocontrol guide for distributors
• Traceability based upon lot/serienumber for distribution to other economic operators and health institutions or
healthcare professionals
30Traceability – MDR
Article 25 – Identification within the supply chain
1. Distributors and importers shall co-operate with manufacturers or authorised representatives to achieve an
appropriate level of traceability of devices.
2. Economic operators shall be able to identify the following to the competent authority, for the period referred
to in Article 10(8):
a) any economic operator to whom they have directly supplied a device;
b) any economic operator who has directly supplied them with a device;
c) any health institution or healthcare professional to which they have directly supplied a device.
Article 27 – Unique device identification system
8. Economic operators shall store and keep, preferably by electronic means, the UDI of the devices which they
have supplied or with which they have been supplied, if those devices belong to:
• class III implantable devices;
• the devices, categories or groups of devices determined by a measure referred to in point (a) of
paragraph 11.
31Traceability – Summary
Currently
• Register IN
• Register OUT to economic operators
• Register OUT (lotnr) spec MD to healthcare professionals (MD 18/05/2005)
• Register IN and OUT (lotnr) to economic operators, health institutions and
healthcare professionals
After 26/05/2020
• Register IN
• Register OUT to economic operators, health institutions and healthcare
professionals
• Register IN and OUT (UDI) for Class III implantable devices and Commissions list
• Register IN and OUT (lotnr) to economic operators, health institutions and
healthcare professionals
32General obligations of distributors
33Obligations of distributors – current
situation
MDD
• No obligations for distributors
National legislation (RD ../10(11)/2017)
• Registration at FAMHP via portal
• Adapted storage facilities
Autocontrol guide for distributors
• QMS
• Personnel
• Documentation
• Adapted facilities
• Activities
• Returned goods, complaints, recalls
• Materiovigilance
34Obligations of distributors – MDR
Article 14 – General obligations of distributors
1. When making a device available on the market, distributors shall, in the context of their activities, act with
due care in relation to the requirements applicable.
2. Before making a device available on the market, distributors shall verify that all of the following requirements
are met:
a) the device has been CE marked and that the EU declaration of conformity of the device has been
drawn up;
b) the device is accompanied by the information to be supplied by the manufacturer in accordance
with Article 10(11);
c) for imported devices, the importer has complied with the requirements set out in Article 13(3);
d) that, where applicable, a UDI has been assigned by the manufacturer.
In order to meet the requirements referred to in points (a), (b) and (d) of the first subparagraph the distributor
may apply a sampling method that is representative of the devices supplied by that distributor.
Where a distributor considers or has reason to believe that a device is not in conformity with the
requirements of this Regulation, it shall not make the device available on the market until it has been brought
into conformity, and shall inform the manufacturer and, where applicable, the manufacturer's authorised
representative, and the importer. Where the distributor considers or has reason to believe that the device
presents a serious risk or is a falsified device, it shall also inform the competent authority of the Member
State in which it is established.
35Obligations of distributors – MDR
Article 14 – General obligations of distributors
3. Distributors shall ensure that, while the device is under their responsibility, storage or transport conditions
comply with the conditions set by the manufacturer.
4. Distributors that consider or have reason to believe that a device which they have made available on the
market is not in conformity with this Regulation shall immediately inform the manufacturer and, where
applicable, the manufacturer's authorised representative and the importer. Distributors shall co-operate
with the manufacturer and, where applicable, the manufacturer's authorised representative, and the
importer, and with competent authorities to ensure that the necessary corrective action to bring that device
into conformity, to withdraw or to recall it, as appropriate, is taken. Where the distributor considers or has
reason to believe that the device presents a serious risk, it shall also immediately inform the competent
authorities of the Member States in which it made the device available, giving details, in particular, of the
non-compliance and of any corrective action taken.
5. Distributors that have received complaints or reports from healthcare professionals, patients or users
about suspected incidents related to a device they have made available, shall immediately forward this
information to the manufacturer and, where applicable, the manufacturer's authorised representative, and
the importer. They shall keep a register of complaints, of non-conforming devices and of recalls and
withdrawals, and keep the manufacturer and, where available, the authorised representative and the
importer informed of such monitoring and provide them with any information upon their request.
36Obligations of distributors – MDR
Article 14 – General obligations of distributors
6. Distributors shall, upon request by a competent authority, provide it with all the information and
documentation that is at their disposal and is necessary to demonstrate the conformity of a device.
