Galaxy InteractoMIX: An Integrated Computational Platform for the Study of Protein-Protein Interaction Data
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Database
Galaxy InteractoMIX: An Integrated
Computational Platform for the
Study of Protein–Protein
Interaction Data
Patricia Mirela-Bota 1,2, Joaquim Aguirre-Plans 2, Alberto Meseguer 2,
Cristiano Galletti 1, Joan Segura 3, Joan Planas-Iglesias 4, Javi Garcia-Garcia 2,
Emre Guney 5, Baldo Oliva 2,⇑ and Narcis Fernandez-Fuentes 1,6,⇑⇑
1 - Department of Biosciences, U Science Tech, Universitat de Vic-Universitat Central de Catalunya, Vic, Catalonia 08500, Spain
2 - Structural Bioinformatics Group, Research Programme on Biomedical Informatics, Department of Experimental and Health
Sciences, Universitat Pompeu Fabra, Barcelona, Catalonia 08003, Spain
3 - Research Collaboratory for Structural Bioinformatics, Protein Data Bank, San Diego Supercomputer Center, University
of California, San Diego, La Jolla, CA 92093, USA
4 - Loschmidt Laboratories, Department of Experimental Biology, Faculty of Science, Masaryk University, Kamenice 5/a13, 625
00 Brno, Czech Republic
5 - Integrative Biomedical Informatics Group, Research Programme on Biomedical Informatics, Hospital del Mar
Medical Research Institute, Department of Experimental and Health Sciences, Universitat Pompeu Fabra, Barcelona,
Catalonia 08003, Spain
6 - Institute of Biological, Environmental and Rural Sciences, Aberystwyth University, SY233EB Aberystwyth, United Kingdom
Correspondence to Baldo Oliva and Narcis Fernandez-uentes: Department of Biosciences, U Science Tech,
Universitat de Vic-Universitat Central de Catalunya, Vic, Catalonia 08500, Spain. baldo.oliva@upf.edu, narcis.
fernandez @gmail.com
https://doi.org/10.1016/j.jmb.2020.09.015
Edited by Michael Sternberg
Abstract
Protein interactions play a crucial role among the different functions of a cell and are central to our under-
standing of cellular processes both in health and disease. Here we present Galaxy InteractoMIX (http://ga-
laxy.interactomix.com), a platform composed of 13 different computational tools each addressing specific
aspects of the study of protein–protein interactions, ranging from large-scale cross-species protein-wide
interactomes to atomic resolution level of protein complexes. Galaxy InteractoMIX provides an intuitive
interface where users can retrieve consolidated interactomics data distributed across several databases
or uncover links between diseases and genes by analyzing the interactomes underlying these diseases.
The platform makes possible large-scale prediction and curation protein interactions using the conserva-
tion of motifs, interology, or presence or absence of key sequence signatures. The range of structure-
based tools includes modeling and analysis of protein complexes, delineation of interfaces and the mod-
eling of peptides acting as inhibitors of protein–protein interactions. Galaxy InteractoMIX includes a range
of ready-to-use workflows to run complex analyses requiring minimal intervention by users. The potential
range of applications of the platform covers different aspects of life science, biomedicine, biotechnology
and drug discovery where protein associations are studied.
Ó 2020 Elsevier Ltd. All rights reserved.
