Discovery-Versus Hypothesis-Driven Detection of Protein-Protein Interactions and Complexes - MDPI
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
International Journal of
Molecular Sciences
Review
Discovery–Versus Hypothesis–Driven Detection of
Protein–Protein Interactions and Complexes
Isabell Bludau
Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry,
82152 Martinsried, Germany; bludau@biochem.mpg.de
Abstract: Protein complexes are the main functional modules in the cell that coordinate and perform
the vast majority of molecular functions. The main approaches to identify and quantify the interac-
tome to date are based on mass spectrometry (MS). Here I summarize the benefits and limitations of
different MS-based interactome screens, with a focus on untargeted interactome acquisition, such as
co-fractionation MS. Specific emphasis is given to the discussion of discovery- versus hypothesis-
driven data analysis concepts and their applicability to large, proteome-wide interactome screens.
Hypothesis-driven analysis approaches, i.e., complex- or network-centric, are highlighted as promis-
ing strategies for comparative studies. While these approaches require prior information from public
databases, also reviewed herein, the available wealth of interactomic data continuously increases,
thereby providing more exhaustive information for future studies. Finally, guidance on the selec-
tion of interactome acquisition and analysis methods is provided to aid the reader in the design of
protein-protein interaction studies.
Keywords: protein complexes; protein-protein interactions; interactomics; mass-spectrometry; tar-
geted proteomics; data analysis; databases; systems biology
Citation: Bludau, I.
Discovery–Versus Hypothesis–Driven
Detection of Protein–Protein
1. Introduction
Interactions and Complexes. Int. J.
Mol. Sci. 2021, 22, 4450. https:// Proteins are the main functional molecules within the cell. However, most proteins
doi.org/10.3390/ijms22094450 do not act in solitude. Instead, they often interact with other proteins to form large
macromolecular assemblies, which coordinate and perform the majority of molecular
Academic Editor: Istvan Simon functions inside the cell. Many protein complexes are evolutionarily conserved [1] and their
dysregulation is associated with the development and progression of various diseases [2].
Received: 19 March 2021 Identifying and quantifying protein-protein interaction (PPI) networks and stable protein
Accepted: 21 April 2021 complexes is thus a major goal in basic and translational research.
Published: 24 April 2021 Importantly, this review mainly focuses on the physical interactome. Thus, a PPI
generally refers to a physical interaction between two proteins, unless otherwise stated. A
Publisher’s Note: MDPI stays neutral PPI network consequently represents a set of proteins that are connected to each other via
with regard to jurisdictional claims in PPIs. A protein complex, in turn, refers to a stable group of physically interacting proteins.
published maps and institutional affil-
However, the physicochemical boundaries for differentiating a stable protein complex from
iations.
more dynamic functional modules in the cell still remain to be defined. In context of this
review, clusters of co-regulated PPIs are termed protein complexes.
Over the last decades, mass spectrometry (MS)-based proteomics has emerged as
the main approach for studying both the proteome [3,4] and its interactome [5]. Here, I
Copyright: © 2021 by the author. summarize the main MS-based approaches for acquiring data that provides information
Licensee MDPI, Basel, Switzerland. about PPIs and protein complexes, with an emphasis on the scope of protein complexes
This article is an open access article covered by each technique. I subsequently focus on computational concepts for analyzing
distributed under the terms and
global interactome studies, ranging from discovery- to hypothesis-driven concepts. Finally,
conditions of the Creative Commons
I review state-of-the-art databases that can be leveraged when performing complex- or
Attribution (CC BY) license (https://
network-centric analyses and provide guidance on the selection of appropriate interactome
creativecommons.org/licenses/by/
acquisition and analysis approaches depending on the research question.
4.0/).
Int. J. Mol. Sci. 2021, 22, 4450. https://doi.org/10.3390/ijms22094450 https://www.mdpi.com/journal/ijms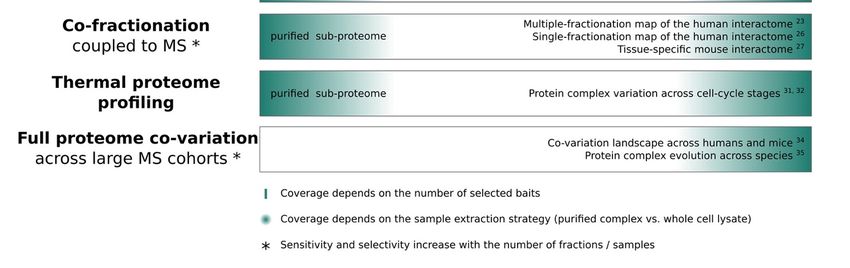
Int. J. Mol. Sci. 2021, 22, 4450 2 of 14
2. Targeted and Untargeted Interactome Screens
MS-based workflows for acquiring interactome data can broadly be categorized into
two groups: (1) targeted approaches, which focus on a single protein and its interactions at
a time, and (2) untargeted interactome screens, which aim to investigate the entire com-
pendium of protein complexes in a given sample. Below I summarize different strategies of
each category, focusing on the benefits and limitations of each approach. Figure 1 further
provides an overview of the discussed methods, setting each approach in context to its
theoretically achievable scope.
The most common MS based technology to study PPIs is affinity purification coupled
to MS (AP-MS) [6,7]. Here, a single protein of interest (bait) is purified from a cell lysate
by using an antibody that is either specific for the bait or, more commonly, specific for
an affinity tag that is fused to the bait. Mild, non-denaturing conditions during the pull-
down ensure that stable PPIs between the bait and its interaction partners (preys) are
preserved. Bait and prey proteins are subsequently identified and quantified by bottom-
up mass spectrometry. This generates a minimal PPI network of a single central node
(bait) with all co-purified interaction partners (preys) connected via edges. Importantly,
a single AP-MS experiment can potentially determine all stable interaction partners of
a given bait. However, it is not possible to directly determine from a single experiment
whether all these interaction partners occur in the same macromolecular complex, or
if multiple alternative modules with different subunits exist. Addressing this question
requires multiple reciprocal pull-downs. Applications of AP-MS range from targeted
analysis of a specific macromolecular machinery, to analyses of entire pathways or classes
of proteins (e.g., kinases [8]), and to even larger scale studies, such as the effort to map PPIs
across all human ORFs [6,9,10]. While the sensitivity and specificity of AP-MS is considered
superior to most other techniques and established protocols and data analysis workflows
are available, it has three main limitations: (1) a dependency on the availability of specific
antibodies, or a requirement for genetic engineering, (2) the necessity to carefully select
appropriate controls to ensure that high confidence prey proteins can be distinguished from
potentially high background noise [7], and (3) a very high cost for global, and especially
differential, analyses. Additionally, only stable interactions that remain intact during
sample processing are detectable by AP-MS.
