Metagenomics 2nd Edition - A review of publications featuring Illumina Technology
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Metagenomics 2nd Edition A review of publications featuring Illumina® Technology
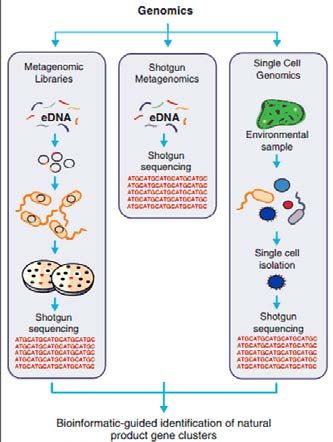
INDEX
Introduction........................................................................................................................................................... 3
Microbial Populations ............................................................................................................................................ 4
16S Ribosomal RNA Gene ..........................................................................................................................................5
Deep Sequencing ........................................................................................................................................................................ 6
Complex Populations .................................................................................................................................................................. 7
Amplification and Cloning Bias ................................................................................................................................................... 7
Mosaicism................................................................................................................................................................................... 8
Intragenomic Heterogeneity ...................................................................................................................................................... 9
Lack of Threshold Identity .......................................................................................................................................................... 9
Metagenome Sequencing .......................................................................................................................................10
Eukaryotes ...............................................................................................................................................................11
Transcriptome Sequencing ......................................................................................................................................12
Plasmids (Plasmidomics) .........................................................................................................................................14
Single-Cell Analysis .............................................................................................................................................. 15
Viruses ................................................................................................................................................................. 17
Human Health...................................................................................................................................................... 18
Antibiotic Resistance ...............................................................................................................................................19
Oral Microbiome .....................................................................................................................................................20
The Human Gut .......................................................................................................................................................21
IBD and Crohn’s Disease ..........................................................................................................................................23
Cystic Fibrosis ..........................................................................................................................................................24
Other Human Microbiota ........................................................................................................................................25
Soil....................................................................................................................................................................... 26
Bioremediation .................................................................................................................................................... 27
Marine Environment............................................................................................................................................ 28
Biofuels and Biocatalysts ..................................................................................................................................... 30
Glossary of Terms ................................................................................................................................................ 32
Bibliography ........................................................................................................................................................ 33
2
INTRODUCTION
Metagenomics refers to the study of genomic DNA obtained from microorganisms that cannot be cultured in the
1
laboratory. This represents the vast majority of terrestrial microorganisms . Microbial populations occur in every
2-3
biological niche on earth . Humans actually carry ten times more bacterial cells than human cells, and 100 times
4
more bacterial genes than the inherited human genome . Microbes also hold the secrets for generating renewable
5 6
biofuels and bioremediation . The new generation of sequencing technology , with its ability to sequence
thousands of organisms in parallel, has proved to be uniquely suited to this application. As a result, the sequencing
of microbial genomes has become routine. Recent technical improvements allow nearly complete genome
assembly from individual microbes directly from environmental samples or clinical specimens, without the need to
7
develop cultivation methods . This accumulation of sequence information has greatly expanded the appreciation
of the dynamic nature of microbial populations and their impact on the environment and human health. With this
extraordinary and powerful set of sequencing tools now available, it is no surprise that metagenomics has become
one of the fastest growing scientific disciplines.
This document highlights recent publications that demonstrate the use of Illumina sequencing technologies in
metagenomics. To learn more about Illumina sequencing and microarray technologies, visit www.illumina.com.
Reviews
Blainey P. C. (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37: 407-427
Lipkin W. I. (2013) The changing face of pathogen discovery and surveillance. Nat Rev Microbiol 11: 133-141
Bik H. M., Porazinska D. L., Creer S., Caporaso J. G., Knight R., et al. (2012) Sequencing our way towards understanding
global eukaryotic biodiversity. Trends Ecol Evol 27: 233-243
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Huntley J., et al. (2012) Ultra-high-throughput microbial
community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621-1624
Foster J. A., Bunge J., Gilbert J. A. and Moore J. H. (2012) Measuring the microbiome: perspectives on advances in
DNA-based techniques for exploring microbial life. Brief Bioinform 13: 420-429
Fuhrman J. A. (2012) Metagenomics and its connection to microbial community organization. F1000 Biol Rep 4: 15
Temperton B. and Giovannoni S. J. (2012) Metagenomics: microbial diversity through a scratched lens. Curr Opin
Microbiol 15: 605-612
Weinstock G. M. (2012) Genomic approaches to studying the human microbiota. Nature 489: 250-256
1
Hugenholtz P., Goebel B. M. and Pace N. R. (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial
diversity. J Bacteriol 180: 4765-4774
2
Rosenthal A. Z., Matson E. G., Eldar A. and Leadbetter J. R. (2011) RNA-seq reveals cooperative metabolic interactions between two termite-
gut spirochete species in co-culture. ISME J 5: 1133-1142
3
Lloyd K. G., Schreiber L., Petersen D. G., Kjeldsen K. U., Lever M. A., et al. (2013) Predominant archaea in marine sediments degrade detrital
proteins. Nature 496: 215-218
4
Barwick B. G., Abramovitz M., Kodani M., Moreno C. S., Nam R., et al. (2010) Prostate cancer genes associated with TMPRSS2-ERG gene fusion
and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer 102: 570-576
5
Desai C., Pathak H. and Madamwar D. (2010) Advances in molecular and "-omics" technologies to gauge microbial communities and
bioremediation at xenobiotic/anthropogen contaminated sites. Bioresour Technol 101: 1558-1569
6
Next Generation Sequencing (NGS) and Massively Parallel Sequencing (MPS) are often used interchangeably to refer to high throughput
sequencing technologies. Sequencing by Synthesis (SBS) refers specifically to Illumina sequencing technology.
7
Lasken R. S. (2013) Single-cell sequencing in its prime. Nat Biotechnol 31: 211-212
3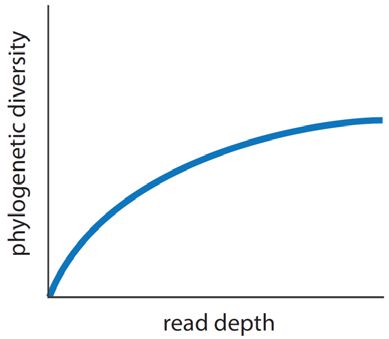
MICROBIAL POPULATIONS
The study of bacterial populations can usefully be divided into two functional areas: metagenomics (“Who is
there?”) and metatranscriptomics (“What are they doing?”). To fully answer those questions, researchers need to
integrate the genomic and transcriptomic sequence analyses, and also look at individual genomes. Transcriptome
sequencing will be discussed in more detail in the section: Transcriptome Sequencing.
Reviews
Muller E. E., Glaab E., May P., Vlassis N. and Wilmes P. (2013) Condensing the omics fog of microbial communities.