Introduction humans, or protein–protein interactions (PPIs) in
cells at molecular level. Indeed, PPIs are central
Interactions are the basis of life, be it gravitational to all cellular processes and so the charting and
forces in the universe, social interactions between description of the network of PPIs, or the
0022-2836/Ó 2020 Elsevier Ltd. All rights reserved. Journal of Molecular Biology 433 (2021) 166656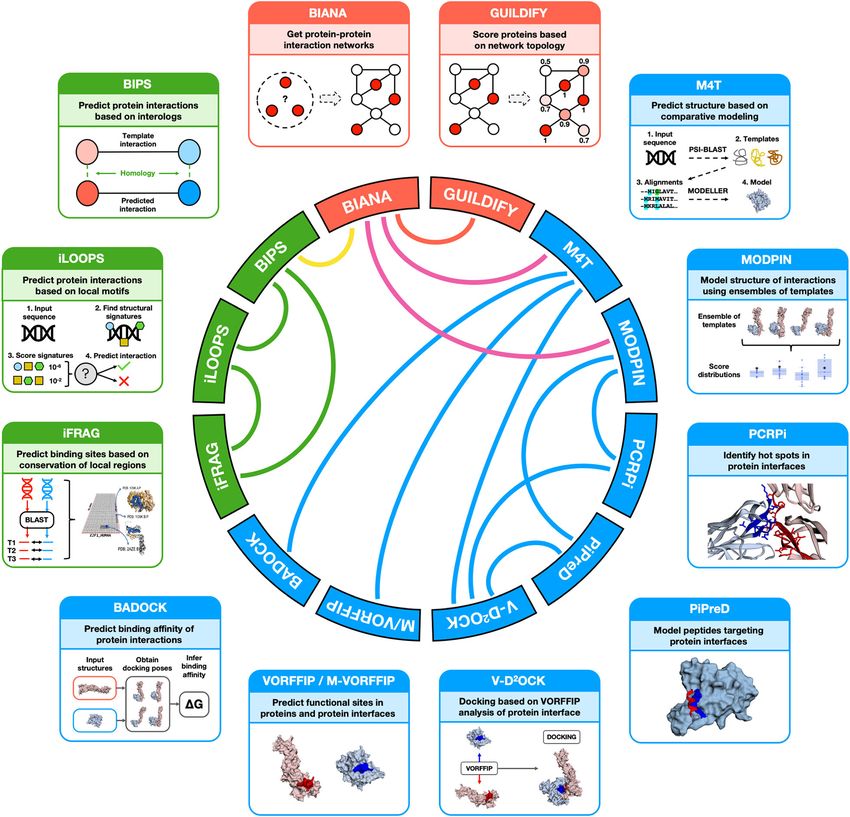
P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
interactome, is a key aspect to fully understand the that Galaxy InteractoMIX will be of interest to a wide
inner workings of cells. The so-called post-genomic target audience in life science and biomedicine but
era was ushered by the advent of high-throughput also in biotechnology and drug discovery.
technologies that represented an exponential
increase of experimental information on PPIs, or
interactomics, but computational tools have still an Results and Discussion
important role to play at different levels.
At the level of data management and integration, Galaxy, a common framework to interface
computational tools and resources
multiple large-scale projects aimed at the discovery
of PPIs in mainly model organisms, sparkled the The types of computational analyses that can be
development of range databases to archive the performed on interactomics data are very wide,
growing body of interactomics data [1–7]. This fact thus requiring access to a range of different
presents the problem of data integration and cura- tools. From a user point of view, and particularly
tion among the different databases. Experimental non-expert users, access and use of
data are also intrinsically noisy, having high error computational tools can present a number of
rates [8], and finally the completeness of the human challenges. Tools might be distributed in different
interactome to date is still low; i.e. there are still a forms: source code, stand-alone applications or
large number of PPIs that are predicted to exist web servers and having different interfaces, i.e.
and have not been identified experimentally [9]. web-based, command line or through a GUI.
These limitations together with the need of holistic Issues of reproducibility, reusability and sharing
and multiscale analyses of protein networks justi- become complicated when requiring complex
fied the use of the computational platform pre- analyses and combinations of different tools
sented here: Galaxy InteractoMIX. (discussed later on).
Galaxy InteractoMIX currently comprises 13 The Galaxy Project, an open source, web-based
different computational tools each addressing a platform for data intensive biomedical research
specific aspect of the study of interactomics data. represents a convenient solution to tackle these
From a very high-level, Galaxy InteractoMIX problems [10]. The overarching goals of Galaxy
addresses the integration and consolidation of project are to provide accessible data analyses
interactomics information shared between serving all scientists regardless of their expertise
databases or in understanding the links between as well as a platform for developers to make tools
diseases and genes, or genotype-to-phenotype available to the wider audience. Firstly, the interac-
associations, by studying underlying protein– tion with any tool implemented in Galaxy is through
protein interactomes. For the prediction of PPIs at the web with underlying computational infrastruc-
genome-wide level Galaxy InteractoMIX uses ture being transparent to users. Galaxy allows
different approaches: conservation of motifs, uploading and storing data, and thus, its handling,
interology or presence/absence of key signatures. exchange and use/reuse between tools are more
Galaxy InteractoMIX provides a range of flexible and easier from a user point of view. Repro-
structure-based tools as well as structure ducibility is also a strong aspect of Galaxy platform
modeling capabilities either by comparative as job runs get archived in users’ history and can be
homology or docking. Finally, structural-based shared and reused as needed.