To capture more transient interactions, proximity labelling approaches such as BioID [11]
and APEX [12] have recently gained increasing attention. The key concept is that a bait
protein is fused to an enzyme, for example a biotin ligase in BioID or a peroxidase in APEX.
Upon activation, the enzymes can label proximal proteins at high spatial (~10 nm) and
temporal resolution (~1 min labelling speed for APEX) [13,14]. Similar to AP-MS, proximity
labelling is a targeted interactome acquisition strategy where experiments are performed
on a per-bait basis, meaning that a large number of experiments are required to generate
a holistic view of all proximal interactions in the cell, or even across conditions. As the
name suggests, the main difference between proximity labelling and other techniques is
that the reported prey proteins do not necessarily interact physically with the bait, but they
may simply be in close proximity. A recent study applied BioID to generate a proximity
map of the human HEK293 cell line, including 35,902 unique high confidence proximity
interactions among 4145 spatially localized proteins [15]. Similar to AP-MS, appropriate
controls are critical to ensure that high confidence prey proteins can be distinguished from
potentially high background labelling.
As discussed above, AP-MS and proximity labelling are inherently targeted ap-
proaches, which can only create whole interactome information through combination
of a large number of experiments. In contrast, there are a number of methods that are
directly geared towards more holistic, untargeted measurements of the interactome, in-
cluding cross-linking or co-fractionation coupled to MS, thermal proteome profiling and
full proteome co-variation analysis. For these techniques, the coverage (number of protein
complexes identified) depends solely on the optional application of prior purification steps
and technical limitations of the MS instrument and acquisition itself.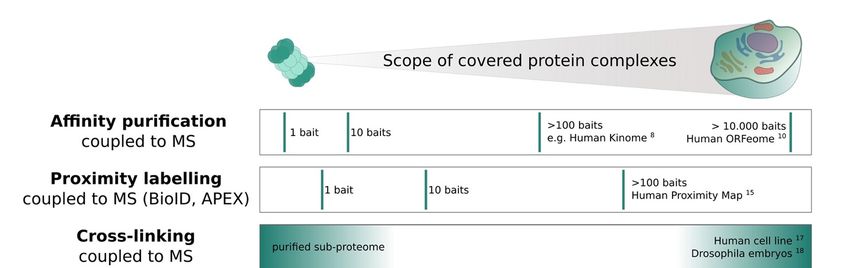
cross-linking or co-fractionation coupled to MS, thermal proteome profiling and full pro-
teome co-variation analysis. For these techniques, the coverage (number of protein com-
Int. J. Mol. Sci. 2021, 22, 4450 plexes identified) depends solely on the optional application of prior purification3 of
steps
14
and technical limitations of the MS instrument and acquisition itself.
Figure 1. The scope of different mass spectrometry (MS)-based interactome screens. State of art MS-based approaches for
Figure 1. The scope of different mass spectrometry (MS)-based interactome screens. State of art MS-based approaches for
investigating the interactome are summarized. Affinity purification and proximity labelling coupled to MS are targeted
investigating the interactome are summarized. Affinity purification and proximity labelling coupled to MS are targeted
strategies for which a high coverage of protein complexes can only be achieved by performing hundreds to ten-thousands
strategies for which a high coverage of protein complexes can only be achieved by performing hundreds to ten-thousands
ofofsingle-bait
single-baitexperiments.
experiments.The Thetheoretically
theoreticallyachievable
achievablecoverage
coverageofofcross-linking,
cross-linking,co-fractionation
co-fractionationandandthermal
thermalproteome
proteome
profiling
profiling strategies in turn depend on the purification level of the sample, e.g., purified sub-proteome (singlecomplex
strategies in turn depend on the purification level of the sample, e.g., purified sub-proteome (single complexoror
organelle)
organelle)vs.vs.
whole
whole cellcell
extract. Finally,
extract. full full
Finally, proteome co-variation
proteome studies
co-variation acrossacross
studies large large
MS cohorts are usually
MS cohorts not acquired
are usually not ac-
quired specifically
specifically with thewith the purpose
purpose of investigating
of investigating proteinprotein interactions,
interactions, butcan
but they theybecan be leveraged
leveraged to global
to find find global patterns
patterns of
of co-variation
co-variation e.g., e.g., to investigate
to investigate evolutionary
evolutionary conservation
conservation of complexes.
of complexes. Exemplary
Exemplary studies
studies fordifferent
for the the different approaches
approaches are
are indicated
indicated at respective
at their their respective proteome
proteome coverage.
coverage.
InIncross-linking
cross-linking coupled
coupled to to MS
MS(XL-MS),
(XL-MS),a across-linking
cross-linking reagent
reagent is added
is added to the
to sam-
the
ple, which
sample, whichcovalently
covalentlylinkslinks
amino acid acid
amino residues (commonly
residues (commonlylysines) that are
lysines) thatclose
are to each
close
toother.
each Traditionally, XL-MS has
other. Traditionally, XL-MSmainly
hasbeen applied
mainly beentoapplied
purifiedtoproteins
purified and protein and
proteins com-
plexes,complexes,
protein with the goal with to the
gaingoal
structural
to gaininformation [16]. However,
structural information [16].recent
However, studies have
recent
demonstrated
studies that XL-MS can
have demonstrated thatalso be applied
XL-MS can also onbe
a more
applied global
on ascale,
morefor example
global scale,inves-
for
example
tigating investigating interactions
interactions between between in
all proteins allthe
proteins
human in HeLa
the human
cell lineHeLa [17]cell
orline
even[17]
the
orentire
evenDrosophila Drosophila melanogaster
the entiremelanogaster embryo [18]. Currently, the main
embryo [18]. Currently, the main limitation of system-wide limitation
ofXL-MS
system-wide
studies isXL-MS studies
the modest is the
depth of modest
interactomedepth of interactome
coverage, reachingcoverage,
only a maximumreachingof
only a maximum
~10,000 of ~10,000
cross-links among across-links
few hundred among a few which
proteins, hundred addsproteins, whichPPIs
up to ~1000 adds upAd-
[19]. to
~1000 PPIs [19].
ditionally, the Additionally, the reported
reported interactions are interactions
strongly biasedare strongly
towardsbiased
highlytowards
abundant highly
pro-
abundant
teins. Theseproteins.
effectsThese
occureffects
due tooccur due limitations
technical to technicalin limitations in MS and
MS acquisition acquisition and
the chemical
the chemical properties of currently available
properties of currently available cross-linkers [19,20]. cross-linkers [19,20].
