Clinical Trials for Huntington Disease - Practical Neurology
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
S P E C I A L R E P OR T
Clinical Trials for
Huntington Disease
The promise for potential disease‑modifying research is strong, although
continued discovery uncovers new questions and challenges to overcome.
By Natalia P. Rocha, PharmD, MSc, PhD; Gabriela D. Colpo, PhD;
Antonio L. Teixeira, MD, PhD, MSc; and Erin Furr Stimming, MD
Huntington disease (HD) is an improving function and quality of life for people with HD.
inherited neurodegenerative Pharmacologic options for the motor symptoms include
disease characterized by a clini‑ neuroleptics, gabaergics, and antiglutaminergics, all of which
cal triad of motor, cognitive, and are used off‑label. The vesicular monoamine transporter 2
psychiatric symptoms. Common (VMAT2) inhibitors, tetrabenazine and deutetrabenazine, are
motor symptoms include cho‑ the only 2 medications currently approved by the Food and
rea, dystonia, and incoordina‑ Drug Administration (FDA) for the treatment of chorea in HD.
tion. The cognitive symptoms Antidepressants, neuroleptics, and mood stabilizers are often
are primarily subcortical, which used to treat neuropsychiatric symptoms. Interdisciplinary,
results in a dysexecutive syn‑ nonpharmacologic treatments are also important, including
drome. Common neuropsychi‑ physical, occupational, and speech therapy, psychotherapy,
atric symptoms include apathy, genetic counseling, nutrition and social work services.4
anosognosia, irritability, impulsivity, depression, and anxiety.1
The autosomal dominant inheritance pattern of HD has Investigational Disease‑Modifying Treatments
been known for decades. The specific mutation, however, was Initial attempts to develop DMTs for HD focused on cel‑
described over 26 years ago and is a highly penetrant CAG lular mechanisms associated with neuronal death, such as glu‑
trinucleotide repeat expansion in the huntingtin (HTT) gene tamate‑mediated excitotoxicity, and apoptosis. Subsequently,
(chromosome 4 (4p16.3)]).2 The CAG repeat expansion in other neurodegenerative mechanisms were targeted, includ‑
exon 1 is translated into an expanded polyglutamine resulting ing inflammatory and immune mechanisms, oxidative stress,
in a mutant HTT protein (mHTT). Ubiquitous at the tissue and mitochondrial dysfunction. Despite promising results
and subcellular level, wild‑type HTT (wtHTT) has numerous in preclinical studies, none of these have yet demonstrated
functions, not all of which are understood, including tissue efficacy as DMTs for HD. Several factors may have influenced
maintenance, cell morphology and survival, and control of these disappointing results, including the lack of appropriate
organelle and vesicle transport. In the nucleus, HTT may act clinical and biologic markers for selecting participants and
as a scaffold for transcriptional complexes. Of note, the HTT tracking disease progression and modification. In addition, the
gene is thought to be necessary for embryonic development.3 power of the studies and clinical scales used as primary end‑
The identification of the huntingtin gene, previously known points may not have been sensitive enough to detect changes
as interesting transcript 15 (IT15) has been a major milestone in disease progression. More recently, strategies have shifted
in HD research and several disease‑modifying therapies from nondisease specific neurodegeneration to interventions
(DMTs) have been studied since its discovery, although none targeting core upstream disease‑specific processes.5 This revo‑
have yet proven efficacy or been approved.4 Various com‑ lutionary change has been made possible by rapid advances
pounds have been tested, including novel approaches target‑ in gene‑modifying technology. Current and future trials are
ing immune dysregulation in HD, mitochondrial‑based pro‑ focused on reducing the amount of mHTT (Figure).
tective strategies, and gene‑modifying technologies (Table).
