Faadiel Essop September 2011 - Stellenbosch University
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Faadiel Essop September 2011
Treading the tightrope of healthy living: pondering heart metabolism’s balancing act Inaugural lecture delivered September 2011 Prof Faadiel Essop Department of Physiological Sciences Faculty of Natural Sciences Stellenbosch University Editor: SU Language Centre Design: Heloïse Davis Printing: rsamprinters@gmail.com ISBN: 978-0-7972-1334-0 Copyright ©Stellenbosch University Language Centre
P rof Faadiel Essop is the current chairperson of the Depart-
ment of Physiological Sciences at Stellenbosch University. He
hails from a strong rural background; he was born in Ceres and
grew up in Paarl (Western Cape, South Africa). He completed
his undergraduate studies (biochemistry and microbiology
majors) and PhD degree (medical biosciences) at the University
of Cape Town (UCT). He also completed (part-time basis) a BA
Hons degree (Arabic) at the University of the Western Cape.
After postdoctoral fellowship stints at UCT and the University of
Leeds, he joined the Hatter Cardiovascular Research Institute at
UCT’s Faculty of Health Sciences (1998). During this time he
focused on mechanisms driving the onset of cardiac hypertrophy and the effects of
hypoxia on the heart. He was also appointed to the board of the Medical Research
Council of South Africa by the Minister of Health (1998–2000). Prof Essop was
awarded the prestigious Fulbright fellowship (2005–2006) to spend time in Prof
Heinrich Taegtmeyer’s laboratory at the University of Texas-Houston Medical School.
During February 2007, he joined the Department of Physiological Sciences at
Stellenbosch University as an associate professor and was promoted to full professor
in 2011. At Stellenbosch University he established the Cardio-Metabolic Research
Group that focuses on altered fuel substrate metabolism and its contribution to the
onset of type 2 diabetes and heart failure. Prof Essop and his students have received
several awards over the last few years. He has published 42 peer-reviewed papers and
supervised six PhD, five MSc and four postdoctoral students. He is married to Dr
Rehana Essop and they have three children, Ziyaad, Aaliyah and Yasin.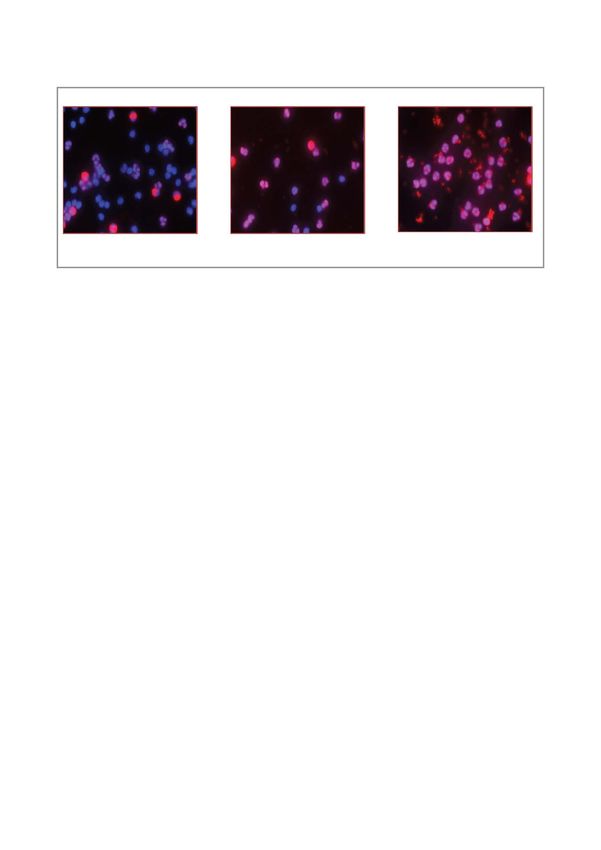
ACKNOWLEDGEMENTS
I wish to acknowledge – in keeping with the African spirit of Ubuntu – all those who played a
significant role in my academic career thus far. Archbishop Desmond Tutu captured the concept
of Ubuntu best with the following words: “Ubuntu speaks particularly about the fact that you can't
exist as a human being in isolation. It speaks about our interconnectedness. You can't be human
all by yourself, and when you have this quality – Ubuntu – you are known for your generosity.”
I am highly appreciative of the efforts of all my former teachers at Paulus Joubert Primary
School and Noorder Paarl High School. I am grateful to my PhD supervisor, Prof Eric Harley, for
introducing me to heart mitochondria and mountain-face climbing! He has also taught me that
the pursuit of the unknown is really an adventure, and to always be on the outlook for the
“outlier” – the data not necessarily fitting your hypothesis. Many thanks to Profs Lionel Opie and
Michael Sack for their outstanding mentorship while I was based at the Hatter Institute at the
University of Cape Town. Prof Opie triggered my interest in heart metabolism and is an excellent
mentor. I miss the visits to his office for stimulating conversations or bouncing off a new idea. I
would also like to express my gratitude to Profs Heinrich Taegtmeyer (University of Texas-
Houston Medical School) and William Stanley (University of Maryland, Baltimore) for their
guidance and mentorship during my career.
I would like to acknowledge former postgraduate students who helped advance my research
endeavours: Joy McCarthy, Siyanda Makaula, Nayna Manga, Kholiswa Ngumbela, Uthra
Rajamani, Makhosazane Zungu, Tasneem Adam, Anna Chan, Aretha van der Merwe, Gordon
Williams, Rebecca Felix, James Meiring, Nihar Singh, Celeste Brand, Sideeqah George, Anique
Jordaan, Carla Pool and Karien Viljoen. Further I am indebted to all my wonderful colleagues at
the Department of Physiological Sciences and to Prof Eugene Cloete, Dean of the Faculty of
Science.
I am most grateful to the current members of the Cardio-Metabolic Research Group: Jamie
Imbriolo, Danzil Joseph, Rudo Mapanga, Kathleen Reyskens, Rinah Harris, Delita Otto, Clare
Springhorn, Kirsty Garson and Burger Symington. You are an incredible group of individuals,
making supervision and mentorship an absolute pleasure!
I value the support of family and friends throughout my career. To conclude, I thank my wife,
Rehana Essop, for her continuous support and love over the course of my academic career. This
is highly appreciated! Thank you to my children, Ziyaad, Aaliyah and Yasin, for their love and
understanding. Lastly, I wish to acknowledge my parents, Yusuf and Fareda, for supplying me with
their DNA, and also for their love, nurturing and support over the years.THE VIRTUE OF MODERATION met) or toxicity (cells poisoned by oversupply).2 As a
T
result of such disruption, organisms trigger various
he quest for balance and moderation is well en-
trenched in major belief systems, ancient traditions pathways within affected cells to limit damage caused.
and philosophical musings. For example, Aristotle However, in the longer term, this disruption may also
(384–322 BC) noted that “the virtue of justice consists in result in maladaptation and disease onset.
moderation’’ while Muslims are reminded in the Quran, The human body consists of various systems (e.g.