AnAnalternative
alternativeapproach
approachtotogain gainholistic
holisticinteractome
interactomeinformation
informationininananuntargeted
untargeted
fashion
fashion is based on complex co-fractionation coupled to MS(CoFrac-MS).
is based on complex co-fractionation coupled to MS (CoFrac-MS).Originally
Originallyde- de-
veloped
velopedand andapplied
appliedwith
withthe thegoal
goaltotomap
mapproteins
proteinstotodifferent
differentcellular
cellularorganelles
organelles[21,22],
[21,22],
CoFrac-MS
CoFrac-MSisisnow nowan anincreasingly
increasinglypopular
popularstrategy
strategytotoprobe
probePPIs
PPIsandandprotein
proteincomplexes
complexes
on a proteome-wide scale, by utilizing high-resolution fractionation strategies [23–25].
Here, protein complexes are extracted under mild conditions to keep them intact, followed
by separation and fractionation according to some physicochemical property. Exemplary
co-fractionation techniques include size-exclusion chromatography (SEC), which sepa-
rates complexes by hydrodynamic radius, and ion-exchange chromatography (IEX), whichInt. J. Mol. Sci. 2021, 22, 4450 4 of 14
separates complexes by charge. While CoFrac-MS can be applied to purified samples
of a sub-proteome (for example, a specific organelle), it is more frequently applied to
whole cell lysates. In this case, interactome coverage depends on the resolution of the
chromatographic separation, as well as the proteome coverage achieved by the selected
MS acquisition of each sampled fraction. Finally, the sensitivity and selectivity of protein
complex information obtained by CoFrac-MS are determined by the applied computa-
tional inference strategy, which is used to differentiate random co-elution from signals
indicating true physical interactions (see next section). In recent years, CoFrac-MS has
been widely applied in different contexts, including human interactome maps generated
from multi-dimensional fractionation [23] or from a single high-resolution fractionation
experiment [26]. A recent study impressively demonstrated interactome mapping across
different mouse tissues [27]. In comparison to XL-MS, CoFrac-MS approaches that utilize
high-resolution fractionation strategies, such as SEC, can obtain a much deeper proteome
and interactome coverage, reaching > 1000 complexes composed of > 10,000 PPIs from
a single two-condition experiment [28]. While coverage is still technically limited by the
proteomic depth and dynamic range that can be captured by the selected MS technology,
CoFrac-MS could, in principle, be used to simultaneously assess interactions occurring
across the entire proteome.
Similar to CoFrac-MS, thermal proteome profiling (TPP) also offers a global strategy
to assess PPIs and protein complexes. TPP is a method to systematically measure protein
thermal stability and abundance [29]. Here, a proteomic sample is heated to increasing tem-
peratures, thereby inducing protein denaturation. At each temperature, soluble proteins
are quantified by high-resolution MS, a process which yields denaturation curves for all
detectable proteins. TPP studies have shown that proteins that are part of the same protein
complex show similar thermostability profiles [30–32]. While CoFrac-MS is expected to
provide more depth and sensitivity in detecting changes in the composition of protein com-
plexes and their abundance across conditions, TPP has the potential to identify more subtle
changes in protein stability [33]. These could for example arise from allosteric regulation
by small molecule binding to a protein complex, thus influencing its thermostability.
A further, more indirect strategy for investigating protein complexes in an untargeted
and holistic fashion is to perform full proteome co-variation analysis. Here, data is typi-
cally not specifically acquired for protein complex analysis. The basis for full proteome
co-variation studies are large MS datasets, which can be computationally leveraged to in-
vestigate co-variation of proteins. This strategy has been applied to identify evolutionarily
conserved or highly variable protein complexes across species and individuals [34,35]. The
sensitivity of such analyses largely depends on the number of samples at hand. Further-
more, it is important to keep in mind that co-variation (i.e., co-expression) is not necessarily
equivalent to co-complex membership, but could also refer to functional, indirect interac-
tions (for example, a transcription factor regulating the abundance of another protein). A
recent study highlighted the potentially misleading observations from co-expression stud-
ies [27]. Nevertheless, full proteome co-variation analyses still provide an initial estimate
of protein complex information, especially when combined with prior information and
when the investigation of global patterns is the main interest.
In summary, MS-based workflows for interactome profiling can be categorised into
targeted (AP-MS and proximity labelling) and untargeted (XL-MS, CoFrac-MS, TPP and
full proteome co-variation MS) interactome acquisition strategies. The targeted approaches
require a large number of reciprocal experiments to obtain a holistic view of the interactome
of a single sample. In contrast, XL-MS, CoFrac-MS and TPP, in principle, enable the
inference of a PPI network from a single sample in one experiment (typically including
multiple MS runs). Full proteome co-variation analysis, by definition, cannot be performed
on a single sample, but requires large MS cohorts of hundreds of individual samples. Here,
multiplexed data acquisition strategies such as stable isotope labeling with amino acids in
cell culture (SILAC) provide the only possibility to reduce the number of required MS runs.
Differently to targeted interactome acquisition strategies, the proteome and interactomeInt. J. Mol. Sci. 2021, 22, 4450 5 of 14
coverage of untargeted interactome screens is solely determined by current technical state
of the art, but not by the acquisition concept itself.
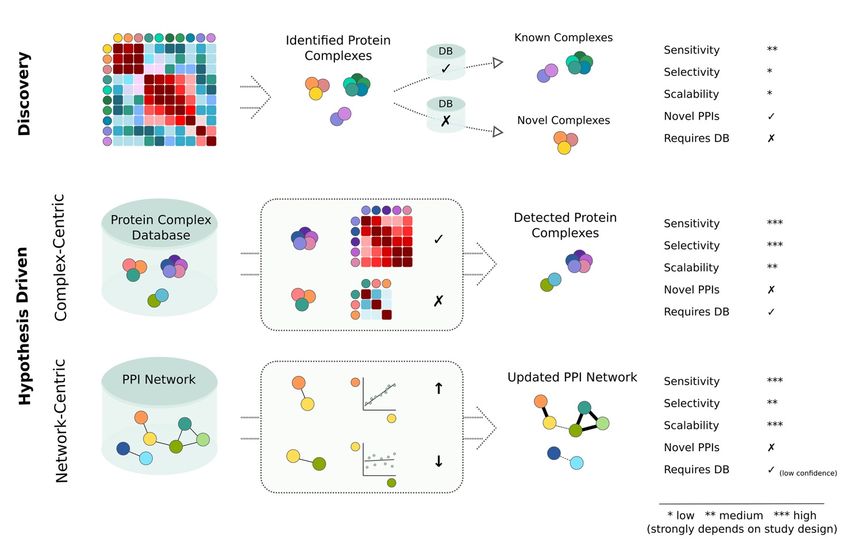
3. Discovery and Hypothesis Driven Data Analysis Strategies
A major challenge in untargeted interactome screens, especially in CoFrac-MS, TPP
and full proteome co-variation analysis, is to differentiate between signals originating from
protein complexes and signals derived purely from random co-elution/co-variation. In this
section I describe the two main approaches for analyzing data from untargeted interactome
screens: (1) discovery- and (2) hypothesis-driven data analysis. Figure 2 illustrates their
conceptual differences and summarizes their benefits and limitations.