Lowering Mutant Huntingtin Levels
Symptomatic Treatments Lowering mHTT levels is among the most promising
Current treatments are purely symptomatic, aimed at strategies for developing DMTs for HD. These strategies are
JUNE 2020 PRACTICAL NEUROLOGY 69
SP E C I A L R EPO R T
TABLE. CLINICAL TRIALS OF POTENTIAL DISEASE‑MODIFYING THERAPIES FOR HUNTINGTON’S DISEASE
Phase/designa Intervention/treatment Study status; Outcome measures NCT
Huntingtin‑Lowering Therapies
Phase 1/2a 13‑wk with Intrathecal second‑gener‑ Completed; showed a dose‑dependent reduction in CSF levels of NCT02519036
14‑month OLE (n=46) ation 2’‑O‑meth‑oxyethyl mutant HTT; no serious adverse events were reported6 NCT03342053
chimeric ASOs (10, 30, 60,
90, 120 mg) every 4 or 8 wks
Phase 3 5‑year OLE Tominersen every 8 or Recruiting; long‑term safety/tolerability of tominersen in people NCT03842969
(n=1,050) 16 wks with HD who were in prior studies
Phase 3 (n=909) Tominersen every 8 or Recruiting; efficacy and safety of tominersen in manifest HD NCT03761849
16 wks
Phase 1b/2a (n=48) Intrathecal stereopure ASO Recruiting; safety, tolerability, pharmacokinetics, and pharmacody‑ NCT03225833
(single and multiple doses) namics of single/multiple doses of stereopure ASO in adults with
early HD with single nucleotide polymorphism (SNP) rs362307
Phase 1b/2a (n=48) Intrathecal injections of Recruiting; safety, tolerability, pharmacokinetics, and pharmacody‑ NCT03225846
stereopure ASO (single and namics of single/multiple doses of stereopure ASO in adults with
multiple doses) early HD with SNP rs362331
Phase 1/2 5‑year with Striatally‑administered Recruiting; evaluate the safety and potential efficacy NCT04120493
3.5‑year OLE (n=26) recombinant HTT
(rAAV5‑miHTT)
Huntingtin Lowering and Modulation Therapies
Phase 2 26‑week Second‑generation Completed; showed improvement in executive function with NCT01590888
(n=109) 8‑hydroxyquinoline analog, 250 mg treatment vs placebo; well tolerated and safe15
100 or 250 mg/d
Phase 1 14‑day (n=55) Selisistat 10 or 100 mg/d Completed; showed safety and tolerability in adults with early NCT01485952
stage HD at plasma concentrations beneficial in animal models18
Phase 2 12‑week Selisistat 50 or 200 mg/d Completed; showed safety and tolerability with a trend toward NCT01521585
(n=144) modulated plasma levels of soluble mutant huntingtin19
Phase 1b 14‑day Selisistat 100 mg/d Completed; no results published or posted NCT01485965
open‑label paral‑ Groups are fasting vs nonfasting
lel‑group (n=26)
Immunomodulatory and Anti‑inflammatory Therapies
Phase 2 12‑month Laquinimod 0.5, 1.0 or Completed; reduced caudate and whole‑brain atrophy (most NCT02215616
(n=352) 1.5 mg/d evident in participants with early HD) but no change in UHDRS
Phase 2 (n=301) Pepinemab (humanized Active, not recruiting; safety, tolerability, immunogenicity, and NCT02481674
antiSEMA4D MAb) efficacy of pepinemab in prodromal and early manifest HD
Metabolic‑ and Mitochondrial‑Based Protective Strategies
Phase 3 60‑month Coenzyme Q10 (CoQ10) Terminated; futility analysis failed to show likelihood of benefit; NCT00608881
(n=609) 240 mg/d generally safe and well tolerated24
Phase 1/2 16‑week Creatine 8 g/d Completed; showed safety and tolerability, reduced levels of NCT00026988
(n=64) serum 8‑hydroxy‑2’‑deoxyguanosine levels (indicatory of DNA
oxidative injury), increased creatine (serum/brain) that returned
to baseline after washout, but no change in UHDRS
Phase 2 6‑month Creatine monohydrate Completed; showed safety and tolerability; slowed cortical and NCT00592995
(n=64); 12‑ (n=38) and 30 g/d striatal atrophy at 6 and 18 months but no clinical improvement NCT01411150
24‑month OLEs (n=24) NCT01411163
Table continued on page 71
70 PRACTICAL NEUROLOGY JUNE 2020
S P E C I A L R E P OR T
TABLE. CLINICAL TRIALS OF POTENTIAL DISEASE‑MODIFYING THERAPIES FOR HUNTINGTON’S DISEASE (CONT.)
Phase/designa Intervention/treatment Study status/outcome measures NCT
Metabolic‑ and Mitochondrial‑Based Protective Strategies (cont.)