“and thus We willed you to be a community of the nervous, digestive, endocrine and cardiovascular) that
middle way’’. The Buddha (563–483 BC) also under- function in concert to ensure overall well-being. Since
stood this when overhearing a lute player and realizing no system operates as an independent unit, disruption
that the harmonious sounds produced depended on the of one or more systems may result in serious conse-
lute strings’ not being tuned too tightly or too loosely. quences for other systems of the body and threaten
Likewise, the well-being of individual cells and organisms overall well-being.2 For example, if homeostasis of the
depends on a constant internal environment, referred cardiovascular system is compromised due to internal
to as ‘homeostasis’’. The term ‘homeostasis’ derives and/or external exposures, it will result in serious
from the Greek homoios (‘same’) and stasis (‘condition’) consequences for the host organism.
and was coined in 1929 by the American physiologist
Walter Cannon.1,2 However, the concept was earlier IMPACT OF CONTEMPORARY
recognized by others. The French scientist Claude
Bernard (1813–1878) used the term milieu interieur and EXTERNAL EXPOSURES
noted that “all the vital mechanisms, varied as they are,
have only one object, that of preserving constant the
conditions of life in the internal environment’’.1,2 More-
A World Health Organization report warns of the
escalating global burden of cardiovascular diseases
(CVD),5 and a recent study investigating the worldwide
over, some suggest that the celebrated physician Ibn prevalence of CVD projects a marked increase for its
Sina (Avicenna) (980–1037) was aware of intrinsic and incidence in developing nations (including South Africa)
extrinsic factors and their role in the development of by 2020.6 Data suggest that the dramatic surge in CVD
disease, and also of the body’s interior milieu and rates in developing countries is due to accelerated
homeostasis.3,4 urbanization and associated lifestyle changes (e.g. poor
External environmental exposures depend largely on nutritional choices, obesity and decreased physical acti-
lifestyle choices (e.g. nutrition and physical activity) and vity).7 For example, urbanization is a growing problem
the outside environment (e.g. urbanization and pollu- in Africa, drastically altering lifestyle and thereby leading
tion). Organisms employ numerous mechanisms to to changes in body composition and shape, which
ensure that their internal milieu is exquisitely balanced, ultimately increases the risk for insulin resistance,
for example through continuous monitoring of internal type 2 diabetes and CVD.8 Demographic change in
and external environments. If required, the body elicits South Africa is also a major contributing factor; for
the necessary adjustments to remain near its ‘set point’. example, the number of individuals aged 35–64 years
Here the organism functions inside particular limits old is projected to rise three-fold by 2025 despite the
within specific environments/contexts. If these set limits threat of HIV/AIDS.9 Moreover, antiretroviral treat-
are exceeded or not adequately met, it is sensed and ment (protease inhibitors) is associated with increased
organisms initiate the necessary adjustments to again onset of insulin resistance, dyslipidemia and lipodys-
operate within their set confines.2 However, organisms trophy.9 Patients on chronic antiretroviral treatment
are sometimes unable to maintain homeostasis, leading will further swell the growing burden of cardio-meta-
to cellular dysfunction and the onset of diseases. This bolic diseases in developing countries such as South
may occur either due to deficiency (needs of cells not Africa. This sentiment is aptly echoed by Alafia Samuels
5(University of the West Indies, Barbados) who state interventions to blunt the onset of insulin resis-
that “we have to be patient-focused. We cannot cure (a tance/type 2 diabetes and CVD and 4) develop novel
patient’s) HIV and then send them off to die with methods to detect the onset of diabetes and CVD.
diabetes.’’10 Projections show that the global incidence The rest of this document will concentrate on
of type 2 diabetes will continue to rise, with the African selected research work conducted in our laboratory
continent facing ~50% increase in numbers.11 Since over the last years, emphasizing perturbed metabolism
cardiovascular complications are common in patients and its contribution to the onset of cardio-metabolic
with type 2 diabetes, this will further increase the over- diseases. Our work focused particularly on unraveling
all burden of disease.12 The higher mortalities and the mechanisms underlying heart metabolism and
morbidities associated with increased type 2 diabetes function within the setting of type 2 diabetes and cardiac
and CVD rates will have serious socio-economic im- hypertrophy (thickened cardiac muscle). Here our
plications, including disruption of family units, greater rationale is that such an understanding will allow for the
health care costs and diminished productivity. This will development of novel diagnostic tools and therapies to
eventually threaten the sustainable development of treat the growing burden of cardio-metabolic diseases
South Africa and the rest of the African continent. in developed and developing nations.
THE METABOLIC SYNDROME ONSET OF METABOLIC SYNDROME
A n emerging paradigm suggests that a cluster of IN SOUTH AFRICAN POPULATIONS
O
metabolic abnormalities, referred to as the meta- f concern is that relatively limited data are availa-
bolic syndrome, is associated with increased risk for the ble for the prevalence of the metabolic syndrome
development of both type 2 diabetes and CVD.13,14 in South Africa and the African continent, thus limiting
Here an underlying rationale is to provide a clinically the development of effective strategies to deal with the
accessible diagnostic tool that will allow for the early increasing burden of disease for type 2 diabetes and
identification of individuals at risk for the development CVD. In light of this, we began investigating the inci-
of type 2 diabetes and/or CVD. In broad terms, the dence of metabolic risk factors in a younger student
metabolic syndrome is characterized by a ‘deadly quar- population (< 30 years old) at Stellenbosch University.
tet’, including impaired glucose regulation (hyper- Here risk factors (4% of student population) presented
glycemia), poor lipid profile (dyslipidemia), hypertension at a much younger age than commonly expected.15 Our
and obesity.13,14 Since abdominal fat deposition is data showed some gender-based differences, with
associated with more serious health implications than women displaying a greater prevalence of increased
fat accumulating elsewhere,7 the International Diabetes waist circumference while men exhibited higher blood
Federation now includes increased waist circumference pressures. Waist circumference in the female study
as a prerequisite for the diagnosis of the metabolic population was positively associated with blood pres-
syndrome.13,14 Of note, central obesity is thought to be sures and cholesterol levels. We propose that these
the single most important factor contributing to the differences are related to student behavioural patterns,
development of the metabolic syndrome. Moreover, in other words, male students displaying poor lifestyle
adipose tissue is now recognized to be an active meta- choices (e.g. increased consumption of ‘junk food’) and
bolic organ, secreting hormones and cytokines that may female students exercising less than would normally be
have both paracrine and endocrine effects on different expected. We therefore recommend that it is impera-
tissue types. tive to screen young students in order to identify meta-
In our opinion, the alarming projections for the onset bolic risk profiles relatively early on and to thereafter
of type 2 diabetes and CVD necessitate a comprehen- initiate appropriate lifestyle changes.
sive strategy to deal with this problem and therefore We are presently expanding our initial work in Stel-
our laboratory is coordinating a multi-pronged project lenbosch to include a greater number of students and
to evaluate the metabolic syndrome within the southern also the more senior campus population (> 30 years
African context. Here our focus is to 1) investigate the old). Our data show a marked incidence of metabolic
onset of metabolic risk factors in southern African risk factors in the older Stellenbosch campus population
communities, 2) gain insight into the basic mechanisms (38–58 years old) (see Table 1), with elderly men the
whereby metabolic risk factors actually trigger type 2 most vulnerable group (unpublished data). Since obesity
diabetes and CVD onset, 3) establish unique therapeutic and poor lifestyle choices (older men) appear to be key
6mediators of metabolic risk factor onset in this group, THE REDUCTIONIST APPROACH
we advise improved body weight management (e.g. in-
creased exercise) and sound lifestyle choices (e.g. lower
AND BASIC SCIENCE
alcohol intake). Together, these data show that meta-
bolic risk factors present a) at a much earlier age than T o better understand the underlying molecular
mechanisms driving the onset of disease states, bio-
medical scientists currently adopt ‘reductionism’ as
what would be commonly expected and b) at alarming
rates in the older population. This therefore necessi- their operating paradigm. The basic rationale of re-
tates effective strategies to counter the predicted onset ductionism is that a complex system, for example the
of type 2 diabetes and CVD. Moreover, it also requires onset of a particular disease state within a human being,
further investigation and identification of mechanisms can be understood by examining its basic elements. The
whereby risk factors eventually cause the development approach is to reduce a specific disease condition to its
of cardio-metabolic diseases. basic cellular and molecular elements (e.g. genes, pro-
teins and enzymes). Moreover, biomedical scientists
Female Female P-values employ various experimental systems (e.g. animal and
students Staff cell-based models) in their laboratories to investigate
(< 30 (> 30
years) years) and identify molecular events that may ultimately result
Systolic BP 119 132.96 in the onset of a particular disease.