Trends Microbiol 21: 325-333
Carlos N., Tang Y.-W. and Pei Z. (2012) Pearls and pitfalls of genomics-based microbiome analysis. Emerg Microbes
Infect 1: e45
Shade A., Gregory Caporaso J., Handelsman J., Knight R. and Fierer N. (2013) A meta-analysis of changes in bacterial
and archaeal communities with time. ISME J 7: 1493-1506
Weinstock G. M. (2012) Genomic approaches to studying the human microbiota. Nature 489: 250-256
Wrighton K. C., Thomas B. C., Sharon I., Miller C. S., Castelle C. J., et al. (2012) Fermentation, hydrogen, and sulfur
metabolism in multiple uncultivated bacterial phyla. Science 337: 1661-1665
Metagenomics strives to determine the abundance and identity of microbes in a sample. There are two
approaches: amplicon sequencing and shotgun sequencing. In amplicon sequencing, an informative marker—such
as the 16S rRNA gene—is amplified by polymerase chain reaction (PCR) and sequenced. Shotgun sequencing refers
to DNA that has been extracted and randomly sheared into smaller fragments before sequencing. These
approaches provide different types of information and each offers unique advantages and trade-offs.
16S rRNA (Amplicon Sequencing) Shotgun Sequencing
Type of information produced The taxonomic composition and Functional and process-level
phylogenetic structure of a microbial characterization of microbial communities as
8
community expressed as OTUs a whole, and the reconstruction of draft
genome sequences for individual community
members.
Application Monitor populations Detect new members, new genes, and
resolve complex taxonomies.
Ability to detect rare members Highly sensitive. rRNA makes up 80% of Requires much deeper sequencing to
of the community (sensitivity) total bacterial RNA achieve the same level of sensitivity
9
Biases Bias produced by the probes and the PCR Sequence content bias
10
itself . The amplified region may not
accurately represent the whole genome
11
due to horizontal transfer or mutations .
Gene content The gene inventory and the encoded Generate extensive gene inventories and
functionality of most microbial species are partial genomes. Discover new genes and
largely unknown and may also vary biological pathways.
considerably among strains.
8
Operational taxonomic units (OTUs)
9
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., et al. (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical
and next-generation sequencing-based diversity studies. Nucleic Acids Res 41: e1
10
Soergel D. A., Dey N., Knight R. and Brenner S. E. (2012) Selection of primers for optimal taxonomic classification of environmental 16S rRNA
gene sequences. ISME J 6: 1440-1444
11
Asai T., Zaporojets D., Squires C. and Squires C. L. (1999) An Escherichia coli strain with all chromosomal rRNA operons inactivated: complete
exchange of rRNA genes between bacteria. Proc Natl Acad Sci U S A 96: 1971-1976
416S RIBOSOMAL RNA GENE
The new era of metagenomics was ushered in by studies using 16S rRNA as a phylogenetic marker of microbial
12
taxa . The 16S rRNA gene occurs in all living organisms, with the notable exception of viruses, and represents
more than 80% of total bacterial RNA. The 16S rRNA gene includes interspersed conserved and variable regions,
which makes it well suited for PCR amplification and sequencing. In this process, probes are designed to hybridize
to the conserved regions, allowing for amplification and sequencing of the variable regions. Focusing on a small
part of the microbial genome lowers sequencing costs dramatically. This approach has been particularly effective
13
for monitoring fluctuations in microbial populations .
Schematic representation of the 16S rRNA gene. Location of variable (purple) and conserved
(brown) regions in a canonical bacterial 16S rRNA. The black region is invariable in all bacteria.
The impact of read length, read depth, and sequencing errors of the various next-generation sequencing
14
technologies has been extensively studied . For example, Illumina technology is capable of 250 bp paired-end
15
reads, effectively interrogating 500 bases at a time .
References
McCafferty J., Muhlbauer M., Gharaibeh R. Z., Arthur J. C., Perez-Chanona E., et al. (2013) Stochastic changes over
time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J
Studying the microbiome in mouse gut is a powerful model for the human gut microbiome. A common experimental
model includes raising mice in a germ-free environment and then inoculating with specific gut samples under study.
This system has however been challenged by the potential effect of microbiome adaptation from the cage
microenvironment, the "cage effect." In this study, the extent and impact of the cage effect was investigated. The
authors find that, while there are long-term effects of the founding community, these effects are mitigated by the
cage microenvironment.
Illumina technology: HiSeq
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., et al. (2013) Evaluation of general 16S ribosomal RNA gene
PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41: e1
Degnan P. H. and Ochman H. (2012) Illumina-based analysis of microbial community diversity. ISME J 6: 183-194
Loman N. J., Misra R. V., Dallman T. J., Constantinidou C., Gharbia S. E., et al. (2012) Performance comparison of
benchtop high-throughput sequencing platforms. Nat Biotechnol 30: 434-439
12
Pace N. R. (1997) A molecular view of microbial diversity and the biosphere. Science 276: 734-740
13
Caporaso J. G., Paszkiewicz K., Field D., Knight R. and Gilbert J. A. (2012) The Western English Channel contains a persistent microbial seed
bank. ISME J 6: 1089-1093
14
Luo C., Tsementzi D., Kyrpides N., Read T. and Konstantinidis K. T. (2012) Direct comparisons of Illumina vs. Roche 454 sequencing
technologies on the same microbial community DNA sample. PLoS ONE 7: e30087
15
Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Huntley J., et al. (2012) Ultra-high-throughput microbial community analysis on the
Illumina HiSeq and MiSeq platforms. ISME J 6: 1621-1624
5Luo C., Tsementzi D., Kyrpides N., Read T. and Konstantinidis K. T. (2012) Direct comparisons of Illumina vs. Roche 454
sequencing technologies on the same microbial community DNA sample. PLoS ONE 7: e30087
Mende D. R., Waller A. S., Sunagawa S., Jarvelin A. I., Chan M. M., et al. (2012) Assessment of metagenomic assembly
using simulated next generation sequencing data. PLoS ONE 7: e31386
Deep Sequencing
One of the most important advantages of next-generation sequencing is the wealth of sequence information it can
produce. Deep sequencing refers to the sequencing of a genomic region multiple times—typically hundreds or
even thousands of times. This makes it possible to detect organisms that exist in very low abundance within
complex populations. These sub-populations can constitute a genetically diverse pool that will survive under
changing environments or environmental stress. As a result, the ability to detect low-abundance populations can
profoundly impact the interpretation of microbiological changes
16
. The actual sequencing read depth required for this type of analysis will depend on the desired sensitivity as well
as the complexity of the population.
Panels A and B represent the same microbial population sampled at two different depths. In panel
A, it appears that the microbes were reintroduced from an external source. However, deep
sequencing in panel B reveals that the microbes were present at all time-points, but dropped below
the detection level used in panel A.