The most widely applied data analysis strategy for untargeted interactome screens is
the discovery approach. Similar to discovery proteomics, where the goal is the identifica-
tion of a protein, discovery analyses in interactomics are focused on the identification of
novel protein complexes in a given dataset. The analysis concept is based on calculating
pairwise scores (for example, based on correlation) between all detected proteins, followed
by a classification task that determines which protein-pairs likely represent a true interac-
tion. The classification is commonly achieved by machine learning (ML) algorithms that
are trained based on a positive and negative reference set of interactions [28,36,37]. Proteins
in the resulting interaction network can be grouped into defined protein complexes by
applying a graph partitioning algorithm. The inferred protein complexes can then be
classified as either known or novel by cross-referencing to available complex databases.
Previous studies have successfully implemented different versions of the discovery concept
for analyzing interactome data, mainly derived from CoFrac-MS studies [23–25,38–41]. The
key benefit of the discovery approach is that only limited prior knowledge is necessary
(only enough to train the ML algorithm) and that novel protein complexes can be readily
identified. In practice, high proteomic coverage coupled to limited biochemical resolution
(determined by the resolution of fractionation strategies in CoFrac-MS, sampled tempera-
tures in TPP and the number of individual samples in full proteome co-variation analysis)
results in a high degree of random correlation of non-interacting proteins [42]. This has
negative effects on the sensitivity and selectivity of purely discovery-based approaches.
This challenge can be addressed by integrating information from multiple orthogonal
fractionations [23] or datasets [43], but this comes at the cost of large experimental efforts.
Targeted (i.e., hypothesis-driven) data analysis strategies provide an alternative ap-
proach to address the challenges associated with untargeted interactome screens. Prior
knowledge about known PPIs or protein complexes is leveraged to increase the sensitivity,
selectivity, and consistency of quantitative information. In complex-centric data analysis,
a protein complex database is used as prior information to directly test for evidence of
specific protein complexes in the dataset at hand. Here, only a small fraction of the pairwise
comparisons are necessary, thereby markedly reducing the search space and the risk of
false positives. Complex-centric analysis of interactome data can be regarded as analo-
gous to peptide-centric data analysis of proteomics data generated by data-independent
acquisition (DIA) [44,45]. In peptide-centric DIA analysis, a spectral library is utilized to
perform a targeted extraction of fragment-ion chromatograms from highly convoluted MS2
spectra. This improves the sensitivity, selectivity and consistency of peptide detections.
In complex-centric interactome analysis, the protein complex database serves an equiva-
lent purpose as the spectral library for DIA. Despite this general analogy, it is important
to keep in mind that peptide to protein associations are much stronger than protein to
complex associations. While spectral libraries in DIA commonly contain only high con-
fidence identifications and are specific to a given experiment, PPI and protein complex
databases are usually generic and information of various confidence could be included.
The higher the confidence of protein complexes in the reference database and the more
representative it is for a given experiment, the closer complex-centric analysis resembles
the characteristics of the peptide-centric workflow. As stated above, previous studies have
shown significant benefits in the sensitivity and selectivity of complex-centric approachesportant to keep in mind that peptide to protein associations are much stronger than pro-
tein to complex associations. While spectral libraries in DIA commonly contain only high
confidence identifications and are specific to a given experiment, PPI and protein complex
databases are usually generic and information of various confidence could be included.
The higher the confidence of protein complexes in the reference database and the more
Int. J. Mol. Sci. 2021, 22, 4450 representative it is for a given experiment, the closer complex-centric analysis resembles 6 of 14
the characteristics of the peptide-centric workflow. As stated above, previous studies have
shown significant benefits in the sensitivity and selectivity of complex-centric approaches
compared to discovery analyses [26]. However, this comes at the cost of not being able to
compared to discovery
identify completely analyses
novel protein[26]. However,
complexes and this
beingcomes at theoncost
dependent of not being
trustworthy priorable to
identify
knowledge, which might not always be available. Complex-centric analysis will therefore prior
completely novel protein complexes and being dependent on trustworthy
knowledge, which
substantially might
benefit fromnot alwayswith
databases be available.
increasing Complex-centric analysis will therefore
confidence and modularity.
substantially benefit from databases with increasing confidence and modularity.
2. Discovery-
FigureFigure vs. hypothesis-driven
2. Discovery- vs. hypothesis-driven analysis
analysisconcepts
conceptsfor
forinteractome analysis.The
interactome analysis. Thetwo
two main
main strategies
strategies for for analyzing
analyz-
ing untargeted interactome screens are discovery and hypothesis driven approaches. To focus on
untargeted interactome screens are discovery and hypothesis driven approaches. To focus on the conceptual differences,the conceptual differ-
ences, all approaches
all approaches are illustrated
are illustrated by a correlation-based
by a correlation-based analysis.
analysis. The colored
The colored circles
circles indicate
indicate differentproteins.
different proteins. Corre-
Correlation
lation matrices are illustrated by square grids with high correlations indicated in shades of red and low correlations indi-
matrices are illustrated by square grids with high correlations indicated in shades of red and low correlations indicated in
cated in shades of blue. In the context of this figure the term scalability refers to the ability of the method to provide
shadesconsistent
of blue. Inandthe context ofresults
comparable this figure the term
at modular scalability
resolution refers
across to thedatasets
multiple ability and
of the method to
conditions. Noteprovide consistent
that this figure and
comparable results ata schematic
only represents modular resolution across
illustration of multiple
the different datasets
analysis and conditions.
concepts which can be Note that this in
implemented figure only
various represents a
different
flavors
schematic and whichof
illustration will
thealso influence
different the compared
analysis concepts performance
which canmetrics. The check signs
be implemented indicate
in various ‘yes’ and
different cross signs
flavors and which
indicate ‘no’. The arrows indicate an increase (up) and decrease (down) in correlation.
will also influence the compared performance metrics. The check signs indicate ‘yes’ and cross signs indicate ‘no’. The
arrows indicate an increase (up) andWe decrease
recently(down)
applied in correlation.
complex-centric analysis to map the interactome of the human
HEK293 cell line in a single CoFrac-MS experiment [26,42]. We used protein complexes
We recently applied complex-centric analysis to map the interactome of the hu-
annotated in CORUM [46], or derived via graph partitioning from BioPlex [9] and
man HEK293
STRING [47],cell
andline in a single
searched CoFrac-MS
for evidence of theirexperiment
presence in [26,42]. We used
the CoFrac-MS protein
data. False com-
plexes
discovery rates were determined based on a set of randomly generated complex decoys.[9] and
annotated in CORUM [46], or derived via graph partitioning from BioPlex
STRING [47], and analysis
Complex-centric searched hasfor evidence
also of their
been applied presence
to analyze in generated
data the CoFrac-MS
by TPPdata.