(mmHg) Although this particular worldview has resulted in
significant strides being made to develop novel thera-
Diastolic BP 77.65 88.61
(mmHg) P < 0.001 pies, its shortfalls are also increasingly being highlighted.
For example, scientists are now facing ‘information
Glucose (mmol/L) 4.6 7.1 overload’ with the overspecialization of disciplines often
resulting in a plethora of molecular pathways with sig-
Triglycerides 1.47 2.78
(mmol/L) nificant cross-talk a compounding factor. This may im-
pair information flow and/or meaningful and coherent
Total Cholesterol 4.65 5.19 p < 0.05 synthesis/understanding of the various molecular path-
(mmol/L)
ways and regulators continuously being identified.
BMI (kg/m2) 20.98 27.99 P < 0.001 Oversimplification is another potential problem since
therapeutic advances tested within the laboratory setting
are not always effective when employed in the clinic. In
Table 1: Metabolic risk factors for female students
agreement, Dr Claude Lenfant, former director of the
and staff members at Stellenbosch University
National Heart, Lung, and Blood Institute (NHLBI),
Older Stellenbosch University female staff exhibits a states, “Enormous amounts of new knowledge are
marked increase in metabolic risk factors and anthro-
barrelling down the information highway, but they are
pometric measures compared to the younger population.
Here we found higher blood pressures and fasting levels
not arriving at the doorsteps of our patients.’’16 As a
of several metabolites, namely increased glucose, result, major institutions such as the National Institutes
triglycerides and total cholesterol. Note: systolic blood of Health in the United States of America have adjusted
pressures ≥ 130 and/or diastolic pressures ≥ 85, fasting their strategic approach with a renewed interest in the
triglyceride ≥ 1.7 mmol/L and fasting glucose levels ≥ 5.6 more rapid ‘translation’ of laboratory-based science in-
mmol/L make up some of the criteria that constitute the to the clinic and to also foster greater co-operation be-
metabolic syndrome, according to the International tween basic scientists and clinicians. Furthermore, recent
Diabetes Federation’s definition. We are currently in the
approaches such as ‘systems biology’ are also attemp-
process of evaluating low-density lipoprotein and high-
density lipoprotein levels in these populations. The body
ting to deal with the shortcomings of molecular reduc-
mass index (BMI) – marker of obesity – of older women tionism. However, Joyner17 correctly points out that
was also higher than the cut-off value of 25 kg/m2 and systems biology still employs a cell-centric focus with
falls within the range that is regarded as pre-obese limited understanding and application beyond the cell.
(n = 117). Since some of these approaches are still rooted in
the existing scientific worldview, it is likely that the
current paradigm will need to be revised in future to
reflect a more integrative approach. Physiology as a
7discipline may indeed be able to help bridge the gap We also performed studies on transcriptional
between molecular reductionism and complexity. Here mechanisms that regulate ACCβ g ene expres s ion to
the idea is put forward that physiology may fill the iden tify novel ‘on’ a nd ‘off’ s witches tha t reg ula te
current intellectual void since its focus is to understand this proces s . This a pproa ch wa s a dopted s ince
the integrated functions of organs/organisms within the both decrea s ed a nd increa s ed fa tty a cid β-oxidation
larger context of homeostasis and regulation.17,18 rates are implicated in the onset of insulin resistance
and diabetes-related CVD. We hypothesized that this
THE ABCS OF HEART METABOLISM apparent contradiction may be clarified when viewing
such findings within particular contexts, for example ex-
T he main focus of our research work over the last
years was on the role of altered metabolism of the
heart and its impact on myocardial energy (adenosine
perimental models employed and also the stage of
disease progression. Thus the identification of both acti-
vators and inhibitors of ACCβ gene expression offers
triphosphate [ATP]) production and contractile func- therapeutic promise depending on the particular con-
tion. The normal human heart requires between 3.5 and text. Here we identified a unique transcriptional acti-
5 kg of ATP per day to sustain its function.19 An ade- vator, upstream stimulatory factor 1 (USF1), that in-
quate supply of oxygen and fuel substrates allows for creases cardiac ACCβ g ene expres s ion. 25 Further-
their metabolism to generate enough ATP to keep the more, we also identified nuclear respiratory factor-1
heart pumping. Fatty acids serve as the major fuel sub- (NRF-1) as a novel inhibitor of ACCβ g ene expres-
strate for the normal, fasting adult mammalian heart, sion.26 We therefore propose that both USF1 and NRF-
providing the majority of its energetic requirements.19 1 are useful targets to control malonyl-CoA levels and
The rest of the heart’s ATP is derived from glucose and thereby up- or downregulate cardiac mitochondrial
lactate in nearly equal proportions, while ketone bodies fatty acid β-oxida tion, depending on the pa rticula r
are also utilized as a fuel substrate under certain condi- intra cellula r milieu (s ee F ig ure 1).
tions. After uptake by specific cardiac fatty acid trans-
Upstream, 5΄-AMP -a ctiva ted protein kina s e
porters, long-chain fatty acids are esterified by fatty
(AMP K) is a pivota l fuel s ens or tha t is a ctiva ted in
acyl-CoA synthetase and may either be stored as trig-
res pons e to environmenta l s tres s (e.g . oxyg en la ck
lycerides or transported into the mitochondrion for
a nd nu trient depriva tion) with the prima ry a im to
fatty acid β-oxida tion. res tore both cellu la r a nd whole body energ y ba-
Mitochondrial fatty acid uptake is controlled by lance.27 AMPK activates both myocardial glucose and
malonyl-CoA, a potent allosteric inhibitor of carnitine fatty acid metabolic pathways to ultimately ensure in-
palmitoyltransferse-1 (CPT-1), the rate-limiting enzyme creased production of myocardial ATP when required.
regulating this process.20 The rates of synthesis and de- It is well established that AMPK can phosphorylate and
gradation of malonyl-CoA in the heart are stringently inhibit ACCβ a ctivity, thereby lowering ma lonyl-CoA
controlled by acetyl-CoA carboxylase (ACC) and malo- levels a nd s timula ting mitochondria l fa tty a cid β-
nyl-CoA decarboxylase (MCD), respectively. Two ACC oxida tion. We a ls o found, for the firs t time a s fa r a s
isoforms with distinct functional roles have been identi- we a re a wa re, tha t AMP K dos e-de pen dently redu-
fied in mammals, namely ACCa and ACCβ.21,22 ACCa ces ACCβ g ene promoter a ctivity 26 (see Figure 1).