16
Caporaso J. G., Paszkiewicz K., Field D., Knight R. and Gilbert J. A. (2012) The Western English Channel contains a persistent microbial seed
bank. ISME J 6: 1089-1093
6Complex Populations
17
The exhaustive analysis of a complex population is a significant technical challenge . The primary goal is to
sequence deep enough to distinguish low-abundance members of the population from sequencing errors. A low
18
sequencing error rate is important, as well as strict filters to remove sequencing errors .
Rarefaction curve. Phylogenetic diversity increases with read depth. The optimal read depth for
discovery applications would be the read depth where phylogenetic diversity no longer increases.
Amplification and Cloning Bias
,
There are PCR and cloning biases inherent in 16S rRNA protocols 19 20. The primers are designed to hybridize to
conserved regions, but these regions may change over long evolutionary periods, resulting in a loss of
hybridization to the probe. This will lead to an underestimation of evolutionarily distant members of the
population. The variable regions are different sizes and can change at different rates; for example, V6 tags appear
to systematically overestimate species richness 21. Paired-end Illumina sequencing of the larger V4 region has been
successfully used to build phylogenetic trees 22.
17
Bartram A. K., Lynch M. D., Stearns J. C., Moreno-Hagelsieb G. and Neufeld J. D. (2011) Generation of multimillion-sequence 16S rRNA gene
libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol 77: 3846-3852
18
Mende D. R., Waller A. S., Sunagawa S., Jarvelin A. I., Chan M. M., et al. (2012) Assessment of metagenomic assembly using simulated next
generation sequencing data. PLoS ONE 7: e31386
19
Haas B. J., Gevers D., Earl A. M., Feldgarden M., Ward D. V., et al. (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and
454-pyrosequenced PCR amplicons. Genome Res 21: 494-504
20
Soergel D. A., Dey N., Knight R. and Brenner S. E. (2012) Selection of primers for optimal taxonomic classification of environmental 16S rRNA
gene sequences. ISME J 6: 1440-1444
21
Youssef N., Sheik C. S., Krumholz L. R., Najar F. Z., Roe B. A., et al. (2009) Comparison of species richness estimates obtained using nearly
complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ
Microbiol 75: 5227-5236
22
Werner J. J., Zhou D., Caporaso J. G., Knight R. and Angenent L. T. (2012) Comparison of Illumina paired-end and single-direction sequencing
for microbial 16S rRNA gene amplicon surveys. ISME J 6: 1273-1276
7Mosaicism
Many bacteria have a history of horizontal gene transfer. Horizontal transfer of functional genes, or even
23
significant genomic rearrangements, may not be reported by the 16S rRNA region . In addition, bacteria can
24
tolerate transfer of complete 16S genes . Any species identification using 16S gene-based probes or homology-
based analysis of partial 16S sequences may lead to misidentification, because the marker may represent a
25
transferred gene structure .
Horizontal transfer of functional genes, or even significant genomic rearrangements, may not be
reported by the 16S rRNA region
Some bacteria can tolerate the transfer of complete 16S genes, which could impact phylogenetic
interpretation based on 16S rRNA metagenomics analysis.
23
Altermann E. (2012) Tracing lifestyle adaptation in prokaryotic genomes. Front Microbiol 3: 48
24
Asai T., Zaporojets D., Squires C. and Squires C. L. (1999) An Escherichia coli strain with all chromosomal rRNA operons inactivated: complete
exchange of rRNA genes between bacteria. Proc Natl Acad Sci U S A 96: 1971-1976
25
Schouls L. M., Schot C. S. and Jacobs J. A. (2003) Horizontal transfer of segments of the 16S rRNA genes between species of the Streptococcus
anginosus group. J Bacteriol 185: 7241-7246
8Intragenomic Heterogeneity
The copy number of rRNA operons per bacterial genome varies from 1 to 15 26. The sequences of multi-copy rRNA
can vary by as much as 6.4% 27. This will impact abundance estimates based on the rRNA and limit the phylogenetic
resolution of species based on those sequences.
The copy number of rRNA operons per bacterial genome varies from 1 to 15 and the sequences of
the copies can vary by as much as 6.4%.
Lack of Threshold Identity
There is no consistent relationship between the conservation of 16S rRNA and the rest of the bacterial genome.
For example, the 16S rRNA genes of type strains B. globisporus and B. psychrophilus share 99.8% sequence identity
but, at the genome level, exhibit only 23–50% relatedness 28. This can be due to many factors, such as mosaicism
and differences in evolutionary pressure. As a result, species identification solely based on the 16S rRNA sequences
may lead to misidentifications 29.
There is no consistent relationship between the conservation of 16S rRNA and the rest of the
bacterial genome. As a result, species identification solely based on the 16S rRNA sequences may
lead to misidentifications.
26
Klappenbach J. A., Dunbar J. M. and Schmidt T. M. (2000) rRNA operon copy number reflects ecological strategies of bacteria. Appl Environ
Microbiol 66: 1328-1333
27
Wang Y., Zhang Z. and Ramanan N. (1997) The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16S
rRNA genes. J Bacteriol 179: 3270-3276
28
Fox G. E., Wisotzkey J. D. and Jurtshuk P., Jr. (1992) How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species
identity. Int J Syst Bacteriol 42: 166-170
29
Rajendhran J. and Gunasekaran P. (2011) Microbial phylogeny and diversity: small subunit ribosomal RNA sequence analysis and beyond.
Microbiol Res 166: 99-110
9METAGENOME SEQUENCING
Metagenome sequencing, also called shotgun sequencing, refers to sequencing DNA fragments extracted from
microbial populations. Because this approach captures the complete genomes of all the organisms in the
30
population, mosaicism and biases have little impact . The comprehensive information obtained by this approach
enables accurate phylogenetic inferences of close and distant relatives. However, the most substantial advantage
is the information it provides about the genes present in the bacterial population, without assembling the
individual bacterial genomes. Functional gene groupings can be more informative and more stable than a record of
bacterial species.
Reviews
Droge J. and McHardy A. C. (2012) Taxonomic binning of metagenome samples generated by next-generation
sequencing technologies. Brief Bioinform 13: 646-655
Gonzalez A. and Knight R. (2012) Advancing analytical algorithms and pipelines for billions of microbial sequences.
Curr Opin Biotechnol 23: 64-71
References
Erkus O., de Jager V. C., Spus M., van Alen-Boerrigter I. J., van Rijswijck I. M., et al. (2013) Multifactorial diversity
sustains microbial community stability. ISME J
A complex cheese starter culture with a long history of use was characterized as a model system to study simple
microbial communities. Given that cheese is manufactured under strictly controlled conditions, this study asked "Why
is there a high degree of biodiversity in the culture, and what is the mechanism that maintains it?" The genetic
lineages were found to be collections of strains with variable plasmid content and phage sensitivities. An active
suppression of the fittest strains by density-dependent phage predation—"kill the winner"—was determined as the
predominant mechanism maintaining heterogeneity.