[31]. False
discovery rates were determined based on a set of randomly generated complex decoys.
Complex-centric analysis has also been applied to analyze data generated by TPP [31].
Here, a set of 279 manually curated, largely non-overlapping protein complexes [48] was
taken as prior information for a targeted analysis of subunit correlation in comparison to
a random set of complex decoys. The same hypothesis set was applied in a study by Ro-
manov et al., who analyzed protein complex co-variation across 11 full proteome datasets
from humans and mice [34]. Stalder et al. further applied complex-centric analysis based
on ortholog mapping to provide evidence that complex covariance profiles are conserved
across species [35].
An alternative hypothesis-driven approach for untargeted interactome screens follows
a network-centric rationale. Here, an entire PPI network is leveraged as prior information.
Instead of looking at groups of proteins together (as is the case in complex-centric analy-
sis), the network-centric approach evaluates individual edges in a network to test if the
edge is supported by the given data, for example, by a positive correlation of intensitiesInt. J. Mol. Sci. 2021, 22, 4450 7 of 14
across fractions. The reference PPI network is thereby updated during the process, finally
representing only condition-specific interactions. The main benefit of the network-centric
analysis concept compared to discovery approaches is that its search space is significantly
reduced, thus boosting sensitivity and selectivity. However, in contrast to complex-centric
analysis, no protein complex inference (i.e., grouping of proteins to defined protein com-
plexes) is conducted, which improves robustness against technical effects and allows
prior knowledge to be less strictly defined, also including low confidence interactions in
a PPI network. These properties make the network-centric approach particularly scal-
able across multiple conditions and larger datasets, where protein complex subunits may
not be equally measurable across all conditions. We recently developed and applied a
network-centric strategy for analyzing CoFrac-MS data across different cell cycle stages
in the HeLa cell line [49]. Although prior knowledge is still required for the successful
application of network-centric analysis, the approach also works well for predicted PPI
networks such as PrePPI [50]. This indicates that network-centric analyses can be utilized
to confirm predicted interactions using actual data. The network-centric concept is still
new in proteomics-based interactomics, but these characteristics promise a high potential
for it to gain increasing momentum in the future.
In summary, analysis strategies for untargeted interactome screens can be divided into
discovery- and hypothesis-driven approaches. The main benefit of discovery approaches
is that they enable the identification of novel complexes without requiring prior knowl-
edge. However, they suffer from reduced sensitivity and selectivity in single experiments.
Hypothesis-driven strategies overcome this challenge by using prior knowledge to reduce
the search space and allow consistent detection and quantification of interactions and pro-
tein complexes across conditions. The main limitation of hypothesis-driven strategies is the
necessity to have access to prior knowledge of sufficient depth and quality. Complex-centric
analysis might require the use of graph partitioning algorithms, such as ClusterOne [51],
to derive defined protein complex modules from a PPI network such as STRING [52].
Network-centric approaches can directly utilize such PPI networks and are thereby less
dependent on variable parameter selection. Although both complex- and network-centric
approaches benefit from high-confidence priors, strong protein co-variation patterns, espe-
cially in CoFrac-MS, can serve as important evidence to also verify low-confidence priors,
for example derived from prediction tools. In this context it is important to note that
experimental data acquisition and data analysis are not fully independent. The lower the
resolution of the experimental data, the more confident the prior information has to be for
deriving meaningful results. However, it is critical to keep in mind that poorly designed
experiments at low resolution cannot purely be compensated by elaborate targeted analysis
strategies. Importantly, discovery and targeted approaches are not mutually exclusive.
Similar to recent attempts [28], large datasets can first be analyzed by a discovery approach
to determine a set of tentative protein complexes, followed by targeted re-extraction and
quantification of protein complexes. This is analogous to current hybrid approaches for DIA
analysis, exemplified by directDIA in Spectronaut [53] or workflows that couple library
generation by DIA-Umpire [54] with targeted extraction by OpenSWATH [55]. Such hybrid
approaches have the potential to combine the benefits of both discovery and targeted
analysis concepts and provide a promising future direction.
4. Protein Complex and PPI Databases
As discussed above, hypothesis-driven analysis approaches offer many benefits. How-
ever, their successful application depends on the availability of prior knowledge based on
PPI or protein complex databases. Here, I review the most commonly applied databases,
including the type of information they contain (for example, curated or predicted), the
organisms they cover and their comprehensiveness. Please note that I cover only a se-
lection of the most comprehensive, widely used and recently updated PPI databases (for
an inclusive review see [56]). Table 1 summarizes information about the selected protein
complex and PPI databases.Int. J. Mol. Sci. 2021, 22, 4450 8 of 14
Table 1. Summary of protein complex and PPI databases.
DB Information Interaction type Organisms Size Website
Human (67%)
4274 complexes based on 4473 genes http://mips.helmholtz-
Manually curated from Direct (physical) Mouse (15%)
CORUM 3.0 (including 22% of human protein muenchen.de/corum/
experimental data interactions Rat (10%)
coding genes) (accessed on 9 March 2021)
& other mammals
Complexes
• Human: 985 complexes
• Mouse: 740 complexes http://www.ebi.ac.uk/
Complex Portal Manually curated from Direct (physical) 26 organisms across all • Yeast: 607 complexes complexportal (accessed on 9
(accessed 9 March 2021) experimental data interactions domains of life • E. coli: 321 complexes March 2021)
• Others
Integration of over Direct (physical)
6969 complexes consisting of 57,178 http://humap2
15,000 mass interactions
huMap 2.0 Human unique interactions among .proteincomplexes.org/
spectrometry (and proximity
9,968 proteins (accessed on 9 March 2021)
experiments interactions)
Human (61%)
Yeast (12%)
IntAct Manually curated from Direct (physical) 1,130,596 interactions among http://www.ebi.ac.uk/intact
Mouse (8%)
(accessed 11 March 2021) experimental data interactions 119,281 proteins (accessed on 9 March 2021)
& other organisms across all
domains of life
PPIs
• Budding yeast: 755,000 PPIs
• Fission yeast: 79,000 PPIs
Direct (physical) • Human: 670,000 PPIs
Manually curated from https://thebiogrid.org/
BioGRID 4.3 interactions and genetic 70 species • Worm: 29,000 PPIs
experimental data (accessed on 9 March 2021)
interactions • Fly: 78,000 PPIs
• All other organisms:
300,000 PPIsInt. J. Mol. Sci. 2021, 22, 4450 9 of 14
Table 1. Cont.