Phase 2 44‑month OLE Creatine monohydrate Completed ; showed safety and tolerability in adults with HD NCT01412151
(n=10) ≤30 g/d
Phase 3 48‑month Creatine monohydrate Terminated; interim analysis showed lack of promise for slow‑ NCT00712426
(n=553) ≤40 g/d ing disease progression in patients with early‑HD27
Phase 3 6‑month study Ethyl‑EPA, 2 g/d Completed; showed that ethyl‑EPA did not improve motor NCT00146211
(n=316) function, global functioning cognition, or global impression28
Abbreviations: ASO, antisense oligonucleotide; EPA, eicosapentaenoic acid; HD, Huntington disease; HTT, huntingtin; MAb, monoclo‑
nal antibody; OLE, open‑label extension; SEMA4D, semaphorin 4D; UHDRS, Unified Huntington Disease Rating Scale.
a Studies are double‑blind randomized placebo‑ or sham‑controlled unless otherwise noted.
focused on inhibiting messenger RNA (mRNA) synthesis by conserved protein with several important cellular functions,3
blocking transcription with zinc finger motif proteins, avoid‑ it is unclear if wtHTT reduction has long‑term safety. In this
ing posttranscriptional processing to increase mRNA degra‑ regard, the use of an allele‑specific ASO is an interesting alterna‑
dation with antisense oligonucleotides (ASOs), and inhibit‑ tive. There are 2 randomized double‑blind placebo‑controlled
ing mRNA translation with small interfering RNA (siRNA).5 phase 1a/2b trials studying safety, tolerability, pharmacokinetics,
Antisense Oligonucleotides. Designed to target mRNA, ASOs and pharmacodynamics of intrathecally administered allele‑spe‑
are synthetic, short, single‑stranded DNA sequences that form cific ASOs in people with early manifest HD (Table). Allele
a hybridized complex with specific mRNAs causing them to be specificity has been achieved by developing stereopure ASOs
degraded through an RNAse‑H1 mechanism. The first human directed at specific single nucleotide polymorphisms (SNPs).
clinical trial with ASO therapy for lowering HTT evaluated the Both trials are enrolling participants with specific HTT SNPs on
safety, tolerability, pharmacokinetics, and pharmacodynam‑ the same allele as the pathogenic CAG expansion.
ics of multiple ascending doses of tominersen administered Small Interfering RNAs. The siRNA and artificial microRNA
intrathecally to 46 participants with early manifest HD at (miRNA) neutralize mRNA molecules to inhibiting mRNA
4‑week intervals for 13 weeks. Tominersen is nonallele‑specific translation, a process known as RNA interference (RNAi).7,8
and thus targets both wtHTT and mHTT. Preliminary results Using the adeno‑associated viral (AAV) vector to deliver
suggest that tominersen is safe, and well tolerated. At the an siRNA targeting mHTT mRNA in a mouse model of HD,
90‑mg and 120‑mg doses, tominersen was associated with an more than 80% of the cells in the striatum were successfully
approximately 40% reduction of mHTT levels in cerebrospinal transduced (ie, expressed the siRNA). Of note, there were also
fluid (CSF), which may correspond to a 55% to 85% reduction significant improvements in HD‑associated behavioral deficits
in cortical mHTT levels. There was also a correlation between and the reduction of striatal HTT aggregates in the transfected
reduced CSF mHTT levels and improved composite Unified mice.9 A cholesterol‑conjugated siRNA targeting HTT (cc‑siR‑
Huntington Disease Rating Scale (cUHDRS) scores.6 NA‑HTT) ameliorated HD‑related neuropathology and motor
There are 2 open‑label extensionsa underway for partici‑ deficits in a different mouse model.8 An artificial miRNA deliv‑
pants who have previously received tominersen and a phase 3 ered via the AAV vector to a sheep model of HD also reduced
double‑blind placebo‑controlled interventional trial evaluat‑ 50% to 80% of HTT mRNA and protein in the striatum and