Illumina Technology: Illumina mate-pair sequencing
Harris S. R., Clarke I. N., Seth-Smith H. M., Solomon A. W., Cutcliffe L. T., et al. (2012) Whole-genome analysis of
diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat
Genet 44: 413-419, S411
This study presents a detailed phylogeny based on whole-genome sequencing of representative strains of C.
trachomatis from both trachoma and lymphogranuloma venereum (LGV) biovars. It shows that predicting
phylogenetic structure using the ompA gene, which is traditionally used to classify Chlamydia, is misleading because
extensive recombination in this region masks the true relationships.
Illumina Technology: Genome AnalyzerII
Squire M. M., Carter G. P., Mackin K. E., Chakravorty A., Noren T., et al. (2013) Novel molecular type of Clostridium
difficile in neonatal pigs, Western Australia. Emerg Infect Dis 19: 790-792
Altermann E. (2012) Tracing lifestyle adaptation in prokaryotic genomes. Front Microbiol 3: 48
Coelho A. C., Boisvert S., Mukherjee A., Leprohon P., Corbeil J., et al. (2012) Multiple mutations in heterogeneous
miltefosine-resistant Leishmania major population as determined by whole genome sequencing. PLoS Negl Trop Dis 6:
e1512
30
Harris S. R., Clarke I. N., Seth-Smith H. M., Solomon A. W., Cutcliffe L. T., et al. (2012) Whole-genome analysis of diverse Chlamydia
trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet 44: 413-419, S411
10Martin J., Sykes S., Young S., Kota K., Sanka R., et al. (2012) Optimizing read mapping to reference genomes to
determine composition and species prevalence in microbial communities. PLoS ONE 7: e36427
EUKARYOTES
Microscopic eukaryotic taxa are abundant and diverse, playing a globally important role in the functioning of
31
ecosystems and host-associated habitats . The term "microscopic eukaryotes" generally refers to individuals less
than 1 mm in size and encompasses meiofaunal metazoans, microbial representatives of fungi, protists, and algae,
32
as well as eggs and juvenile stages of some larger metazoan species .
Reviews
Bik H. M., Porazinska D. L., Creer S., Caporaso J. G., Knight R., et al. (2012) Sequencing our way towards understanding
global eukaryotic biodiversity. Trends Ecol Evol 27: 233-243
This is a comprehensive review of how advances in DNA sequencing and bioinformatics now allow accurate, en masse
biodiversity assessments of microscopic eukaryotes from environmental samples. The review also contains a table of
online resources and popular software tools for eukaryotic marker gene surveys.
Andersen L. O., Vedel Nielsen H. and Stensvold C. R. (2013) Waiting for the human intestinal Eukaryotome. ISME J 7:
1253-1255
Munoz J. F., Gallo J. E., Misas E., McEwen J. G. and Clay O. K. (2013) The eukaryotic genome, its reads, and the
unfinished assembly. FEBS Lett 587: 2090-2093
31
Bik H. M., Porazinska D. L., Creer S., Caporaso J. G., Knight R., et al. (2012) Sequencing our way towards understanding global eukaryotic
biodiversity. Trends Ecol Evol 27: 233-243
32
Bik H. M., Porazinska D. L., Creer S., Caporaso J. G., Knight R., et al. (2012) Sequencing our way towards understanding global eukaryotic
biodiversity. Trends Ecol Evol 27: 233-243
11TRANSCRIPTOME SEQUENCING
The metatranscriptome is the identity and quantity of a complete set of transcripts in a population of cells. While
metagenomics tells us who is there and what they are capable of, based on their gene complement,
metatranscriptomics tells us what they are doing at that moment. Unlike hybridization-based techniques—such as
PCR, Northern blotting, or microarrays—RNA-Seq information is matched to genes by sequence alignment. This
approach offers the following advantages:
Characteristic Application
No prior knowledge of the genome sequence • Discover novel transcripts and genetic features (gene mining).
is required • Annotate functional domains in the genome.
Accurate mapping • Mapping of sequences with an aligner is more precise than hybridization
in solution.
• Transcription can be studied at a much higher resolution and specificity
without interference from non-specific cross-hybridization.
Dynamic range • Greater dynamic range than fluorescence-based measurements.
• Better discrimination at high and low levels of expression.
Gene mining has become one of the fastest growing applications of this technology. Transcription analyses of
microbial populations in the rumen of various animals, termite gut, and biodigesters have led to the discovery of a
33
staggering array of novel biocatalysts for the biofuel and related industries .
Reviews
Mutz K. O., Heilkenbrinker A., Lonne M., Walter J. G. and Stahl F. (2013) Transcriptome analysis using next-generation
sequencing. Curr Opin Biotechnol 24: 22-30
van Hijum S. A., Vaughan E. E. and Vogel R. F. (2012) Application of state-of-art sequencing technologies to indigenous
food fermentations. Curr Opin Biotechnol
References
Maurice C. F., Haiser H. J. and Turnbaugh P. J. (2013) Xenobiotics shape the physiology and gene expression of the
active human gut microbiome. Cell 152: 39-50
The microorganisms in the human gut microbiome are known to impact digestive health, but their influence on health
by the metabolism of xenobiotics, including antibiotics and drugs, is still unclear. In this metagenomics study,
xenobiotic-responsive genes were found across multiple bacterial phyla involved in various metabolic and stress-
response pathways. The results suggest that xenobiotics may have important implications for the human gut
microbiome.
Illumina technology: HiSeq to sequence the V4 region of the 16S rRNA gene
33
Warnecke F. and Hess M. (2009) A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. J Biotechnol 142: 91-95
12Baker B. J., Sheik C. S., Taylor C. A., Jain S., Bhasi A., et al. (2013) Community transcriptomic assembly reveals
microbes that contribute to deep-sea carbon and nitrogen cycling. ISME J
The biochemical properties of deep-sea microbial communities are an important component of the retention and
recycling of carbon and nitrogen in our environment. This study collected microbial populations from the deep Gulf of
California and analyzed the microbial community using shotgun sequencing of whole-genome RNA. The functional
groups of mRNAs revealed many components of nitrite oxidation pathways and showed that Nitrospirae are minor
yet widespread members of deep-sea microbial communities.
Illumina technology: HiSeq 2000 for RNA-Seq
He S., Ivanova N., Kirton E., Allgaier M., Bergin C., et al. (2013) Comparative metagenomic and metatranscriptomic
analysis of hindgut paunch microbiota in wood- and dung-feeding higher termites. PLoS ONE 8: e61126
Termites effectively feed on many types of lignocellulose, assisted by their gut microbial symbionts. This study aimed
to determine if system-specific differences exist between hindgut paunch microbiota from higher termites with
different diets. The metagenomic and metatranscriptomic profiles were determined for two termite species. The
results showed that the overrepresented functions in each (hemicellulose vs. cellulose breakdown) were consistent
with the dietary differences in carbohydrate composition between the species.