is enriched in lipogenic tissues where it plays a key role AMPK is therefore able to inhibit ACCβ a t both
in fatty acid biosynthesis. Conversely, ACCβ is abun- tra ns crip tiona l a nd enzyme a ctivity levels . After its
dantly expres s ed in oxida tive tis s ues (e.g . hea rt a nd mitochon dria l up ta ke, the fa tty a cyl-CoA enters the
s keleta l mus cle) a nd is phys ica lly a s s ocia ted with β-oxida tion s pira l tha t s ucces s ively s hortens fa tty
mitochondria . 23 ACCβ is therefore s trong ly impli- a cids by two ca rbons (per cycle) a nd g ene ra tes
cated in the control of mitochondria l fa tty a cid β-oxi- NADH a nd FADH2. The reducing equivalents generated
dation. In support, we found de crea sed myoca rdia l subsequently donate electrons to the mitochondrial
ma lonyl-CoA levels a nd in crea s ed ca rdia c mito- electron transport chain for ATP production. Mito-
chondrial fa tty a cid β-oxida tion in ACCβ muta nt mice chondrial fatty acid β-oxida tion provides ~60–90% of
(dis pla ying reduced ACCβ a ctivity). 24 the tota l energ y require ments of the hea rt. 19
8Glucose
Insulin Fatty acids
GLUT4
Insulin
FAT
receptor
Signaling Glycolysis
(e.g. PI3K, Akt,
AS160)
AMPK
Activated
fatty acids
Pyruvate
GLUT 4 USF1 NRF-1
NRF 1
ACCǃ
ACC ǃ gene
CPT-1 Malonyl-CoA
ACCǃ mRNA
Nucleus
Glucose Fatty acid
o idation
oxidation Acetyl-CoA
Acetyl CoA ǃ o idation
ǃ-oxidation
Kreb’s cycle ETC ATP
C di
Cardiomyocyte
t
Mitochondrion
Figure 1: Basic scheme of cardiac fatty acid and glucose metabolism
Fatty acids are taken up by specific transporters (e.g. FAT – fatty acid transporter) and activated by the addition of
coenzyme A. Mitochondrial uptake is controlled by carnitine palmitoyltransferase-1 (CPT-1). Fatty acid β-oxida tion sub-
sequently occurs within the mitochondria l ma trix to produce reducing equiva lents tha t ca n enter the electron
tra ns port cha in (E TC) to g enera te ATP for work. Mitochon dria l fa tty a cid upta ke is reg ula ted by ma lonyl-CoA, a
potent inhibitor tha t is s ynthes ized by a cetyl-CoA ca rboxyla s e-beta (ACCβ). We found that upstream stimulatory
factor 1 (USF1) and nuclear respiratory factor-1 (NRF-1) can increase or decrease ACCβ g ene expres s ion, res pectively,
a nd thus control ca rdia c ma lonyl-CoA levels . This , in turn, will reg ula te CP T-1 a ctivity a nd ca rdia c mitochon dria l
fa tty a cid upta ke. We a ls o found tha t AMP K a ctiva tion ca n decrea s e ACCβ g ene expres s ion. Circula ting ins ulin
reg ula tes g lucos e upta ke by promoting GLUT4 s tora g e ves icles to mig ra te to the cell membra ne where in-
creased GLUT4 a va ila bility ca n now media te g lucos e upta ke. After its upta ke, g lucos e is ca ta bolized (in s evera l
s teps ) into pyruva te tha t ca n be ta ken up by mitochondria to be further meta bolized. R educing equiva lents
produced by the Krebs cycle ca n g enera te ATP via the E TC.
Myocardial glucose uptake is mediated by glucose its conversion to glycogen (for rapid utilization at a later
transporter (GLUT) isoforms, namely GLUT1 and GLUT4. stage) and flux through the polyol, hexosamine bio-
GLUT1 is enriched during the fetal stages of develop- synthetic and pentose phosphate pathways. Pyruvate
ment, while GLUT4 is postnatally induced, insulin- subsequently enters the mitochondrion where it is
dependent and the major transporter in the adult further catabolized via the Krebs cycle to generate re-
heart.19 After insulin binds to the membrane-bound in- ducing equivalents (NADH and FADH2) that can
sulin receptor, it triggers signaling cascades (e.g. PI3 eventually enter the electron transport chain to
kinase, Akt and AS160) that allow GLUT4 to migrate produce mitochondrial ATP. The complete breakdown
from intracellular vesicular stores to the sarcolemma, of glucose (glycolysis and glucose oxidation) accounts
thereby increasing myocardial glucose uptake1,19 (see for ~10–40% of the heart’s energy requirements.19
Figure 1). Glucose subsequently enters the glycolytic NADH and FADH2 generated by glucose and fatty acid
pathway and is ultimately converted to pyruvate with metabolism can initiate electron transfer through the
the production of two ATP molecules (per glucose mitochondrial electron transport chain. Electrons are
molecule) without any oxygen requirements (anaerobic passed through complex I (NADH dehydrogenase) and
glycolysis). Additional metabolic fates of glucose include complex II (succinate dehydrogenase) to coenzyme Q.
9Reduced CoQ then transfers electrons to complex III left ventricle.33–40 However, these findings will not be
(ubiquinol cytochrome c reductase) that donates it to discussed in this particular document. Rats were typi-
oxidized cytochrome c. Thereafter reduced cytochrome cally exposed to hypobaric hypoxia (11% oxygen) for
c passes electrons to complex IV (cytochrome c oxi- various lengths of time (1–12 weeks), and we found a
dase) that reduces molecular oxygen to water in the robust hypertrophic response (increased right ventri-
final step. During such electron transfer (between cular weight and myocyte size) as early as one week. No
complex I and complex IV), a proton gradient is esta- fibrosis was detected in the hypertrophied right
blished across the inner mitochondrial membrane and ventricle (see Figure 2).
exploited for ATP production. As protons move back We found coordinate induction of several genes
into the mitochondrial matrix via complex V (ATP syn- regulating mitochondrial function, and increased citrate
thase) mitochondrial ATP is produced. synthase activity and mitochondrial DNA levels in the
Cardiac fuel substrate utilization is dynamic and may hypertrophied right ventricle (at the two-week and
be altered according to the prevailing physiologic or four-week time points).31,32 In parallel, mitochondrial
pathophysiologic milieu. For example, after meal intake respiratory capacity and right ventricular contractile
(fed state), circulating insulin levels rise to promote function were both sustained (see Figure 3). Thus the
myocardial glucose uptake. During the fasted state physiologic hypertrophied right ventricle maintains its
(several hours after a meal), fatty acid utilization predo- output by increasing mitochondrial respiratory capacity
minates with a concomitant decrease in carbohydrate and ATP production. We propose that pathophysio-
utilization and circulating insulin levels. Furthermore, logic hypertrophy may occur when mitochondrial
high-altitude natives, such as the Himalayan Sherpas and respiratory capacity diminishes, thereby leading to de-
the Andean Quechuas, display enhanced myocardial creased cardiac output and eventually heart failure. In
glucose uptake.28 Here increased glucose utilization at agreement, Neubauer41 proposed that during end-stage
high altitude (relatively hypoxic environment) may occur heart failure, the heart runs out of fuel since both glu-
since it is a more oxygen-efficient fuel substrate to cose and fatty acid oxidation are downregulated. These
generate ATP compared to fatty acids. In certain in- data therefore indicate that the restoration of mito-
stances, myocardial fuel substrate switches may also chondrial respiratory capacity in failing hearts may be a
result in maladaptive effects, thereby leading to im- useful therapeutic strategy to pursue. However, the
paired cardiac contractility. The latter has been the pitfalls of reductionism should be borne in mind and
focus of our laboratory during the last years, namely to such treatments be tailored to be employed within spe-
delineate the effects of altered metabolism in hyper- cific contexts, for example end-stage heart failure in this
trophied and diabetic hearts. We employed several ani- instance.
mal and cell-based experimental systems to investigate
our hypothesis that chronically high fuel substrate (fatty
acids and glucose) and lower oxygen levels trigger mal-
THE DIABETIC HEART
adaptive pathways, leading to decreased mitochondrial
ATP generation and increased cell death. This, in turn, W e investigated cardiac contractile function and
mitochondrial respiratory capacity in a rat
model of diet-induced prediabetes, in other words,
would be expected to lead to impaired cardiac con-
tractile function. “metabolic syndrome-like’’.42 Here intake of a sub-
optimal diet induced obesity together with insulin
resistance, increased visceral fat, dyslipidemia and higher
THE PHYSIOLOGIC plasma insulin levels. Moreover, prediabetic rats ex-
HYPERTROPHIED HEART hibited reduced cardiac contractile function at baseline
T o gain insight into the link between physiologic and
pathophysiologic cardiac hypertrophy, we established
and characterized a rat model of hypobaric hypoxia-
together with decreased mitochondrial bioenergetic
capacity. We also found increased myocardial damage
when isolated rat hearts were challenged with an
induced right ventricular hypertrophy (physiologic).29–32 ischemic insult (representing a simulated heart attack).