ing safety and efficacy in individuals with manifest HD along prevented subsequent neuron loss at 1 and 6 months after
with a natural history study. The primary outcome measure injection. The sheep model results show that effective silencing
for the pivotal trial is the total functional capacity (TFC) in of mHTT by AAV‑delivered artificial miRNA can be achieved
the US and the cUHDRS outside the US. Secondary outcomes and sustained in a large‑animal brain.10 Another mouse model
include additional components of the UHDRS, including cog‑ injected with an AAV‑delivered microRNA (miRNA) for
nitive and behavioral assessments, the clinical global impres‑ human HTT mRNA selectively knocks down expression and is
sion score, adverse events, pharmacokinetic markers, CSF most effectively delivered to the putamen and thalamus with
mHTT, neurofilament (NfL) levels, and brain volume on MRI. MRI‑guided injection and reduced HTT levels in deep brain tis‑
Tominersen has demonstrated reduction in CSF HTT levels, sues, such as the caudate, putamen, and thalamus, in addition
and efficacy of the treatment is under investigation. It is unclear, to the cortex. This treatment was well tolerated by the animals
however, if residual mHTT levels and lowering both wtHTT and for up to 5 weeks and phase 1 clinical trials in humans with HD
mHTT levels in the CSF are clinically relevant. HTT is a highly are expected to begin soon.11
a. See table for NCT identifiers of all clinical trials mentioned without published results
JUNE 2020 PRACTICAL NEUROLOGY 71
SP E C I A L R EPO R T
ed HTT and reversed the HD‑associated neuropathology and
behavioral phenotypes. Decreased mHTT expression did not
affect cell viability, but alleviated motor deficits.13 Despite the
positive results in preclinical studies, however, several issues
must be addressed before bringing CRISPR/Cas9 technologies
into humans, including that it is irreversible with ethical con‑
cerns regarding germline alteration and that there are delivery
problems with viral vectors and concerns about the possible
immunogenicity of bacterial proteins.14
Huntingtin Protein Modulation
Strategies for HTT protein modulation include clearance
and inhibition of protein aggregation. Excessive metal concen‑
trations induce mHTT aggregation and promote formation of
reactive oxygen species (ROS). Therefore, it has been hypoth‑
esized that 8‑hydroxyquinoline transition metal ligand, which
redistributes metals (eg, copper, zinc, and iron) from locations
where they are abundant to subcellular locations where they
might be deficient, could attenuate mHTT effects by inhib‑
iting metal‑induced aggregation.15 In preclinical studies in
Caenorhabditis elegans and mouse models of HD, 8‑hydroxy‑
quinoline transition metal ligand was effective in ameliorat‑
ing the HD phenotype.16 In a 26‑week phase 2 randomized
double‑blind placebo‑controlled clinical trial, 109 participants
with HD had significant improvement in cognition (Trail
Making Test Part B) with a 250‑mg dose, but no effects in
other domains were seen compared with the 100‑mg dose
or placebo. This isolated finding was thought to be of limited
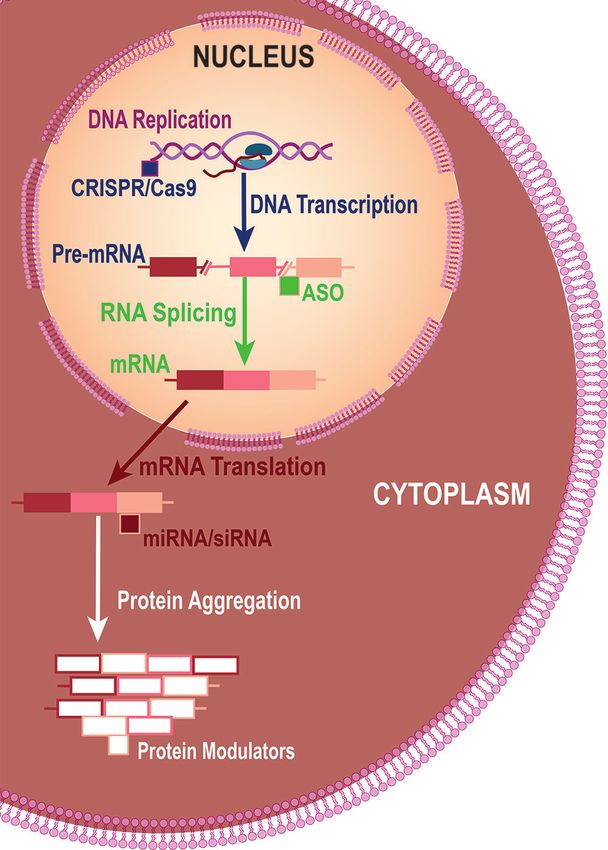
Fig. 1: Strategies for Lowering Mutant Huntingtin Levels. Different clinical relevance.15 Although the treatment was considered
approaches for lowering mutant huntingtin (mHTT) include safe and well tolerated, the FDA issued a partial clinical hold
using clustered regularly interspaced short palindromic repeats because of safety concerns for the 250‑mg dose and required
(CRISPR)‑Cas9 to edit DNA so mHTT is not transcribed, antisense more neurotoxicity data. There have been no recent updates.