DB Information Interaction type Organisms Size Website
https:
Direct (physical) 118,162 interactions among 14,586
BioPlex 3.0 Experimental Human //bioplex.hms.harvard.edu/
interactions proteins
(accessed on 9 March 2021)
Direct (physical) and http://bhapp.c2b2.columbia.
PrePPI contains 1.35 million PPIs for
PrePPI Predicted indirect (functional) Human edu/PrePPI (accessed on 9
~85% of the human proteome
interactions March 2021)
Direct (physical) and
Experimental & >2000 million unique interactions https://string-db.org/
STRING v11 indirect (functional) 5090 different organisms
predicted among 24.6 million proteins (accessed on 9 March 2021)
interactionsInt. J. Mol. Sci. 2021, 22, 4450 10 of 14
Protein complex databases generally include modules of physically interacting pro-
teins that have been experimentally observed. The most commonly used, gold-standard
database for protein complex information is the CORUM database, which contains 4274
manually-curated protein complexes across different mammals, mainly human (67%),
mouse (15%) and rat (10%) [57]. An alternative protein complex database is the Complex
Portal [58]. This covers fewer mammalian complexes compared to CORUM, but contains
complex information for other organisms, for example, A. thaliana, S. cerevisiae, E. coli,
C. elegans and SARS-CoV-2. HuMap is a third protein complex database, containing only
human complexes. In contrast to CORUM and Complex Portal, HuMap is not manually
curated, but contains protein complex information derived from the integrative analysis
of over 15,000 mass spectrometry experiments covering AP-MS, proximity labelling and
CoFrac-MS measurements [59]. HuMap covers 6969 protein complexes consisting of 57,178
unique interactions among 9968 human proteins.
Among available PPI databases solely based on experimental data, IntAct [60] and
BioGrid [61] offer very comprehensive networks. IntAct contains 1,130,596 interactions
among 119,281 proteins, mainly from human (61%), yeast (12%) and mouse (8%), but
also covering other organisms across all domains of life [60]. IntAct provides all PPI
contributions to the Complex Portal database discussed above [58]. BioGrid is another
manually-curated PPI database, containing information about physical, as well as genetic,
interactions between proteins [61]. BioGrid covers more than 70 species, with a total of
907,858 physical and 694,730 genetic, non-redundant interactions. In contrast to these
organism-wide and multi-experiment databases, BioPlex is a resource of PPIs generated
from a single experimental AP-MS study of human cell lines [10]. The most recent release,
3.0, contains 118,162 interactions among 14,586 proteins. At the other end of the spec-
trum, PrePPI is a large prediction-based PPI database, containing 1.35 million predicted
interactions for ~85% of the human proteome [50]. Finally, STRING is the largest and
most common PPI database, and integrates experimental and predicted PPI information
for 5,090 different organisms across all domains of life, covering a total of 24.6 million
proteins [52]. A key asset of STRING is its intuitive usability and the possibility to filter
PPIs based on various confidence criteria, which will determine the overall confidence of
the resulting network.
5. Considerations for the Selection of Interactome Acquisition and
Analysis Approaches
Many different interactome acquisition and analysis approaches, as well as available
prior knowledge, have been discussed, but the question remains how to decide between
these different strategies. To guide the reader in experimental design, I here present
different scenarios based on alternative research questions and provide advice for the most
appropriate interactome acquisition and analysis approaches.
Targeted interactome acquisition approaches are usually the method of choice for
studies aiming to investigate the interaction partners of a single protein, a specific pathway
or a submodule in the cell. If stable, physical interactions are of interest, AP-MS should be
used. If transient interactions or short, time-resolved processes are of interest, proximity
labelling may be preferred. Given the constraints of keeping experimental costs and time in
a feasible range, a trade-off must be made between the number of investigated bait proteins
and the number of samples (for example, conditions).
XL-MS is the leading MS-based method for structural studies in which the interest
is not only the protein complexes themselves, but also their three-dimensional structures
and binding interfaces. However, system-wide XL-MS studies are still technically limited
in their achievable proteome and interactome coverage [19]. Therefore, CoFrac-MS is
currently the method of choice for proteome-wide screening of interactome rewiring across
samples or conditions. In this context, different fractionation strategies should be selected
(reviewed in [62]) depending on the desired resolution and type of protein complexes
under investigation (for example, soluble or membrane bound). TPP is another alternative
to CoFrac-MS. It could be of specific interest if the research question is to evaluate subtleInt. J. Mol. Sci. 2021, 22, 4450 11 of 14
changes in protein complex stability, for example upon small molecule perturbation. For MS
data acquisition, DIA has proven superior to data-dependent acquisition (DDA) when label-
free quantification is used [26]. Once data is acquired, the choice of an appropriate data
analysis approach is crucial. If the focus of the research question is on the identification of
novel complexes, or if prior information is sparce, a discovery approach should be selected.
On the other hand, hypothesis-driven approaches are the preferred choice when confident
detection and quantification of defined PPIs or protein complexes within a sample or across
conditions is desired. The prerequisite of a hypothesis-driven analysis is the availability of
appropriate prior knowledge. Table 1 summarized some of the most comprehensive and
commonly used PPI and protein complex databases. To extend the scope of hypothesis-
driven approaches, these existing databases can also be supplemented by appending
manually-curated protein complex entries. Importantly, different experimental interactome
acquisition strategies can also be combined to increase confidence in discovered PPIs and
to reduce false positives. This can be achieved by integrating respective analysis results or
by directly coupling workflows, exemplified by cross-linking coupled to CoFrac-MS [63].