Relatively few studies have separately examined the In agreement, isolated heart mitochondria from predia-
metabolism and function of the right and left ventricles betic rats displayed decreased mitochondrial respiration
in response to hypertrophy. This model also allowed us in response to acute oxygen deprivation. In a separate
to separately examine the effects of hypoxia per se on study, we also found attenuated mitochondrial respira-
the heart, namely by focusing on the non hypertrophied tory capacity in a mouse model of type 1 diabetes.43
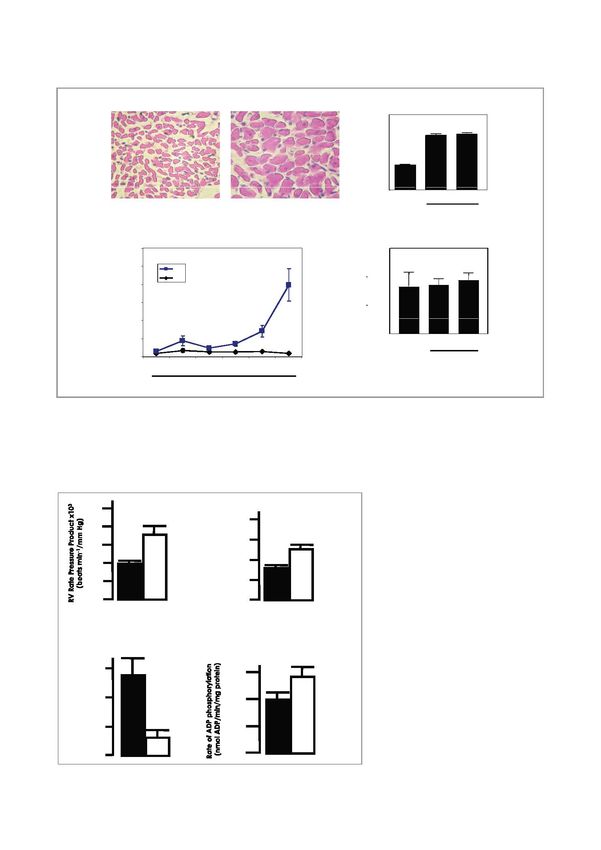
10a) RV myocyte diameter a) RV myocyte diameter
0.08
** **
0.07
0.06
0 05
0.05
mm
m
0.04
0.03
0.02
0.01
0.00
Normoxia 7 days 14 days
Normoxia Hypoxia (14 days) Hypoxia
b) IIncreased
d ANP gene expression
i iin RV c)) N
No fib
fibrosis
i iin RV (Si
(Sirius
i R Red)
d)
1.2 30000
ANP mRNA/18S rRNA
1 RV
nits
arbitrary un
LV 20000
0.8 **
*
0.6
10000
0.4 **
0
0.2
Normoxia 7 days 14 days
0 Hypoxia
0 hrs 12 hrs 24 hrs 2 days 7 days 14 days
Duration of hypoxia
Figure 2: Robust hypertrophic response in right ventricles exposed to hypobaric hypoxia
Male Wistar rats were exposed to 14 days of hypobaric hypoxia (45 kPa, 11% O2), and the degree of right ventricular
(RV) cardiac hypertrophy was assessed versus normoxic controls. Histologic analysis of RV heart cells revealed markedly
increased cell size – refer a) (magnification 40 x). Moreover, gene expression levels of atrial natriuretic peptide (ANP),
a well-known marker for cardiac hypertrophy, increased significantly after 14 days – refer b). Sirius Red staining of heart
tissues detected no fibrosis in the hypertrophied RV. *p < 0.05 vs. matched controls.
20
** 80
RV Developed Prressure
16
Figure 3: Improved heart function
60 ** and mitochondrial respiratory
12
(mmHg)
capacity in the hypertrophied right
8 40 ventricle
20 After 14 days, hearts were isolated and
4
ex vivo Langendorff perfusions performed
0 to assess contractile function. Here the
hypertrophied RV displayed increased
function – refer a). In separate experi-
b) Improved mitochondrial function with RV hypertrophy ments, mitochondria were isolated and
purified to evaluate respiratory capacity.
* These data show decreased mitochondrial
6 300
n)
proton leak and increased ADP phos-
(nmolO2/min/mg protein
phorylation – refer b). Thus these
on Leak
4 200 mitochondria become more efficient in
terms of mitochondrial ATP production.
*
Proto
2 100 *p < 0.05 and **p < 0.01 vs. matched
controls, respectively.
0 0
11These data therefore demonstrate that attenuated pression of respiratory chain complex components and
mitochondrial respiratory function and bioenergetic contractile proteins) that contribute to impaired res-
capacity contribute to the onset of decreased cardiac piratory capacity and contractile function observed in
contractile function at baseline and in response to an the diabetic heart.
ischemic insult in the prediabetic state.
The effects of increased free fatty acids
Proteomic studies To further understand how altered circulating fuels may
To gain further insight into the mechanisms responsible alter cardiac metabolism and heart function, we also
for impaired mitochondrial function and contractile focused on the effects of elevated free fatty acids usually
dysfunction, we examined alterations in the mitochon- found during the prediabetic and the diabetic state. We
drial proteome with the onset of type 2 diabetes. We determined whether cardiac efficiency, that is the ratio
employed the db/db transgenic mouse model that is of cardiac work to myocardial oxygen consumption
characterized by deficient leptin receptor activity, which (MVO2), is diminished in diabetic hearts. Our data
subsequently leads to obesity and the onset of type 2 showed decreased cardiac efficiency in db/db hearts,
diabetes. The db/db mice display hyperphagia (overeating) namely an 86% increase in myocardial oxygen consump-
since the fat-specific hormone leptin is now unable to tion (increased mitochondrial respiration and cardiac
perform its usual function, in other words, acting on lep- fatty acid β-oxidation).43 The extra oxygen cost of
tin receptors in the hypothalamus to suppress appetite. increased fatty acid β-oxidation relative to glucose oxi-
We employed 2D-polyacrylamide gel electrophoresis dation will, however, make a minor contribution since
(PAGE) studies and found several differences between there is only a theoretical 11% decrease in the efficiency
control and obese hearts that broadly fall into two of hearts shifting from complete glucose oxidation to
categories, namely related to energy metabolism and complete fatty acid β-oxidation. We therefore propose
related to contraction/cytoskeleton, respectively.44 that intracellular futile metabolic cycles and upregu-
Here we found a significant decrease in peptide levels of lation of mitochondrial uncoupling proteins (induced by
ubiquinol cytochrome-c reductase core protein 1, a elevated plasma fatty acids) could dissipate the proton
subunit of complex III of the electron transfer chain that gradient across the inner mitochondrial membrane.48
catalyzes transfer of electrons from coenzyme Q to Since an early increase in fatty acid β-oxida tion pre-
cytochrome c. Despite attempts by the obese heart to cedes the ons et of contra ctile dys function s uch pro-
augment mitochondrial ATP production, attenuated nounced ‘oxyg en wa s ta g e’ ma y compromis e hea rt
ubiquinol cytochrome-c reductase core protein 1 pep- func tion when oxyg en dema nd is hig h (e.g . eleva-
tide levels likely contribute to impaired mitochondrial ted workloa ds ) or when oxyg en delivery is limited
ATP production. We also found co-ordinated down- (e.g . a n is chemic ins ult).