modeling of peptide inhibitors to target PPIs is Over the years, we developed a range of tools,
also among the repertoire offered by Galaxy which will be described briefly below, that were
InteractoMIX. compiled in a single portal: InteractoMIX [11].
Besides the wide range and breadth of the tools Albeit valuable as a common portal, tools
included in Galaxy InteractoMIX, the unique and remained disconnected, only working as individual
key point is its integration within the Galaxy applications. We have now integrated the various
platform [10]. Running under a common framework tools of InteractoMIX within the Galaxy Project
such Galaxy facilitates the use in a truly integrated, framework creating the Galaxy InteractoMIX plat-
one stop, manner. Galaxy uncouples data and form. The current Galaxy InteractoMIX platform
applications in the way that users can upload and is composed of 13 different tools allowing network,
store data, and thus, the exchange and use/reuse sequence- and structure-based data analyses
of data between tools are more agile, transparent (Figure 1). Moreover, these tools can be con-
and flexible (e.g. some tools outputs can be used nected between themselves as the type of analy-
as inputs for others and vice versa). Galaxy Inter- sis performed individually can be complemented
actoMIX also features a range of workflows each or because inputs and outputs are related (Fig-
addressing a different set of analyses that otherwise ure 1). This interdependence between the different
would need to be done manually and individually. computational tools highlights the strong and key
Besides, these tools being part of Galaxy, users aspect of Galaxy InteractoMIX as a coherent plat-
can easily develop their own workflows using a sim- form for integration. Thus, Galaxy InteractoMIX
ple graphical interface (drag-and-drop). We foresee provides a single-stop portal to access these tools
2
P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
Figure 1. Schematic depiction of the different tools implemented in Galaxy InteractoMIX. Tools are color coded as
follow: network-, sequence-, and structure-based analyses are shown in orange, green and light blue, respectively.
The CIRCOS plot shown in the center illustrates the different interrelationships between the different tools.
but also a platform for their integration. Moreover, BIANA: integration of biological entities
we capitalize on all different advantages that
Galaxy Project offers as a platform. BIANA is a platform to integrate biological
interaction data into a unique knowledge database
[12]. The platform facilitates the integration of bio-
logical elements (e.g. genes, proteins, drugs, dis-
Tools in Galaxy InteractoMIX: integration of
eases, phenotypes, etc.) gathered from different
interactomics data and network-based
databases and assigned with distinct identifiers into
analyses
a database where they are assigned a unique
At the genome-wide level, Galaxy InteractoMIX BIANA identifier. BIANA provides solutions to com-
provides the resources to handle both the mon annotation errors due to human and experi-
integration of interactomics information, BIANA mental slips. BIANA presents a unification
[12], and a network-based tool to predict genes protocol, allowing for crosschecking and
associated with diseases, GUILDify [13,14]. traceability.
3P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
Within the Galaxy InteractoMIX platform, BIANA Tools in Galaxy InteractoMIX: sequence-based
can be accessed to retrieve PPIs associated with tools included in InteractoMIX galaxy
specific species. Currently, BIANA contains the
interactomes of seven model species: Homo There are currently three different sequence-
sapiens, Mus musculus, Rattus novergicus, Danio based tools in Galaxy InteractoMIX (Table 1). The
renio, Drosophila melanogaster, Saccharomyces goal of these tools is the prediction of PPIs each
cerevisiae and Arabidopsis thaliana. Upon presenting their unique features complimenting
selecting the protein targets of interest, specie and each other; while iLoops [15,16] and BIPS [17] pre-
the radius of expansion (e.g. 1 would imply a dict PPIs by pairs or within proteomes, i.e. a yes/no
direct interaction to any targets of interest), BIANA answer based on quantitative scores; iFrag [18] pre-
will return the associated subnetwork in the format dicts the likely binding region(s), i.e. interface, in the
of choice. sequences.