Distributors shall be considered to have fulfilled the obligation referred to in the first subparagraph when the
manufacturer or, where applicable, the authorised representative for the device in question provides the
required information. Distributors shall co-operate with competent authorities, at their request, on any action
taken to eliminate the risks posed by devices which they have made available on the market. Distributors,
upon request by a competent authority, shall provide free samples of the device or, where that is
impracticable, grant access to the device.
37Obligations of distributors – Summary
Currently
• Registration at FAMHP
• Adapted storage facilities
• Good Distribution Practices
After 26/05/2020
• Current obligations
• Check conformity before making a device available on the market. Not conform
not on the market
• Respect storage and transport conditions
• Report non-conformities and cooperate in case of corrective actions
• Immediately forward complaints / reports about suspected incidents
• Register complaints, non conforming devices, recalls and withdrawals
• Provide CA with information and documentation. Provide free samples or grant
access to the device
• Good Distribution Practices
38Repackaging/relabelling
39Repackaging/relabelling – MDR
Article 16 – Cases in which obligations of manufacturers apply to importers, distributors of other
persons
1. A distributor, importer or other natural or legal person shall assume the obligations incumbent on
manufacturers if it does any of the following:
a) makes available on the market a device under its name, registered trade name or registered
trade mark, except in cases where a distributor or importer enters into an agreement with a
manufacturer whereby the manufacturer is identified as such on the label and is responsible for
meeting the requirements placed on manufacturers in this Regulation;
b) changes the intended purpose of a device already placed on the market or put into service;
c) modifies a device already placed on the market or put into service in such a way that
compliance with the applicable requirements may be affected.
The first subparagraph shall not apply to any person who, while not considered a manufacturer as
defined in point (30) of Article 2, assembles or adapts for an individual patient a device already on
the market without changing its intended purpose.
One shall assume all obligations of a manufacturer when:
He makes MD available on the market under its own name / trade mark
without mentioning the legal manufacturer
He changes the intended use of a device
He modifies a device in a way that might affect compliance with the MDR
40Repackaging/relabelling – MDR
Article 16 – Cases in which obligations of manufacturers apply to importers, distributors of other
persons
2. For the purposes of point (c) of paragraph 1, the following shall not be considered to be a
modification of a device that could affect its compliance with the applicable requirements:
a) provision, including translation, of the information supplied by the manufacturer, in
accordance with Section 23 of Annex I, relating to a device already placed on the market and of
further information which is necessary in order to market the device in the relevant Member
State;
b) changes to the outer packaging of a device already placed on the market, including a change
of pack size, if the repackaging is necessary in order to market the device in the relevant
Member State and if it is carried out in such conditions that the original condition of the device
cannot be affected by it. In the case of devices placed on the market in sterile condition, it shall
be presumed that the original condition of the device is adversely affected if the packaging that
is necessary for maintaining the sterile condition is opened, damaged or otherwise negatively
affected by the repackaging.
Not regarded as manufacturing:
Provision of information incl translation (relabelling)
Changes to outer packaging (repackaging)
41Repackaging/relabelling – MDR
Article 16 – Cases in which obligations of manufacturers apply to importers, distributors of other
persons
3. A distributor or importer that carries out any of the activities mentioned in points (a) and (b) of
paragraph 2 shall indicate on the device or, where that is impracticable, on its packaging or in a
document accompanying the device, the activity carried out together with its name, registered
trade name or registered trade mark, registered place of business and the address at which it
can be contacted, so that its location can be established. Distributors and importers shall ensure that
they have in place a quality management system that includes procedures which ensure that the
translation of information is accurate and up-to-date, and that the activities mentioned in points (a) and
(b) of paragraph 2 are performed by a means and under conditions that preserve the original condition
of the device and that the packaging of the repackaged device is not defective, of poor quality or
untidy. The quality management system shall cover, inter alia, procedures ensuring that the distributor
or importer is informed of any corrective action taken by the manufacturer in relation to the device
in question in order to respond to safety issues or to bring it into conformity with this Regulation.