Illumina technology: Genome AnalyzerII for RNA-Seq
Jung J. Y., Lee S. H., Jin H. M., Hahn Y., Madsen E. L., et al. (2013) Metatranscriptomic analysis of lactic acid bacterial
gene expression during kimchi fermentation. Int J Food Microbiol 163: 171-179
Kimchi is a traditional Korean food made through fermentation of vegetables. Kimchi is usually processed without the
use of starter cultures at low temperatures by spontaneous fermentation. In this study, the metagenomic diversity of
kimchi was determined using 16S rRNA sequencing, identifying six major bacterial strains. Total mRNA was extracted
at five time points during fermentation and mapped to the six identified strains, revealing the dynamic changes of
gene expression during the fermentation process.
Illumina technology: Genome AnalyzerII for RNA-Seq
Anantharaman K., Breier J. A., Sheik C. S. and Dick G. J. (2013) Evidence for hydrogen oxidation and metabolic
plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc Natl Acad Sci U S A 110: 330-335
This study of microbial chemosynthesis in hydrothermal vents used a combination of DNA, cDNA, small-subunit
ribosomal RNA (SSU rRNA) amplicon sequencing, and thermodynamic modeling to characterize the genetic potential,
transcriptional activity, and distribution of SUP05 bacteria. The bioenergetic model suggests hydrogen fixation can
contribute significantly to the SUP05 energy budget, revealing a potential high importance of hydrogen as an energy
source in the deep ocean.
Illumina technology: HiSeq 2000
Poulsen M., Schwab C., Jensen B. B., Engberg R. M., Spang A., et al. (2013) Methylotrophic methanogenic
Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat Commun 4: 1428
Rumen methanogens in livestock are major sources of anthropogenic methane emissions. This study examined the
methylotrophic methanogens in rumen metagenomes using 16S rRNA and whole-transcriptome sequencing. The
most abundant Thermoplasmata transcripts, further examined by in vitro incubations, showed enhanced growth of
Thermoplasmata. The authors suggest Thermoplasmata as a potential target for future strategies to reduce methane
emissions.
Illumina Technology: HiSeq 2000 mRNA-Seq 160bp paired-end reads
13PLASMIDS (PLASMIDOMICS)
Plasmids often serve as mediators of lateral gene transfer, a strong and sculpting evolutionary force in microbial
34,35,36
environments .
References
Brown Kav A., Sasson G., Jami E., Doron-Faigenboim A., Benhar I., et al. (2012) Insights into the bovine rumen
plasmidome. Proc Natl Acad Sci U S A 109: 5452-5457
The authors describe the rumen plasmidome as having a highly mosaic nature that can cross phyla. The rumen
plasmidome profile codes for functions that are enriched in the rumen ecological niche. This profile could confer
advantages to the hosts, suggesting that mobile genetic elements play a role in adaptation to the environment.
Illumina Technology: Genome AnalyzerIIx paired-end reads
Fondi M., Rizzi E., Emiliani G., Orlandini V., Berna L., et al. (2013) The genome sequence of the hydrocarbon-degrading
Acinetobacter venetianus VE-C3. Res Microbiol 164: 439-449
Ho P. L., Lo W. U., Yeung M. K., Li Z., Chan J., et al. (2012) Dissemination of pHK01-like incompatibility group IncFII
plasmids encoding CTX-M-14 in Escherichia coli from human and animal sources. Vet Microbiol 158: 172-179
34
Brown Kav A., Sasson G., Jami E., Doron-Faigenboim A., Benhar I., et al. (2012) Insights into the bovine rumen plasmidome. Proc Natl Acad Sci
U S A 109: 5452-5457
35
Podell S., Ugalde J. A., Narasingarao P., Banfield J. F., Heidelberg K. B., et al. (2013) Assembly-driven community genomics of a hypersaline
microbial ecosystem. PLoS ONE 8: e61692
36
Penders J., Stobberingh E. E., Savelkoul P. H. and Wolffs P. F. (2013) The human microbiome as a reservoir of antimicrobial resistance. Front
Microbiol 4: 87
14SINGLE-CELL ANALYSIS
The sequencing and analysis of the genomes of individual
cells should provide the ultimate resolution of a microbial
population. In addition, the analysis of the RNA from …approximately 70% of all known
individual cells should provide insight into the activities of bacterial phyla do not have a single
the cells and task division within the population. There
cultured representative…
has been substantial progress in individual cell analysis in
37,38,39 Wilson et al. 2013
areas such as cancer research , which will inevitably
find their way into metagenomics research.
Review
Blainey P. C. (2013) The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev 37: 407-427
Bryant J. M. (2013) Culture-free club. Nat Rev Microbiol 11: 434
Lasken R. S. (2012) Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 10: 631-
640
Multiple displacement amplification (MDA) of genomic DNA. The φ29 DNA polymerase is efficient
at strand displacement. As single-stranded DNA is displaced, it becomes available for yet more
annealing of primers resulting in exponential amplification 40.
References
Seth-Smith H. M., Harris S. R., Skilton R. J., Radebe F. M., Golparian D., et al. (2013) Whole-genome sequences of
Chlamydia trachomatis directly from clinical samples without culture. Genome Res 23: 855-866
The use of whole-genome sequencing as a tool for the study of infectious bacteria is of growing clinical interest.
Culture of Chlamydia trachomatis has, until now, been a prerequisite to obtain DNA for whole-genome sequencing.
Unfortunately, this is a technically demanding and time-consuming procedure. This study presents a new approach
combining immunomagnetic separation (IMS) and multiple displacement amplification (MDA) for whole-genome
sequencing of bacterial genomes directly from clinical samples.
Illumina technology: Genome AnalyzerII and HiSeq with 100 bp read lengths
37
Islam S., Kjallquist U., Moliner A., Zajac P., Fan J. B., et al. (2011) Characterization of the single-cell transcriptional landscape by highly
multiplex RNA-seq. Genome Res 21: 1160-1167
38
Navin N., Kendall J., Troge J., Andrews P., Rodgers L., et al. (2011) Tumour evolution inferred by single-cell sequencing. Nature 472: 90-94
39
Moon S., Kim Y. G., Dong L., Lombardi M., Haeggstrom E., et al. (2011) Drop-on-demand single cell isolation and total RNA analysis. PLoS ONE
6: e17455
40
Lasken R. S. (2012) Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 10: 631-640
15Lloyd K. G., Schreiber L., Petersen D. G., Kjeldsen K. U., Lever M. A., et al. (2013) Predominant archaea in marine
sediments degrade detrital proteins. Nature 496: 215-218
The single-cell sequencing of both miscellaneous crenarchaeotal group (MCG) and marine benthic group-D (MBG-D)
showed the encoding of several protein-degrading enzymes known to be abundant and active in marine sediments.