The above-mentioned scenarios are geared towards studies that aim to identify or
quantitatively compare interactomes across specific samples, with the capacity to acquire
exclusive data for the analysis. For more general systems biology questions, where patterns
rather than explicit examples are of interest (for example, concerning general complex
co-variation across individuals or species), full proteome co-variation analysis can provide
sufficient resolution to gain valuable insights. Importantly, systematic co-variation studies
across full-proteome datasets can enable the inference of protein complex level information
for cohorts with sample amounts that are too limited for specific interactome data acquisi-
tion, for example, in clinical studies. While discovery analysis in such datasets is hampered
by an exploding search space, complex-centric analysis approaches have been shown to
provide valuable results [34,35].
6. Summary and Future Perspectives
The identification of protein complexes and investigation of their dynamic rewiring
across biological conditions is an active area of research that is of interest in both basic
and translational science. To date, AP-MS remains the gold standard for generating high
confidence interactome maps. However, the necessity to perform a separate experiment for
every bait provides limited scalability for systems biology studies. Due to its multiplexed
experimental design, CoFrac-MS has become a very promising strategy to perform compar-
ative interactome screens. Technical improvements, especially to MS instrumentation, data
acquisition and analysis, will continue to improve the proteome coverage achievable in
single MS runs, exemplified by recent break-throughs with novel DIA technologies [64,65].
Recent developments in the LC setup for MS data acquisition, exemplified by micro-flow
applications [66] or the Evosep One system [67], will further increase the throughput of in-
teractome screens. When coupled with highly sensitive MS instruments that can operate on
small sample amounts, CoFrac-MS studies could also collect fractions at higher frequency,
thus increasing the resolution for protein complex analysis. Such technical improvements
have a direct impact on the interactome coverage and throughput that can be achieved
by CoFrac-MS and should promote its wider use in the future. The main data analysis
strategy for CoFrac-MS data follows the discovery rationale. As highlighted above, this has
the benefit of enabling the identification of novel complexes, but often comes at the cost of
compromised sensitivity and selectivity. Alternative hypothesis-driven approaches that
leverage prior information from public PPI and protein complex databases were developed
and benchmarked only recently [26,49]. While the use of prior knowledge improves the
sensitivity and selectivity of hypothesis-driven analysis, its applications are limited by
the availability of appropriate databases. Over the last years, the number and compre-
hensiveness of PPI and protein complex databases grew markedly, including information
generated by low-throughput experiments, high-throughput screens, and fully predicted
PPI networks. It is expected that available interactome information will continue to growInt. J. Mol. Sci. 2021, 22, 4450 12 of 14
and improve over time, thus providing increasingly confident and modular information
that can be leveraged for hypothesis-driven analysis. Together with continuously improv-
ing experimental data, and increasing sample sizes of proteomic and interactomic studies,
I predict that these developments will cause a paradigm shift towards hypothesis-driven
data analysis in interactome screens. It will remain interesting to observe the directions in
which the field will develop.
Funding: I.B. acknowledges funding support from her Postdoc.Mobility fellowship granted by the
Swiss National Science Foundation (P400PB_191046, starting date: 01.02.2020).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Acknowledgments: I thank my mentor Matthias Mann for his support and for providing feedback on
the review. I would also like to thank Alexandra K. Davies, Christian Doerig, George Rosenberger and
Julia Schessner for discussions and for providing critical feedback. I further thank Ruedi Aebersold
and Ben Collins for their continuous guidance and support.
Conflicts of Interest: The author declares no conflict of interest.
References
1. Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.W.; Xiong, X.; Kagan, O.; Kwan, J.; Berzginov, A.; et al. Panorama of
ancient metazoan macromolecular complexes. Nat. Cell Biol. 2015, 525, 339–344. [CrossRef] [PubMed]
2. Bergendahl, L.T.; Gerasimavicius, L.; Miles, J.; Macdonald, L.; Wells, J.N.; Welburn, J.P.I.; Marsh, J.A. The role of protein complexes
in human genetic disease. Protein Sci. 2019, 28, 1400–1411. [CrossRef] [PubMed]
3. Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nat. Cell Biol. 2003, 422, 198–207. [CrossRef] [PubMed]
4. Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nat. Cell Biol. 2016, 537, 347–355.
[CrossRef] [PubMed]
5. Bludau, I.; Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol.
Cell Biol. 2020, 21, 327–340. [CrossRef] [PubMed]
6. Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex
Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [CrossRef]
7. Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A
Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723.
[CrossRef]
8. Buljan, M.; Ciuffa, R.; van Drogen, A.; Snijder, B.; Aebersold, R.; Gstaiger, M.; Lee, S.; Varjosalo, M.; Pernas, L.E.; Spegg, V.; et al.
Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases. Mol. Cell 2020, 79, 504–520.e9. [CrossRef]
9. Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al.
Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [CrossRef]
10. Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam,
S.; et al. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome. BioRxiv 2020. [CrossRef]
11. Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins
in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [CrossRef]
12. Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate
peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [CrossRef]
13. Trinkle-Mulcahy, L. Recent advances in proximity-based labeling methods for interactome mapping [version 1; referees: 2
approved]. F1000Research 2019, 8, 135. [CrossRef]
14. Gingras, A.-C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize
protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019, 48, 44–54. [CrossRef] [PubMed]
15. Go, C.D.; Knight, J.D.R.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.-Y.; Samavarchi-Tehrani, P.; Zhang, H.;
Zhu, L.Y.; et al. A proximity biotinylation map of a human cell. BioRxiv 2019. [CrossRef]
16. Holding, A.N. XL-MS: Protein cross-linking coupled with mass spectrometry. Methods 2015, 89, 54–63. [CrossRef] [PubMed]
17. Liu, F.; Lössl, P.; Scheltema, R.; Viner, R.; Heck, A.J.R. Optimized fragmentation schemes and data analysis strategies for
proteome-wide cross-link identification. Nat. Commun. 2017, 8, 15473. [CrossRef] [PubMed]
18. Goetze, M.; Iacobucci, C.; Ihling, C.H.; Sinz, A. A Simple Cross-Linking/Mass Spectrometry Workflow for Studying System-wide
Protein Interactions. Anal. Chem. 2019, 91, 10236–10244. [CrossRef] [PubMed]
19. Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr.
Opin. Biotechnol. 2020, 63, 48–53. [CrossRef] [PubMed]Int. J. Mol. Sci. 2021, 22, 4450 13 of 14
20. Chavez, J.D.; Bruce, J.E. Chemical cross-linking with mass spectrometry: A tool for systems structural biology. Curr. Opin. Chem.