oligonucleotides (ASOs) to reduce production of mRNA during Selisistat has been studied for promotion of HTT protein
processing (splicing) of pre-mRNA, microscopic or small interfering clearance. Selisistat is an inhibitor of the silent information reg‑
RNA (miRNA or siRNA) to reduce production of mHTT protein dur‑ ulator T1 (SirT1), which is a member of the sirtuin deacetylase
ing mRNA translation, and protein modulation to reduce aggrega‑ family that can remove acetyl groups from mHTT. Inhibition
tion or increase clearance. of SirT1 alleviated pathology in animal and cell models of HD.17
Clinical trials of selisistat demonstrated that it is safe and well
Similarly, an engineered miRNA delivered via AAV vector tolerated but failed to show efficacy.18,19 Another trial has been
serotype 5 (AAV5‑miHTT) in a single intrastriatal injection into registered that will assess selisistat in fasted vs fed conditions
a minipig model of HD resulted in a strong, widespread and (ie, the effect of food on the pharmacokinetics of selisistat in
sustained (up to 12 months) reduction of mHTT levels and a patients with HD). The study was designed to explore poten‑
phase 1 trial of this agent in humans with HD is also beginning. tial biomarkers for use in subsequent phase 2/3 studies, but no
Genome Editing. The clustered regularly interspaced short results have been published to date. There are no phase 3 trials
palindromic repeats (CRISPR)‑associated (Cas)9 system has planned; further development appears to be on hold.
emerged as a promising option because of allele‑specificity and
potency. The CRISPR/Cas9 technology was used in fibroblasts Neuroprotective Approaches
of an HD patient to delete the promoter regions, transcription Immune dysregulation and mitochondrial dysfunction are
start site, and the CAG mutation expansion of the mHTT gene. regarded as important contributors to neurodegeneration in
Complete inactivation of the mutant allele occurred without HD and have been targets for HD‑modifying therapies.
effects on the normal allele.12 The CRISPR/Cas9 method has Immunomodulatory Agents. The immunomodulatory
also been tested in an HD rodent model and efficiently deplet‑ strategies for HD include laquinimod, an immunomodulatory
72 PRACTICAL NEUROLOGY JUNE 2020
S P E C I A L R E P OR T
drug originally studied for the treatment of relapsing‑remit‑ eration, immune/inflammatory mechanisms, glutamate‑medi‑
ting multiple sclerosis; minocycline, a second‑generation tetra‑ ated excitotoxicity, the dopamine pathway, and mitochondrial
cycline that has been in therapeutic use for over 30 years; and dysfunction. None have yet been successful in identifying a
pepinemab, an investigational antibody to semaphorin 4D drug with proven efficacy to modify HD progression; however,
(SEM4D). Neuroprotective and HD‑ameliorating effects of pending results may be positive. Several factors might influence
laquinimod in preclinical studies20‑22 motivated clinical trials the previous disappointing results, including underpowered
in participants with HD. In randomized placebo‑controlled studies and the lack of appropriate clinical and biological
double‑blind trials, neither 12 weeks of laquinimod or 8 weeks markers capable of tracking disease modification. The clini‑
of minocycline vs placebo showed efficacy as measured by cal scales currently used as primary endpoints might not be
change in the UHDRS or cognitive test scores. The laquini‑ sensitive enough at detecting signs and symptoms of disease
mod treatment, however, was correlated with a significant progression. New innovative tools to objectively assess disease
reduction in caudate and whole‑brain atrophy that was most signs and symptoms and the development of neuroimag‑
evident in people with early manifest HD. At this time, it is ing‑based or biological fluid‑based markers of disease progres‑
unclear how a positive MRI‑based secondary outcome in sion will be imperative to the success of clinical trials in HD.