regulation of key contractile/cytoskeletal proteins in the
obese heart, namely α-s mooth mus cle a ctin, α-ca r-
diac a ctin, myos in hea vy cha in (MHC)-α a nd MyBP- The effects of elevated glucose levels
C. Thes e peptides pla y a crucia l role to ens ure sus- We are also focusing on the damaging effects of ele-
tained myoca rdia l contra ctile function a nd cyto- vated glucose levels (hyperglycemia), a risk factor that
skeletal s up port. Thes e da ta a re cons is tent with forms part of the metabolic syndrome, on the heart.
previous work tha t found a n MHC is oform s witch There is a growing shift in the paradigm for the role of
during the ons et of dia betes , na mely decrea s ed dietary macronutrient composition in the incidences of
MHCα a nd increa s ed MHCβ expres s ion. 45 MyBP-C CVD, as studies show that there is no reduction in CVD
is a thick filament-associated protein and provides an with a low-fat/high-carbohydrate diet but there is rather
additional regulatory step to myocardial contraction. reduced risk with a diet low in sugar and rapidly ab-
MyBP-C gene mutations can cause hypertrophic sorbed starches and high in polyunsaturated fatty
cardiomyopathy46 while its absence (cMyBP-C null acids.49,50 Moreover, there is increasing evidence from
mice) significantly attenuates in vivo left ventricular animal studies showing that a diet with a high glycemic
function.47 We therefore propose that the prediabetic load, typical of highly processed foods, accelerates the
milieu elicits transcriptional changes (decreased ex- development and progression of CVD50 (see Figure 4).
12Diet rich in sugars,
high glycemic starch Hypertension
& saturated fat Figure 4: Flow diagram
depicting the interaction
between a ‘junk food’ diet,
Obesity obesity and hypertension
The diagram depicts an evolving
paradigm characterizing the
Myocardial pathology effects of diet on left ventricular
remodelling. A diet high in sugar
pathophysiologic cardiac hypertrophy and saturated fats promotes
↑ glucose uptake & ↓ mitochondrial function myocardial dysfunction through
↑ oxidative damage hypertension-dependent and
↑ hexosamine biosynthetic pathway flux independent pathways (adapted
from 50).
↑ cardiomyocyte apoptosis
Heart failure
Glutamine
Figure 5: The hexosamine
biosynthetic pathway (HBP)
Gl
Glucose Gl
Glucose-6-P
6 P F
Fructose-6-P
t 6 P Gl
Glucosamine-6-P
i 6 P Production of glucosamine-6-
GFAT phosphate is catalyzed by
glutamine: fructose-6-phosphate
amidotransferase (GFAT).
Glucosamine 6-phosphate is
UDP ultimately converted to UDP-N-
UDP GlcNAc
UDP-GlcNAc acetylglucosamine (UDP-
O OGT GlcNAc), the HBP’s end
GlcNAc product. O-GlcNAc transferase
protein (OGT) catalyzes the O-linked
protein OGA
transfer of GlcNAc to target
proteins while O-GlcNAcase
GlcNAc (OGA) catalyzes the reverse
reaction.
Our major focus is on the detrimental effects of in- a series of reactions, glucosamine 6-phosphate is ulti-
creased activation of a glucose-based metabolic path- mately converted to UDP-N-acetylglucosamine (UDP-
way, the hexosamine biosynthetic pathway (HBP). The GlcNAc), the end product of the HBP. UDP-GlcNAc is
HBP usually acts as a ‘fuel sensor’ under normal condi- a substrate for O-GlcNAc transferase (OGT) that cata-
tions, in other words, sensing glucose and free fatty acid lyzes the O-linked transfer of GlcNAc to specific serine/
availability and repartitioning fuel substrates into
threonine residues of target proteins. Conversely, O-
suitable storage depots within the body (reviewed in
GlcNAcase (OGA) catalyzes the reverse reaction,
51 and 52). After uptake, glucose is rapidly converted
namely the removal of O-GlcNAc residues from target
to glucose-6-phosphate and thereafter to fructose-6-
phosphate. Fructose-6-phosphate is the entry point for proteins. O-GlcNAc modification is implicated in a di-
the HBP, forming glucosamine-6-phosphate. Production verse range of intracellular reactions and pathways, in-
of glucosamine-6-phosphate is catalyzed by glutamine: cluding altering protein degradation and intracellular
fructose-6-phosphate amidotransferase (GFAT), the localization, modulating protein-protein interactions and
rate-limiting enzyme of the HBP (see Figure 5). Through regulating transcriptional mechanisms.51,52
13Figure 6: Working model
BAD showing how increased
Hyperglycemia Oxidative stress HBP
BAD O-GlcNAcylation
O-GlcNAc
may trigger apoptosis
Hyperglycemia-induced HBP
flux results in increased
O-GlcNAcylation of BAD and
decreased BAD phosphory-
BAD
BAD O-GlcNAc
Bax Bcl-2
Bcl-2 lation (inactive state). Thus
increased ‘active’ BAD levels
lead to greater dimerization of
BAD P O-GlcNAcylated BAD and
Bcl-2. This, in turn, results in
Bcl-2
Bcl-2
Bcl 2 Bcl-2
Bcl
Bcl 2
Bcl-2 Inactive
reduced Bcl-2 availability and
Bax BAD -
O-GlcNAc
less dimerization with Bax.
Free Bax are now able to form
Bax
Bax Apoptosis homodimers, leading to
Bax disruption of the mitochondrial
Disruption of mitochondrial membrane membrane, thereby inducing
apoptosis.
However, we propose that chronically activated HBP creased O-GlcNAcylation of cardiac apoptotic (e.g.
flux is maladaptive and can contribute to pathophysio- BAD) and insulin signaling proteins, thereby leading to
logic phenotypes. For example, our laboratory recently cell death and impaired insulin signaling, respectively.
delineated a novel signaling pathway whereby hyper- This, in turn, will accelerate the development and pro-
glycemia triggers oxidative stress, the HBP and cell gression of insulin resistance and CVD. For example, we
death (apoptosis) in heart cells and in insulin-resistant currently hypothesize that hyperglycemia-mediated ele-
rats.53,54 We also found increased O-GlcNAcylation of vation of HBP flux increases O-GlcNAcylation of regu-
an apoptotic peptide, BAD, together with higher BAD- lators of the insulin signaling pathway (PKB/Akt, AS160),
Bcl-2 dimer formation (pro-apoptotic). We propose thereby impairing its function. Here our data show that
that O-GlcNAcylation maintains BAD in its active form, increased O-GlcNAcylation of PKB/Akt and AS160 is
in other words, dimerizing with Bcl-2 and resulting in associated with decreased GLUT4 translocation to the
increased apoptosis (see Figure 6). Previous work sarcolemma (unpublished data). Furthermore, our
showed that a ‘yin-yang’ relationship may prevail be- current work also demonstrates that HBP inhibition
tween phosphorylation and O-GlcNAcylation whereby significantly blunts the hyperglycemia-induced decrease
they compete for the same site or closely located sites in heart function usually observed in response to ische-
on target proteins.55 Hence with increased HBP flux, we mia-reperfusion (unpublished data).
found greater O-GlcNAcylation of BAD and reduced Together our studies show that high circulating levels
phosphorylation. We propose that O-GlcNAcylation of of fuel substrates (glucose and fatty acids) lead to
BAD occurs in the near vicinity or at the same site(s) as damaging effects on the heart’s function at baseline and
BAD phosphorylation, resulting in reduced access to in response to ischemia-reperfusion. Moreover, these
upstream kinases. data have identified several therapeutic targets that may
We are of the opinion that increased cell death will help alleviate heart diseases that occur in diabetic pa-
lead to impaired cardiac contractile function and tients, for example how to limit free fatty acid-induced
eventually result in ischemic heart disease and/or heart ‘oxygen wastage’ and attenuated mitochondrial ATP
failure. Our postulate is that poor nutritional choices production, or finding ways to decrease HBP activation
(high glycemic diet) activate the HBP leading to in- and its contribution to the onset of insulin resistance
14and CVD. In light of this, our work during the last few functioning of the heart, muscles and nerves.56 Defi-
years has focused on developing novel therapies to ciencies in the B-series vitamins are among the key
blunt the effects of chronic HBP activation and its causative factors leading to diabetic organ damage.57
downstream effects, namely onset of insulin resistance Hence, thiamine and its lipophilic analogue benfotiamine
and cardiac dysfunction. can reduce some of the complications associated with
diabetes, for instance cardiomyopathy, retinopathy and
THE DEVELOPMENT OF neuropathy.58 Since benfotiamine can decrease HBP
flux, we hypothesized that it is a cardioprotective agent.