Illumina Technology: HiSeq 2000
McLean J. S., Lombardo M. J., Ziegler M. G., Novotny M., Yee-Greenbaum J., et al. (2013) Genome of the pathogen
Porphyromonas gingivalis recovered from a biofilm in a hospital sink using a high-throughput single-cell genomics
platform. Genome Res 23: 867-877
Single-cell genomics is becoming an accepted method to capture novel genomes, primarily in the marine and soil
environments. This study shows, for the first time, that it also enables comparative genomic analysis of strain
variation in a pathogen captured from complex biofilm samples in a health-care facility. The authors present a nearly
complete genome representing a novel strain of the periodontal pathogen Porphyromonas gingivalis using the single-
cell assembly tool SPAdes.
Illumina Technology: Genome AnalyzerIIx
Kamke J., Sczyrba A., Ivanova N., Schwientek P., Rinke C., et al. (2013) Single-cell genomics reveals complex
carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J
Many marine sponges are hosts to dense and phylogenetically diverse microbial communities that are located in the
extracellular matrix of the animal. Single-cell sequencing was used to investigate the metabolic potential of five
individual poribacterial cells, representing three phylogenetic groups almost exclusively found in sponges.
Illumina Technology: HiSeq 2000
Rusch D. B., Lombardo M. J., Yee-Greenbaum J., Novotny M., Brinkac L. M., et al. (2013) Draft Genome Sequence of a
Single Cell of SAR86 Clade Subgroup IIIa. Genome Announc 1:
This genome announcement paper presents the whole genome from single-cell sequencing of bacterial clade SAR86.
This clade has been observed in oceans around the world, based on 16S rRNA sequencing. With the complete
genome, the authors report the gene content in comparison to previous metagenomic analysis.
Illumina Technology: Genome AnalyzerIIx
Campbell J. H., O'Donoghue P., Campbell A. G., Schwientek P., Sczyrba A., et al. (2013) UGA is an additional glycine
codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci U S A 110: 5540-5545
Many human-cohabitating microbes belong to phylum-level divisions for which there are no cultivated
representatives. One such taxon is SR1, which includes bacteria with an elevated abundance in periodontitis. In this
single-cell sequencing from a healthy oral sample, SR1 is found to use a unique genetic code: UGA does not function
as a stop codon, but is translated to glycine. The codon reassignment renders SR1 genes untranslatable by other
bacteria, which impacts the potential for horizontal gene transfer within human microbiota.
Illumina Technology: HiSeq
16VIRUSES
In virology, next-generation sequencing has developed into a
powerful tool that can be used to detect, identify, and quantify
41
novel viruses in one step . It is proving to be a sensitive method for …60–99% of the sequences
detecting putative infectious agents associated with human tissues. generated in different viral
At a modest depth of sequencing, viral transcripts can be detected at
42 metagenomic studies are not
frequencies less than 1 in 1,000,000 . One of the fortunate
consequences of deep sequencing is the coincidental sequencing of
homologous to known viruses.
viral DNA or RNA, which has led to the discovery of several Mokili et al. 2012
43
new viruses .
The use of Illumina technology is reviewed in Viral Detection and
Research.
Reviews
Chiu C. Y. (2013) Viral pathogen discovery. Curr Opin Microbiol 16: 468-478
Colson P., Fancello L., Gimenez G., Armougom F., Desnues C., et al. (2013) Evidence of the megavirome in humans. J
Clin Virol 57: 191-200
Lipkin W. I. and Firth C. (2013) Viral surveillance and discovery. Curr Opin Virol 3: 199-204
Lipkin W. I. (2013) The changing face of pathogen discovery and surveillance. Nat Rev Microbiol 11: 133-141
Malboeuf C. M., Yang X., Charlebois P., Qu J., Berlin A. M., et al. (2013) Complete viral RNA genome sequencing of
ultra-low copy samples by sequence-independent amplification. Nucleic Acids Res 41: e13
Mokili J. L., Rohwer F. and Dutilh B. E. (2012) Metagenomics and future perspectives in virus discovery. Curr Opin Virol
2: 63-77
41
Dunowska M., Biggs P. J., Zheng T. and Perrott M. R. (2012) Identification of a novel nidovirus associated with a neurological disease of the
Australian brushtail possum (Trichosurus vulpecula). Vet Microbiol 156: 418-424
42
Moore R. A., Warren R. L., Freeman J. D., Gustavsen J. A., Chenard C., et al. (2011) The sensitivity of massively parallel sequencing for
detecting candidate infectious agents associated with human tissue. PLoS ONE 6: e19838
43
Li S. C., Chan W. C., Lai C. H., Tsai K. W., Hsu C. N., et al. (2011) UMARS: Un-MAppable Reads Solution. BMC Bioinformatics 12 Suppl 1: S9
17HUMAN HEALTH
Humans carry ten times more bacterial cells than human
cells, and 100 times more bacterial genes than the
44
inherited human genome . The impact on human health Dog ownership significantly increased the
is so significant that the human microbiome can be shared skin microbiota in cohabiting adults
considered an additional organ, or organs. Host-gene-
Song S. J. et al. 2013
microbial interactions are major determinants for the
45
development of some multifactorial chronic disorders .
Reviews
Bertelli C. and Greub G. (2013) Rapid bacterial genome sequencing: methods and applications in clinical microbiology.
Clin Microbiol Infect 19: 803-813
Foxman B. and Rosenthal M. (2013) Implications of the human microbiome project for epidemiology. Am J Epidemiol
177: 197-201
Koser C. U., Ellington M. J., Cartwright E. J., Gillespie S. H., Brown N. M., et al. (2012) Routine use of microbial whole
genome sequencing in diagnostic and public health microbiology. PLoS Pathog 8: e1002824
Kuczynski J., Lauber C. L., Walters W. A., Parfrey L. W., Clemente J. C., et al. (2012) Experimental and analytical tools
for studying the human microbiome. Nat Rev Genet 13: 47-58
Morgan X. C., Segata N. and Huttenhower C. (2012) Biodiversity and functional genomics in the human microbiome.
Trends Genet
Weinstock G. M. (2012) Genomic approaches to studying the human microbiota. Nature 489: 250-256
46
Adapted from .