4. At least 28 days prior to making the relabelled or repackaged device available on the market,
distributors or importers carrying out any of the activities mentioned in points (a) and (b) of paragraph 2
shall inform the manufacturer and the competent authority of the Member State in which they plan
to make the device available of the intention to make the relabelled or repackaged device available
and, upon request, shall provide the manufacturer and the competent authority with a sample or
mock-up of the relabelled or repackaged device, including any translated label and instructions for
use. Within the same period of 28 days, the distributor or importer shall submit to the competent
authority a certificate, issued by a notified body designated for the type of devices that are subject
to activities mentioned in points (a) and (b) of paragraph 2, attesting that the quality management
system of the distributer or importer complies with the requirements laid down in paragraph 3.
42Materiovigilance
43Materiovigilance
• The purpose of materiovigilance is to study and follow incidents that might result from
using medical devices as well as the assessment and follow-up of safety corrective
actions. It enables dangerous devices to be withdrawn from the market and to
eliminate faults in medical devices with the intention of constantly improving the
quality of devices and providing patients and users with increased safety.
• Materiovigilance only refers to medical devices and their accessories whereas
pharmacovigilance refers to medicines
• Incident: “Any malfunction or deterioration in the characteristics and/or performance
of a device, as well as any inadequacy in the labeling or the instructions for use
which, directly or indirectly, might lead to or might have led to the death of a patient,
or USER or of other persons or to a serious deterioration in their state of health.”
• Field safety corrective action (FSCA): is an action taken by a MANUFACTURER to
reduce a risk of death or serious deterioration in the state of health associated with
the use of a medial device that is already placed on the market. Such actions should
be notified via a field safety notice (FSN) to the users of the device.
44Materiovigilance – current situation
MDD
• No obligations for distributors
National legislation
• RD 18/03/1999 art. 11 distributors should immediately notify each incident to the FAMHP
• RD 18/03/1999 art. 11 distributors should notify a local contactpoint materiovigilance to the FAMHP
RD ../10(11)/2017
• Local contactpoint materiovigilance
• Notification of incidents
• Collaboration to investigations / corrective actions
45Materiovigilantie – MDR
Article 87 Reporting of serious incidents and field safety corrective actions
• Manufacturer should notify serious incidents and FSCA in MDR Eudamed
Article 14 – General obligations of distributors
5. Distributors that have received complaints or reports from healthcare professionals, patients or users
about suspected incidents related to a device they have made available, shall immediately forward
this information to the manufacturer and, where applicable, the manufacturer's authorised
representative, and the importer. They shall keep a register of complaints, of non-conforming
devices and of recalls and withdrawals, and keep the manufacturer and, where available, the
authorised representative and the importer informed of such monitoring and provide them with any
information upon their request.
46Fees
47Legal basis
• Article 68 – loi du 18 décembre 2016 portant des dispositions
diverses en matière de santé:
L'article 34 de la même loi, modifié par la loi du 26 décembre 2015, est remplacé par ce qui suit :
Art. 34. Les opérateurs sont redevables d'une contribution annuelle d'un maximum de 0,4026092 % sur leur chiffre d'affaires de
dispositifs médicaux réalisé l'année civile précédente sur le marché belge.
La contribution annuelle visée à l'alinéa 1er est, pour l'année de contribution concernée, multipliée par le quotient du déficit à financer
au compte d'exécution du budget tel que visé à l'article 6, § 2, de la loi du 16 mars 1954 relative au contrôle de certains organismes
d'intérêt public, de l'Agence fédérale des Médicaments et des Produits de Santé pour l'année de contribution concernée et du
dénominateur.
Le dénominateur visé à l'alinéa 2 est la somme de :
- la contribution annuelle maximum, celle-ci ne pouvant être inférieure à 500 euros par distributeur;
- le nombre total d'autorisations qui sont soumises à la contribution forfaitaire visée à l'article 225, § 1er, de la loi du 12 août 2000
portant des dispositions sociales, budgétaires et diverses et multiplié par le montant maximum de celle-ci.
Pour l'application du présent article, le déficit à financer au compte d'exécution est la différence entre les dépenses et les recettes
pour cette année avant l'imputation de la présente contribution annuelle et de la contribution forfaitaire visée à l'article 225, § 1er, de la
loi du 12 août 2000 portant des dispositions sociales, budgétaires et diverses.
Le quotient est positif et est de maximum 1. L'Agence fédérale des Médicaments et des Produits de Santé publie le montant du
quotient sur son site web avant le 31 mai de l'année qui suit l'année de contribution.