the setting of a negative clinical primary outcome should be The recent advances in therapeutic strategies promise an
interpreted. It is possible that laquinimod would be more exciting era for clinical trials in HD. Current and future trials
efficient if it were initiated years (or even decades) before are focused on lowering mHTT by targeting mHTT produc‑
disease onset, thus preventing neurodegeneration. Regarding tion, aggregation, misfolding, and removal. The ASOs are
minocycline, the short‑period treatment could explain the the furthest along in clinical development in the realm of
lack of efficacy; however, no results were seen in another trial huntingtin‑lowering strategies; however, many questions
of 18 months of minocycline treatment.23 remain, particularly long‑term safety and the effect of decreas‑
Pepinemab is an antibody against SEM4D thought to ing wtHTT levels. The results of studies using CRISPR/Cas9
decrease central nervous system inflammation, increase technologies in preclinical models of HD are very encouraging,
neuronal outgrowth, and enhance oligodendrocyte matura‑ yet these techniques are distant from use in humans. In sum‑
tion. Pepinemab is being evaluated in a phase 2 multicenter mary, the landscape is changing for many inherited condi‑
randomized double‑blind placebo‑controlled study assessing tions due to recent technological advances in gene modifying
safety, tolerability, pharmacokinetics, and efficacy in partici‑ approaches. The future for HD disease‑modifying research is
pants with prodromal or early manifest HD. promising, although with continued discovery, new questions
Mitochondrial‑Based Agents. Several clinical trials have eval‑ and challenges emerge. n
uated antioxidant treatment with coenzyme Q10 (CoQ10) 1. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev
and creatinine as potential DMTs for HD. Although both Neurol. 2014;10(4):204‑216.
2. The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and
CoQ10 and creatinine were generally safe and well toler‑ unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971‑983.
ated, neither slowed functional decline in HD and the trials 3. Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89(5):910‑926.
4. Testa CM, Jankovic J. Huntington disease: a quarter century of progress since the gene discovery. J Neurol Sci. 2019;396:52‑68.
were terminated based on interim analyses.24‑27 The effects of 5. Mestre TA. Recent advances in the therapeutic development for Huntington disease. Parkinsonism Relat Disord. 2019;59:125‑130.
ethyl‑eicosapentaenoic acid (ethyl‑EPA) in HD have also been 6. Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with Huntington’s disease. N
Engl J Med. 2019;380(24):2307‑2316.
tested but showed no beneficial effects in measures of motor 7. McBride JL, Boudreau RL, Harper SQ, et al. Artificial miRNAs mitigate shRNA‑mediated toxicity in the brain: implications for
function, global functioning cognition, or global impression.28 the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105(15):5868‑5873.
8. DiFiglia M, Sena‑Esteves M, Chase K, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and
Latrepirdine (dimebon) was also tested as a modulator of cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104(43):17204‑17209.
mitochondrial dysfunction in HD. Treatment with latrepirdine 9. Stanek LM, Sardi SP, Mastis B, et al. Silencing mutant huntingtin by adeno‑associated virus‑mediated RNA interference amelio‑
rates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther. 2014;25(5):461‑474.
vs placebo correlated with a discrete but significant improve‑ 10. Pfister EL, DiNardo N, Mondo E, et al. Artificial miRNAs reduce human mutant huntingtin throughout the striatum in a
ment in the Mini‑Mental State Examination (MMSE), but not transgenic sheep model of Huntington’s disease. Hum Gene Ther. 2018;29(6):663‑673.
11. Zhou P, Carroll J, Chen F, et al. Robust huntingtin knockdown in cortex and putamen in large mammals using a novel
other cognitive outcome measures in a phase 2 trial.29 In a dosing paradigm with VY‑HTT01, an AAV gene therapy targeting huntingtin for the treatment of Huntington’s disease.
subsequent multicenter 26‑week randomized double‑blind Hum Gene Ther. 2018;27:A83.
12. Shin JW, Kim KH, Chao MJ, et al. Permanent inactivation of Huntington’s disease mutation by personalized allele‑specific
placebo‑controlled study, no improvement in cognition or CRISPR/Cas9. Hum Mol Genet. 2016;25(20):4566‑4576.
global function compared to baseline was seen in participants 13. Yang S, Chang R, Yang H, et al. CRISPR/Cas9‑mediated gene editing ameliorates neurotoxicity in mouse model of
Huntington’s disease. J Clin Invest. 2017;127(7):2719‑2724.
with HD who had cognitive impairments,30 and an open‑label 14. Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease [published
extension was terminated. correction appears in Neuron. 2019 May 22;102(4):899]. Neuron. 2019;101(5)801‑819.
15. Huntington Study Group Reach HDI. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2,
randomised, double‑blind, placebo‑controlled trial. Lancet Neurol. 2015;14(1):39‑47.