Biol. 2019, 48, 8–18. [CrossRef] [PubMed]
21. Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human
centrosome by protein correlation profiling. Nat. Cell Biol. 2003, 426, 570–574. [CrossRef] [PubMed]
22. Mann, M. The Origins of Organellar Mapping by Protein Correlation Profiling. Proteomics 2020, 20. [CrossRef] [PubMed]
23. Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A
Census of Human Soluble Protein Complexes. Cell 2012, 150, 1068–1081. [CrossRef] [PubMed]
24. Kirkwood, K.J.; Ahmad, Y.; Larance, M.; Lamond, A.I. Characterization of Native Protein Complexes and Protein Isoform
Variation Using Size-fractionation-based Quantitative Proteomics*. Mol. Cell. Proteom. 2013, 12, 3851–3873. [CrossRef]
25. Kristensen, A.R.; Foster, L.J. Protein Correlation Profiling-SILAC to Study Protein-Protein Interactions; Humana Press: New York, NY,
USA, 2014; pp. 263–270. [CrossRef]
26. Heusel, M.; Bludau, I.; Rosenberger, G.; Hafen, R.; Frank, M.; Banaei-Esfahani, A.; Van Drogen, A.; Collins, B.C.; Gstaiger, M.;
Aebersold, R. Complex-centric proteome profiling by SEC SWATH MS. Mol. Syst. Biol. 2019, 15, e8438. [CrossRef] [PubMed]
27. Skinnider, M.A.; Scott, N.E.; Prudova, A.; Stoynov, N.; Stacey, R.G.; Gsponer, J.; Foster, L. An Atlas of Protein-Protein Interactions
Across Mammalian Tissues. SSRN Electron. J. 2018. [CrossRef]
28. Fossati, A.; Li, C.; Uliana, F.; Went, F.; Frommelt, F.; Sykacek, P.; Heusel, M.; Hallal, M.; Bludau, I.; Capraz, T.; et al. PCprophet: A
framework for protein complex prediction and differential analysis using proteomic data. Nat. Methods 2021. [CrossRef]
29. Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.;
Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [CrossRef]
30. Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al.
Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 359, 1170–1177.
[CrossRef]
31. Becher, I.; Andrés-Pons, A.; Romanov, N.; Stein, F.; Schramm, M.; Baudin, F.; Helm, D.; Kurzawa, N.; Mateus, A.; Mackmull,
M.-T.; et al. Pervasive Protein Thermal Stability Variation during the Cell Cycle. Cell 2018, 173, 1495–1507.e18. [CrossRef]
32. Dai, L.; Zhao, T.; Bisteau, X.; Sun, W.; Prabhu, N.; Lim, Y.T.; Sobota, R.M.; Kaldis, P.; Nordlund, P. Modulation of Protein-Interaction
States through the Cell Cycle. Cell 2018, 173, 1481–1494.e13. [CrossRef] [PubMed]
33. Heusel, M.; Frank, M.; Köhler, M.; Amon, S.; Frommelt, F.; Rosenberger, G.; Bludau, I.; Aulakh, S.; Linder, M.I.; Liu, Y.; et al. A
Global Screen for Assembly State Changes of the Mitotic Proteome by SEC-SWATH-MS. Cell Syst. 2020, 10, 133–155.e6. [CrossRef]
[PubMed]
34. Romanov, N.; Kuhn, M.; Aebersold, R.; Ori, A.; Beck, M.; Bork, P. Disentangling Genetic and Environmental Effects on the
Proteotypes of Individuals. Cell 2019, 177, 1308–1318.e10. [CrossRef]
35. Stalder, L.; Banaei-Esfahani, A.; Ciuffa, R.; Payne, J.L.; Aebersold, R. SWATH-MS Co-Expression Profiles Reveal Paralogue 1
Interference in Protein Complex Evolution 2 3 n.d. BioRxiv 2020. [CrossRef]
36. Hu, L.Z.; Goebels, F.; Tan, J.H.; Wolf, E.; Kuzmanov, U.; Wan, C.; Phanse, S.; Xu, C.; Schertzberg, M.; Fraser, A.G.; et al. EPIC:
Software toolkit for elution profile-based inference of protein complexes. Nat. Methods 2019, 16, 737–742. [CrossRef]
37. Stacey, R.G.; Skinnider, M.A.; Scott, N.E.; Foster, L.J. A rapid and accurate approach for prediction of interactomes from co-elution
data (PrInCE). BMC Bioinform. 2017, 18, 1–14. [CrossRef]
38. Liu, X.; Yang, W.-C.; Gao, Q.; Regnier, F. Toward chromatographic analysis of interacting protein networks. J. Chromatogr. A 2008,
1178, 24–32. [CrossRef]
39. Dong, M.; Yang, L.L.; Williams, K.; Fisher, S.J.; Hall, S.C.; Biggin, M.D.; Jin, J.; Witkowska, H.E. A “Tagless” Strategy for
Identification of Stable Protein Complexes Genome-wide by Multidimensional Orthogonal Chromatographic Separation and
iTRAQ Reagent Tracking. J. Proteome Res. 2008, 7, 1836–1849. [CrossRef] [PubMed]
40. Kristensen, A.R.; Gsponer, J.; Foster, L.J. A high-throughput approach for measuring temporal changes in the interactome. Nat.
Methods 2012, 9, 907–909. [CrossRef]
41. Scott, N.E.; Brown, L.M.; Kristensen, A.R.; Foster, L.J. Development of a computational framework for the analysis of protein
correlation profiling and spatial proteomics experiments. J. Proteom. 2015, 118, 112–129. [CrossRef]
42. Bludau, I.; Heusel, M.; Frank, M.; Rosenberger, G.; Hafen, R.; Banaei-Esfahani, A.; Van Drogen, A.; Collins, B.C.; Gstaiger,
M.; Aebersold, R. Complex-centric proteome profiling by SEC-SWATH-MS for the parallel detection of hundreds of protein
complexes. Nat. Protoc. 2020, 15, 2341–2386. [CrossRef] [PubMed]
43. Drew, K.; Lee, C.; Huizar, R.L.; Tu, F.; Borgeson, B.; McWhite, C.D.; Ma, Y.; Wallingford, J.B.; Marcotte, E.M. Integration of over
9000 mass spectrometry experiments builds a global map of human protein complexes. Mol. Syst. Biol. 2017, 13, 932. [CrossRef]
44. Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the
MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis.
Mol. Cell. Proteom. 2012, 11. [CrossRef] [PubMed]
45. Ting, Y.S.; Egertson, J.D.; Payne, S.H.; Kim, S.; MacLean, B.; Käll, L.; Aebersold, R.; Smith, R.D.; Noble, W.S.; MacCoss, M.J.
Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol. Cell.
Proteom. 2015, 14, 2301–2307. [CrossRef] [PubMed]You can also read

















