44
Barwick B. G., Abramovitz M., Kodani M., Moreno C. S., Nam R., et al. (2010) Prostate cancer genes associated with TMPRSS2-ERG gene fusion
and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer 102: 570-576
45
Vaarala, O., Atkinson, M. A. and Neu, J. (2008) The "perfect storm" for type 1 diabetes: the complex interplay between intestinal microbiota,
gut permeability, and mucosal immunity. Diabetes 57: 2555–2562
46
Weinstock G. M. (2012) Genomic approaches to studying the human microbiota. Nature 489: 250-256
18References
Song S. J., Lauber C., Costello E. K., Lozupone C. A., Humphrey G., et al. (2013) Cohabiting family members share
microbiota with one another and with their dogs. elife 2: e00458
Human-associated microbial communities vary across individuals: possible contributing factors include (genetic)
relatedness, diet, and age. This study examined the microbiomes from 60 families, including children and dogs where
present. Household members, particularly couples, shared more of their microbiota than individuals from different
households, and dog ownership significantly increased the shared skin microbiota of cohabiting adults. The authors
conclude that the composition of the human microbiome is significantly affected by contact with cohabitants.
Illumina technology: Genome AnalyzerIIx to sequence variable region 2 (V2) of the bacterial 16S rRNA genes
Keene K. L., Quinlan A. R., Hou X., Hall I. M., Mychaleckyj J. C., et al. (2012) Evidence for two independent associations
with type 1 diabetes at the 12q13 locus. Genes Immun 13: 66-70
ANTIBIOTIC RESISTANCE
The increase in antimicrobial resistance (AMR) is an important worldwide public health threat. Antibiotic
susceptibility is usually monitored based on indicator microorganisms. The choice of the indicator microorganisms
is based on the clinical relevance—for example, well-known pathogens—but also on the cultivability of these
organisms. Since the vast majority of microorganisms cannot be cultured, this leaves significant gaps in the
assessment of AMR of the microbiome as a whole. With the use of next-generation sequencing, it is now possible
47
to detect the presence of antibiotic-resistance genes with high sensitivity in diverse environments .
Reviews
Penders J., Stobberingh E. E., Savelkoul P. H. and Wolffs P. F. (2013) The human microbiome as a reservoir of
antimicrobial resistance. Front Microbiol 4: 87
This review discusses the variety and functional diversity of the gut microbiota, specifically in the context of the
potential for horizontal gene transfer of AMR genes. The authors discuss metagenomics and functional
metagenomics, and give a comparative overview of the abundance of AMR genes in animal rumen.
References
Roelofs D., Timmermans M. J., Hensbergen P., van Leeuwen H., Koopman J., et al. (2013) A functional isopenicillin N
synthase in an animal genome. Mol Biol Evol 30: 541-548
Folsomia candida is a common and widespread arthropod that occurs in soils throughout the world. Horizontal gene
transfer is widespread among prokaryotes, but less common between microorganisms and animals. In this study,
genome sequencing of F. candida revealed the presence of a gene encoding a functional antibiotic enzyme,
isopenicillin N synthase. The data suggest that F. candida has assimilated the capacity for antibacterial activity by
horizontal gene transfer as an important adaptive trait in its microbe-dominated soil ecosystem.
Illumina technology: HiSeq 2000
47
Penders J., Stobberingh E. E., Savelkoul P. H. and Wolffs P. F. (2013) The human microbiome as a reservoir of antimicrobial resistance. Front
Microbiol 4: 87
19Li B., Zhang X., Guo F., Wu W. and Zhang T. (2013) Characterization of tetracycline resistant bacterial community in
saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res 47: 4207-
4216
Antibiotic-resistant bacteria are a growing problem in public health. One of the most commonly used antibiotics is
tetracycline. The efficient detection of tetracycline-resistant bacteria (TRB) is important for monitoring the prevalence
of these bacterial species. In this study, a new method for TRB identification was devised, utilizing Illumina high-
throughput sequencing for accurate quantification of TRB communities in activated sludge saline sewage.
Illumina technology: HiSeq 2000 for 100 bp paired-end reads
Forslund K., Sunagawa S., Kultima J. R., Mende D. R., Arumugam M., et al. (2013) Country-specific antibiotic use
practices impact the human gut resistome. Genome Res 23: 1163-1169
ORAL MICROBIOME
The oral microbiome is a complex ecosystem that includes several thousands of bacterial types inhabiting the
human mouth. Some bacteria in the oral microbiome may give rise to periodontitis, but the transition from health
to disease is not well understood. Current knowledge of the composition and functional spectrum of the human
oral microbiome is limited by the difficulty to culture the majority of microbes that populate the oral cavity. As a
result, the conventional view of periodontitis is largely based on the action of a small set of well-characterized
pathogens. Advances in next-generation sequencing are making it possible to study the healthy and diseased oral
48
microbiomes in much more detail than before .
Reviews
Wade W. G. (2013) The oral microbiome in health and disease. Pharmacol Res 69: 137-143
References
Wang J., Qi J., Zhao H., He S., Zhang Y., et al. (2013) Metagenomic sequencing reveals microbiota and its functional
potential associated with periodontal disease. Sci Rep 3: 1843
The oral microbiome may have significant impact on oral health. In this study, 16 samples were collected from dental
swabs and plaques to characterize the microbiome composition and variation. A number of functional genes and
metabolic pathways were found to be over-represented in the microbiomes of periodontal disease, revealing new
insight into the formation and progression of oral disease.
Illumina technology: HiSeq 2000 and cBot for 100 bp paired-end reads
Liu B., Faller L. L., Klitgord N., Mazumdar V., Ghodsi M., et al. (2012) Deep sequencing of the oral microbiome reveals
signatures of periodontal disease. PLoS ONE 7: e37919
The oral microbiome, the complex ecosystem of microbes inhabiting the human mouth, harbors several thousands of
bacterial types. Some bacteria in the microbiome may give rise to periodontitis, but the system-level mechanism
transitioning from health to disease is not well understood. This study examined the microbiome of periodontitis and
healthy samples using 16S rRNA sequencing and whole-community DNA sequencing. Diseased samples were seen to
share a common structure of the microbiome, suggesting a specific microbiome composition underlying periodontitis.
Illumina Technology: Genome AnalyzerII
48
Liu B., Faller L. L., Klitgord N., Mazumdar V., Ghodsi M., et al. (2012) Deep sequencing of the oral microbiome reveals signatures of
periodontal disease. PLoS ONE 7: e37919
20THE HUMAN GUT
If expanded, the surface of the small intestine alone can reach roughly
49
the size of a tennis court, or 100 times the area of the skin . Nearly 100
…we carry a natural pharmacy
trillion bacteria are harbored within the human gastrointestinal tract,
with archaea, fungi, and viruses representing smaller numbers of the gut in our guts
microbial community. The human microbiome is relatively stable, but Jacobsen et al. 2013
disturbances in the populations of the gut microbiota have been shown
50 51,52 53
to be associated with diseases such as obesity , diabetes , and inflammatory bowel disease .