Conclusions 16. Cherny RA, Ayton S, Finkelstein DI, Bush AI, McColl G, Massa SM. PBT2 reduces toxicity in a C. elegans model of polyQ
aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of
Based on promising data from preclinical studies, multiple Huntington’s disease. J Huntingtons Dis. 2012;1(2):211‑219.
clinical trials have been conducted. targeting different aspects 17. Smith MR, Syed A, Lukacsovich T, et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and
of HD pathology, including mHTT aggregation, neurodegen‑ (Continued on page 76)
JUNE 2020 PRACTICAL NEUROLOGY 73S P E C I A L R E P OR T
(Continued from page 73)
cell models of Huntington’s disease. Hum Mol Genet. 2014;23(11):2995‑3007.
18. Sussmuth SD, Haider S, Landwehrmeyer GB, et al. An exploratory double‑blind, randomized clinical trial with selisistat, a
SirT1 inhibitor, in patients with Huntington’s disease. Br J Clin Pharmacol. 2015;79(3):465‑476.
19. Reilmann R, Squitieri F, Priller J, et al. N02 Safety and tolerability of selisistat for the treatment of Huntington’s disease:
results from a randomised, double‑blind, placebo‑controlled phase II trial.RJ Neurol Neurosurg Psychiatry. 2014;85:A102.
20. Garcia‑Miralles M, Hong X, Tan LJ, et al. Laquinimod rescues striatal, cortical and white matter pathology and results in
modest behavioural improvements in the YAC128 model of Huntington disease. Sci Rep. 2016;6:31652.
21. Li C, Yuan K, Schluesener H. Impact of minocycline on neurodegenerative diseases in rodents: a meta‑analysis. Rev
Neurosci. 2013;24(5):553‑562.
22. Southwell AL, Franciosi S, Villanueva EB, et al. Anti‑semaphorin 4D immunotherapy ameliorates neuropathology and
some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2015;76:46‑56.
23. Huntington Study Group DOMINO Investigators. A futility study of minocycline in Huntington’s disease. Mov Disord.
2010;25(13):2219‑2224.
24. McGarry A, McDermott M, Kieburtz K, et al. A randomized, double‑blind, placebo‑controlled trial of coenzyme Q10 in
Huntington disease. Neurology. 2017;88(2):152‑159.
25. Ross C, Biglan K, Killoran A, et al. PREQUEL—A multicenter phase II study of coenzyme Q10 in premanifest Huntington
disease. Neurotherapeutics. 2014;11:214.
26. Verbessem P, Lemiere J, Eijnde BO, et al. Creatine supplementation in Huntington’s disease: a placebo‑controlled pilot trial.
Neurology. 2003;61(7):925‑930.
27. Hersch SM, Schifitto G, Oakes D, et al. The CREST‑E study of creatine for Huntington disease: a randomized controlled trial.
Neurology. 2017;89(6):594‑601.
28. Huntington Study Group. Randomized controlled trial of ethyl‑eicosapentaenoic acid in Huntington disease: the TREND‑HD
study. Arch Neurol. 2008;65(12):1582‑1589.
29. Kieburtz K, McDermott MP, Voss TS, et al. A randomized, placebo‑controlled trial of latrepirdine in Huntington disease.
Arch Neurol. 2010;67(2):154‑160.
30. Horizon Investigators of the Huntington Study Group, European Huntington’s Disease Network. A randomized, double‑blind,
placebo‑controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013;70(1):25‑33.
Natalia P. Rocha, PharmD, MsC, PhD
The Mitchell Center for Alzheimer’s disease and Related
Brain Disorders
Department of Neurology, McGovern Medical School
HDSA Center of Excellence
The University of Texas Health Science Center
Houston, TX
Gabriela D. Colpo, PhD
Neuropsychiatry Program
Department of Psychiatry and Behavioral Sciences
McGovern Medical School
The University of Texas Health Science Center
Houston, TX
Antonio L. Teixeira, MD, PhD, MSc
Neuropsychiatry Program
Department of Psychiatry and Behavioral Sciences
McGovern Medical School, HDSA Center of Excellence
The University of Texas Health Science Center
Houston, TX
Erin Furr Stimming, MD
Department of Neurology, McGovern Medical School
HDSA Center of Excellence
The University of Texas Health Science Center
Houston, TX
Disclosures
NPR, GDC, ALT report no disclosures
EFS has disclosures at www.practicalneurology.com
76 PRACTICAL NEUROLOGY JUNE 2020You can also read