METABOLIC THERAPIES Our preliminary data show that benfotiamine treatment
T he development of novel therapies to treat cardio-
metabolic diseases usually takes a considerable
length of time, sometimes decades. This route usually
blunts the damaging effects of hyperglycemia and sig-
nificantly improves the heart’s functional recovery in
response to ischemia-reperfusion (unpublished data).
originates with conception of the original idea, basic ex- Thus these data demonstrate that benfotiamine is a pro-
perimentation and further evaluation in smaller and mising cardioprotective agent that may ultimately bene-
larger clinical settings. When criteria of safety are satis- fit pre- and full-blown diabetic patients suffering from
factorily met, drugs are finally approved for treatment cardiovascular disease complications.
of various illnesses. However, unexpected side-effects
We also tested whether oleanolic acid (a clove ex-
can sometimes occur after chronic treatment with spe-
tract) possesses antioxidant properties. Our in vitro data
cific drugs. For example, although antiretroviral treat-
demonstrated that oleanolic acid blunted hypergly-
ment dramatically decreases HIV/AIDS morbidity and
cemia-induced oxidative stress and apoptosis in heart
mortality, the long-term effects may include metabolic
cells (unpublished data). It also markedly improved func-
derangements and the onset of CVD. This example de-
tional recovery of hearts perfused in response to
monstrates the limitation of the reductionist approach
to biomedical science. As a result, additional studies are ischemia-reperfusion under hyperglycemic conditions.
required to effectively treat associated cardio-metabolic We also performed studies on oxygen lack (hypoxia)
diseases associated with chronic antiretroviral treat- as a cardioprotective strategy. We propose that a mo-
ment. Our laboratory hypothesized that protease inhi- derate lack of oxygen (physiologic hypoxia) triggers
bitor treatment elevates oxidative stress that attenuates protective signaling pathways, unlike a severe impair-
contractile function at baseline and in response to myo- ment of oxygen supply (pathophysiologic hypoxia) that
cardial ischemia. We are currently investigating this may exceed the host organism’s defense apparatus, re-
postulate by establishing a unique rat model of chronic sulting in a maladaptive cardiac phenotype (reviewed in
protease inhibitor utilization (lopinavir/ritonavir treat- 38). Physiologic hypoxia triggers various defense mecha-
ment for eight weeks). Here our data show that pro- nisms, for example erythropoiesis and angiogenesis, to
tease inhibitor-treated rats exhibited increased body increase red blood cell mass and oxygen delivery to the
weights and low-density lipoprotein cholesterol levels. heart. Here we propose that physiologic hypoxia acti-
We also found attenuated contractile function at base- vates regulatory pathways that mediate long-term
line following eight weeks of protease inhibitor treat- cardiac metabolic remodeling, particularly at the
ment and increased myocardial cell death (unpublished transcriptional level. The proposal is that physiologic re-
data). Decreased contractile function persisted during active oxygen species (ROS) play a central role by
the reperfusion period (after an experimentally induced modulating the activity of redox-sensitive transcription
ischemic insult). We are currently testing inhibitors of factors to induce a fetal gene program (increased carbo-
maladaptive pathways that can eventually be coadminis- hydrate and decreased fatty acid metabolism) and
tered with antiretroviral drugs. increased mitochondrial biogenesis in the heart.38 These
In light of these difficulties, we have focused on programs will thus increase the efficiency of energy pro-
natural agents as a potential therapeutic strategy since duction and augment mitochondrial bioenergetic capacity
this may result in a more rapid translation into the clinic to sustain cardiac contractile function. Collectively
and also prove to be more cost-effective, especially these studies show promise, and the next step would be
within the developing world context. Previous work to translate some of our findings into the clinic, in other
demonstrated that diabetes results in decreased levels words, small pilot studies.
of thiamine (Vitamin B1), which is essential for normal
15Normal Pre-diabetic Diabetic
Figure 7: Increased HBP activation parallels fasting glucose levels
Leukocytes stained for O-GlcNAc (Texas red) and cell nuclei (Hoechst blue dye) demonstrate increased red staining
(O-GlcNAc) for prediabetic and diabetic individuals.
THE DEVELOPMENT OF dable morbidity and mortality.’’ Thus there is a need for
DIAGNOSTIC TOOLS FOR the development of novel diagnostic tools to detect
prediabetes and diabetes in a timeous, sensitive and
CLINICAL APPLICATION cost-effective manner.
W e are also pursuing the development of novel
diagnostic tools to allow for more sensitive and
earlier detection of cardio-metabolic diseases. Although
Although rapid protein O-GlcNAcylation occurs
within white blood cells,60 there is no literature regar-
ding O-GlcNAcylated leukocyte proteins within the pre-
there are currently tests available to screen for the
diabetic setting. In light of this, we hypothesized that
onset of diabetes, for example glycosylated hemoglobin
there is increased oxidative stress and O-GlcNAcylation
(HbA1c) levels, oral glucose tolerance and fasting blood
in white blood cells in the prediabetic milieu and that
glucose levels tests, these methods have limitations. For
example, Saudek et al59 propose that HbA1c is not this may offer a novel diagnostic tool to predict the
always an effective diagnostic tool to identify pre- onset of insulin resistance. Here our data (flow cyto-
diabetes. At the end of their article the authors state, metry and fluorescence microscopy generated) show
“There are serious deficiencies in the current criteria that O-GlcNAcylation of leukocyte proteins changes in
for diagnosing diabetes, including the requirement that parallel with increasing fasting glucose levels (see Figure
the patient be fasting, and the lack of agreed-on 7) (unpublished data). The data show early promise for
screening criteria. These deficiencies make it unneces- the establishment of a sensitive diagnostic test that
sarily inconvenient for clinicians to diagnose diabetes, may help improve the detection and management of
thereby delaying the diagnosis and contributing to avoi- type 2 diabetes.