Reviews
Cani P. D. (2013) Gut microbiota and obesity: lessons from the microbiome. Brief Funct Genomics 12: 381-387
Torrazza R. M. and Neu J. (2013) The altered gut microbiome and necrotizing enterocolitis. Clin Perinatol 40: 93-108
Albenberg L. G., Lewis J. D. and Wu G. D. (2012) Food and the gut microbiota in inflammatory bowel diseases: a
critical connection. Curr Opin Gastroenterol 28: 314-320
Lamendella R., VerBerkmoes N. and Jansson J. K. (2012) 'Omics' of the mammalian gut--new insights into function.
Curr Opin Biotechnol 23: 491-500
Moon C. and Stappenbeck T. S. (2012) Viral interactions with the host and microbiota in the intestine. Curr Opin
Immunol 24: 405-410
Reyes A., Semenkovich N. P., Whiteson K., Rohwer F. and Gordon J. I. (2012) Going viral: next-generation sequencing
applied to phage populations in the human gut. Nat Rev Microbiol 10: 607-617
Zarowiecki M. (2012) Metagenomics with guts. Nat Rev Microbiol 10: 674
References
Jacobsen U. P., Nielsen H. B., Hildebrand F., Raes J., Sicheritz-Ponten T., et al. (2013) The chemical interactome space
between the human host and the genetically defined gut metabotypes. ISME J 7: 730-742
This meta-analysis of previously published metagenomic data investigates the relationship between microbiome metabolic
networks and disease-related proteins in humans. Thirty-three metabolites were identified that interacted with disease-relevant
protein complexes. These complexes are associated with adaptive immune response system and with squamous cell carcinoma
and bladder cancer. The conclusions highlight the importance of gut microbiota for human health.
Illumina Technology: Genome AnalyzerII
49
Hooper L. V. and Macpherson A. J. (2010) Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol
10: 159-169
50
Turnbaugh P. J., Ley R. E., Mahowald M. A., Magrini V., Mardis E. R., et al. (2006) An obesity-associated gut microbiome with increased
capacity for energy harvest. Nature 444: 1027-1031
51
Wen L., Ley R. E., Volchkov P. Y., Stranges P. B., Avanesyan L., et al. (2008) Innate immunity and intestinal microbiota in the development of
Type 1 diabetes. Nature 455: 1109-1113
52
Vaarala O., Atkinson M. A. and Neu J. (2008) The "perfect storm" for type 1 diabetes: the complex interplay between intestinal microbiota,
gut permeability, and mucosal immunity. Diabetes 57: 2555-2562
53
Ott S. J., Musfeldt M., Wenderoth D. F., Hampe J., Brant O., et al. (2004) Reduction in diversity of the colonic mucosa associated bacterial
microflora in patients with active inflammatory bowel disease. Gut 53: 685-693
21Markle J. G., Frank D. N., Mortin-Toth S., Robertson C. E., Feazel L. M., et al. (2013) Sex differences in the gut
microbiome drive hormone-dependent regulation of autoimmunity. Science 339: 1084-1088
Microbial exposures and sex hormones exert potent effects on autoimmune diseases. This study examined the effect
of early-life microbial exposure on sex hormone levels and autoimmune disease in a non-obese diabetic (NOD) mouse
model. The microbiome was characterized using 16S rRNA Illumina sequencing. Comparing the effects across both
male and female mice, the results indicate that alteration of the gut microbiome in early life potently suppresses
autoimmunity in animals at high genetic risk for disease.
Illumina Technology: MiSeq
Koren O., Goodrich J. K., Cullender T. C., Spor A., Laitinen K., et al. (2012) Host remodeling of the gut microbiome and
metabolic changes during pregnancy. Cell 150: 470-480
This study presents the first dataset characterizing the fecal microbiome during the course of pregnancy for 91
pregnant women. Interestingly, the microbiome samples within the third trimester (T3) showed similarities to the
microbiomes of individuals with metabolic syndrome. The authors argue that the dysbiosis, inflammation, and weight
gain are not only normal, but highly beneficial for a normal pregnancy.
Illumina technology: HiSeq 2000 for shotgun sequencing
Maslanik T., Tannura K., Mahaffey L., Loughridge A. B., Benninson L., et al. (2012) Commensal bacteria and MAMPs
are necessary for stress-induced increases in IL-1beta and IL-18 but not IL-6, IL-10 or MCP-1. PLoS ONE 7: e50636
The microbiome of the stomach (enteric bacteria) interacts with the enteric mucosal immune system, and an
alteration of the bacterial community can disrupt immune function. In this study, commensal bacteria (non-harmful
enteric bacteria) are shown to contribute to the acute stress-induced inflammatory responses in a rat model system.
The microbiome environment was evaluated using Illumina HiSeq sequencing.
Illumina technology: HiSeq 2000 to sequence the V4 variable region of the 16S rRNA
Schloissnig S., Arumugam M., Sunagawa S., Mitreva M., Tap J., et al. (2013) Genomic variation landscape of the
human gut microbiome. Nature 493: 45-50
Subjects sampled at varying time intervals exhibited individuality and temporal stability of single-nucleotide
polymorphism (SNP) variation patterns, despite considerable composition changes of their gut microbiota. This
observation indicates that individual-specific strains are not easily replaced and that an individual might have a
unique metagenomic genotype, which may be exploitable for personalized diet or drug intake.
54 55
Illumina technology: Illumina reads obtained from European MetaHIT study and US Human Microbiome Project
Castellarin M., Warren R. L., Freeman J. D., Dreolini L., Krzywinski M., et al. (2012) Fusobacterium nucleatum infection
is prevalent in human colorectal carcinoma. Genome Res 22: 299-306
The authors carried out total RNA-Seq of 11 colorectal tumor samples and 11 matched controls. The genome
sequences of Fusobacterium nucleatum subsp. nucleatum were enriched in the tumor samples. Fusobacterium
nucleatum is rare in the normal gut and usually associated with dental plaque and periodontitis. The results were
–6
validated by quantitative PCR analysis from a total of 99 subjects (p = 2.5 X 10 ). The authors proceeded to culture
and sequence a Fusobacterium strain and also developed a PCR screen.
Illumina Technology: Genome AnalyzerIIx for RNA-Seq and HiSeq for bacterial resequencing.
Zhang Q., Widmer G. and Tzipori S. (2013) A pig model of the human gastrointestinal tract. Gut Microbes 4: 193-200
54
Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., et al. (2010) A human gut microbial gene catalogue established by metagenomic
sequencing. Nature 464: 59-65
55
Group N. H. W., Peterson J., Garges S., Giovanni M., McInnes P., et al. (2009) The NIH Human Microbiome Project. Genome Res 19: 2317-
2323
22You can also read

















