La contribution annuelle visée à l'alinéa 2 est réglée par le paiement d'une avance avant le 31 décembre de l'année de contribution
concernée et d'un solde. L'avance est calculée sur la base du maximum de la contribution, avec un minimum de 500 euros. Le solde
est remboursé.
Par dérogation à l'alinéa 6, l'AFMPS calcule l'avance, pour l'année de contribution suivante, en y incluant le solde si le redevable
reste le même.
Le montant de 500 euros du présent chapitre est adapté annuellement, en fonction de l'indice du mois de septembre, à l'évolution de
l'indice des prix à la consommation. L'indice de départ est celui du mois de septembre précédant la publication de la présente loi au
Moniteur belge. Les montants indexés sont publiés au Moniteur belge et sont exigibles à partir du 1er janvier de l'année qui suit celle
durant laquelle l'adaptation a été effectuée.
48Legal Basis
• Article 35 – loi du 15 décembre 2013 – loi en matière de
dispositifs médicaux:
Art. 35. Les distributeurs tiennent un journal particulier, organisé de telle sorte que les détails des opérations de vente, d'exportation et
des services fournis concernant les dispositifs puissent être suivis, en indiquant le montant, le mode et le jour de la perception ainsi que
les prélèvements en nature autres que pour leur entreprise, ainsi que les conséquences de ces opérations pour le chiffre d'affaires de
dispositifs médicaux.
Le journal particulier est tenu en permanence à la disposition de l'AFMPS au siège social du distributeur ou, s'il est différent, au lieu
principal où s'exercent les activités. Si le distributeur n'a pas de siège en Belgique, ou est une personne physique sans domicile en
Belgique, mais dispose d'une ou de plusieurs antennes ou d'un ou plusieurs centres d'activités, la mise à disposition a lieu dans la
principale antenne ou le principal centre d'activités en Belgique, et le distributeur s'assure que l'adresse de ce centre est connue de
l'AFMPS.
Le distributeur introduit chaque année avant le 1er avril auprès de l'AFMPS une déclaration du chiffre d'affaires de dispositifs
médicaux de l'année civile précédente, certifiée par un réviseur ou un expert-comptable sur la base du journal particulier.
Que le distributeur soit soumis ou non à la loi du 17 juillet 1975 relative à la comptabilité des entreprises, les articles 6 à 8, les arrêtés
d'exécution de ceux-ci, et l'article 16 de la loi précitée du 17 juillet 1975 s'appliquent à la tenue du journal particulier et à l'attestation du
chiffre d'affaires.
Le Roi peut fixer un modèle pour la déclaration visée à l'alinéa 3.
49Example – Year X
Ex: turnover of the company – Year X: 1 000 000 €
31 mars
01 Jan
Dec
Mai
Quotient Define fees for
Turnover
publication on the year
declaration
web site (income/ ex: 0,20%
outcome)
=> 1 000 000 €
Billing = 0,20% of Balance =
turnover provision for year
Fees payment with a maximum of
+1
0,4026092%
=> 2000 €
=> 2026,09 €
Fees to pay :
CAx0,4026092%
year X+1
Balance customer:
=> 4026,09 €
4026,09€
[0,4026092% du CA]
50Example – Year X + 1
Ex: turnover of the company – Year X+1 : 1 000 000 €
31 mars
01 Jan
Dec
Mai
Turnover Quotient Define fees for
declaration publication on the year
web site (income/ ex: 0,15%
=> 1 000 000 € outcome)
Billing = 0,15% of Balance =
turnover provision for year
Fees payment with a maximum of
+2
0,4026092%
=> 1500 €
=> 2526,09 €
Fees to pay :
CAx0,4026092% - provision year X
Balance Customer:
Year X+2
4026,09€
=> 4026,09 € - 2026,09€ [0,4026092% du CA]
51
=> 2000 €Contact
Federal agency for medicines and health products -
famhp
Place Victor Horta 40/40
1060 BRUSSELS
tel. + 32 2 528 40 00
fax + 32 2 528 40 01
e-mail meddev@fagg-afmps.be
www.fagg-afmps.be
52Vos médicaments et produits de santé,
notre préoccupation
Uw geneesmiddelen en
gezondheidsproducten, onze zorgYou can also read

















