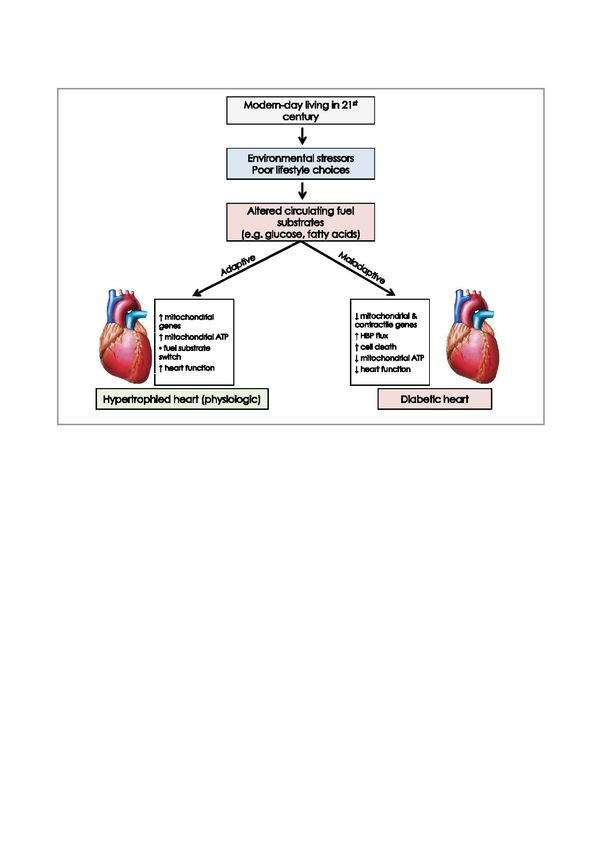
16Modem-day riVing i1211t
centuy
Environmental s1ressors
Poor lifes1yle choices
t mitochondrial l mtochonct1al &
genes co"*actte genes
t mitochondrial AlP jtiiPfUc
o fuei5Ubsl!afe tcelldeath
switch l mitochondrial AlP
theartfuncnon l heart funclton
1 Hyperfrophled heart (physiologic, 1 Dlabeflc heat
Figure 8: The impact of external and internal challenges on the heart’s metabolism
We are of the opinion that markers of the HBP may result in novel diagnostic tools to assess prediabetes, while HBP
inhibition within the prediabetic context may also provide cardioprotection.
HEART METABOLISM’S to successfully adapt, for example increased mitochon-
drial respiratory capacity that sustains the physiologic
BALANCING ACT: QUO VADIS? hypertrophied heart. However, it is hard to continue its
T reading the tightrope of healthy living in the 21st
century is an arduous task. The global economy is
characterized by great wealth disparities with serious
balancing act(s) in the long-term. Chronically high
circulating fuel substrates (e.g. glucose and fatty acids)
trigger maladaptive pathways that result in cell death,
health implications. Moreover, the prevailing economic decreased mitochondrial ATP production and the onset
model struggles to promote equity, efficiency and en- of type 2 diabetes and CVD. Our research work has
vironmental sustainability. Also, production for profit identified several mechanisms whereby high circulating
and related consumerism translate into an environ- fatty acid and glucose levels lead to impaired cardiac
mental imbalance in terms of healthy living, for example contractility (see Figure 8). Moreover, we have dis-
pollution, stress, nutritional excess and sedentary life- covered several molecular targets that can be exploited
styles. Communities are therefore exposed to an in- for diagnostic and therapeutic purposes. If future trans-
creasing number of external stressors that can trigger lational studies are successful, this should ultimately
internal changes, for example altered myocardial fuel improve overall quality of life and well-being and help
substrate metabolism. diminish the growing global burden of type 2 diabetes
It therefore becomes a daunting balancing act for the and CVD. However, it is crucial that such efforts go
heart’s metabolism to sustain its energetic production together with the development of a more integrative
and output in the midst of prolonged external and scientific research approach and a better balanced eco-
internal challenges. In some instances the heart is able nomic framework.
17REFERENCES
1. Silverthorn DU. Human Physiology: an from mechanistic studies. Lancet
integrated approach. 4th edition. San Francisco, 2008;371:1800–9.
CA: Benjamin Cummings; 2007. 13. Zimmet P, Alberti KGMM, Kaufman F, et al.
2. Human Physiology/Homeostasis [Wikibook on The metabolic syndrome in children and
the Internet]. No date [updated 2011, April 25; adolescents. Lancet 2007;369:2059–61.
cited 2011, July 26]. Available from 14. Alberti KGMM, Zimmet P, Shaw J. The
http://en.wikibooks.org/wiki/Human_Physiology/ metabolic syndrome – a new worldwide
Homeostasis definition. Lancet 2005;366:1059–62.
3. Smith RD. Avicenna and the Canon of 15. Smith C, Essop MF. Gender differences in
Medicine: a millennial tribute. West J Med metabolic risk factor prevalence in a South
1980;133:367–70. African student population. Cardiovasc J Afr
4. Sherwani AMK, Navaz M, Ansari AN, Ramesh 2009;20:178–82.
M. Nazla – a well-understood phenomenon of 16. Lenfant C. Clinical research to clinical practice
Arabs misinterpreted by successors. J Int Soc – lost in translation? N Engl J Med
Hist Islamic Med 2006;5:7–10. 2003;349:868–74.
5. Murray CJ, Lopez AD. The global burden of 17. Joyner MJ. Giant sucking sound: can physiology
disease: a comprehensive assessment of fill the intellectual void left by reductionists?
mortality and disability from diseases, injuries, J Appl Physiol 2011;111:335–42.
and risk factors in 1990 and projected to 2020.
18. Wagner PD, Paterson DJ. Physiology: found in
Cambridge, MA: Harvard Press; 2009.
translation. Am J Physiol Endocrinol Metab
6. Leeder S, Raymond S, Greenberg H, Liu H, 2011;301:E427–8.
Esson K. A race against time: the challenge of
19. Opie LH. Heart physiology from cell to
cardiovascular disease in developing economies.
circulation. 4th edition. Philadelphia, PA:
New York: Trustees of Columbia University in
Lippincott Williams & Wilkins; 2004.
the City of New York; 2004.
20. McGarry JD, Brown NF. The mitochondrial
7. Yusuf S, Reddy S, Ounpuu S, Anand S. Global
carnitine palmitoyltransferase system: from
burden of cardiovascular diseases: Part I:
concept to molecular analysis. Eur J Biochem
general considerations, the epidemiologic
1997;244:1–14.
transition, risk factors, and impact of
urbanization. Circulation 2001;104:2746–53. 21. Abu-Elheiga L, Jayakumar A, Baldini A, Chirala
SS, Wakil SJ. Human acetyl-CoA carboxylase:
8. Fezeu L, Balkau B, Kengne A, Sobngwi E,
characterization, molecular cloning, and
Mbanya J. Metabolic syndrome in a sub-Saharan
evidence for two isoforms. Proc Natl Acad Sci
African setting: central obesity may be the key
USA 1995;92:4011–5.
determinant. Atherosclerosis 2007;193:70–6.
22. Abu-Elheiga L, Almarza-Ortega DB, Baldini A,
9. Mayosi BM, Flisher AJ, Lalloo UG, Sitas F,
Wakil SJ. Human acetyl-CoA carboxylase 2,
Tollman SM, Bradshaw D. The burden of non-
Molecular cloning, characterization,
communicable diseases in Africa. Lancet chromosomal mapping, and evidence for two
2009;374:934–47. isoforms. J Biol Chem 1997;272:10669–77.
10. Reardon S. A world of chronic disease. Science 23. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala
2011;333:558–9. SS, Woldegiorgis G, Wakil SJ. The subcellular
11. King H, Aubert R, Herman W. Global burden localization of acetyl-CoA carboxylase 2. Proc
of diabetes, 1995–2025: prevalence, numerical Natl Acad Sci USA 2000;97:1444–9.
estimates and projections. Diabetes Care 24. Essop MF, Camp HS, Choi CS, et al. Reduced
1998;21:1414–31. heart size and increased myocardial fuel
12. Mazzone T, Chait A, Plutzky J. Cardiovascular substrate oxidation in ACC2 mutant mice. Am
disease risk in type 2 diabetes mellitus: insights J Physiol Heart Circ Physiol 2008;295:H256–65.
18You can also read

















