BIPS: prediction of PPIs based on interologs
GUILDify: network-based prioritization of BIPS is a sequence-based predictor of PPI based
genes associated with diseases on interologs [17]. Interolog predictions use the con-
cept of conservation of PPIs across organisms by
GUILDify is a method to extend the information of which two proteins in an organism are predicted to
sets of proteins through the PPI network compiled in interact if their corresponding close homologs in a
BIANA (Figure 1) [13,14]. GUILDify scores proteins different organism are known to interact. BIPS is
according to their proximity with proteins encoded able to handle entire proteomes by comparing to
by genes associated with a phenotype (called the existing interactomes compiled in BIANA [12].
seeds). GUILDify uses four different network- Within Galaxy InteractoMIX, the requirements to
based prioritization algorithms: NetShort, NetZcore, execute the applications are a text file with a list of
NetScore and NetCombo. The top-scoring proteins proteins in different formats (FASTA, Uniprot acces-
are predicted to have the same phenotype or dis- sion or gene symbols) and the taxonomy identifica-
ease than the seeds. tion code for the target organism.
Within the Galaxy InteractoMIX platform,
GUILDify has been adapted so that it can be
iLoops: prediction of PPIs based on local
easily used and integrated with the rest of the
motifs
tools. The user has to provide as input the list of
proteins associated with a phenotype (seeds). The iLoops is a machine-learning predictor of PPIs
user can also specify the species associated with based on the presence of domains and local
the set of proteins, the type of scoring function structural features [19] of paired proteins in the sets
and the cutoff threshold used to define the top- of known interactions [12] and in confirmed non-
scoring proteins. In the output, the Galaxy version interactions [20]. A random forest (RF) yields a bin-
of GUILDify returns the scores of all the ary prediction based on the majority class and in
interactome proteins computed by their scores of the statistical prevalence of domains (or
association with the seeds and the subnetwork of local structures) based on information entropy. Sev-
top-scoring proteins. eral RFs were trained and independently validated
Table 1 List of sequence-based tools in Galaxy InteractoMIX
App Purpose Expected inputs Expected outputs Throughput
iLoops A motif-based predictor of A FASTA file with A file in XML format including all the No limit on the number of pairs
PPIs the sequences of prediction and accessory to predict Recommended not
all proteins information more than 100 pairs per round
A text file defining
the pair of
proteins to test
iFrag A predictor of protein The sequences of A text file containing a matrix with all No limit on the number of pairs
binding site based on two proteins in pairs of residues and corresponding to predict
conserved sequence FASTA format scores Recommended not more than
fragments A link to an external server to 10 pairs per round
visualize the results in a graphic
interface
BIPS An interolog-based A FASTA file with A link to an external server to No limit on the number of pairs
predictor of PPIs the sequences of visualize the results in a graphic to predict including whole
all proteins interface with the predicted proteomes
interactions
4P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
on sets with different ratios of interacting and non- M4T: comparative modeling of proteins
interacting pairs (unbalance ratio), providing the
user with the probability that the output prediction M4T is a structure prediction method based on
is true given a ratio of expected interactions that comparative modeling [24,25]. M4T provides struc-
can be acquired or deduced from experimental tural modeling capabilities to the platform, so pro-
knowledge (i.e. by the percentage of expected inter- tein structures can be used to study features of
actions in the same cellular compartment). Further- interest, e.g. mapping of mutations. M4T is also rel-
more, the relative cost of false predictions in the RF evant to other tools on the platform such as
can be adjusted, so more comprehensive (low BADOCK or V-D2OCK (see Figure 1). To execute
costs) or more precise (high costs) predictions can M4T, the only requirement is the sequence of the
be requested. It has to be noted that the binding out- query protein in raw format. M4T uses multiple tem-
put of previously proposed protein design strategies plates and thus several models can be obtained
based on loop-grafting [21] can uniquely be depending on the number of templates used to
assessed by this method. The current implementa- derive the alignment and the coverage between
tion in Galaxy Interactomix requires the list of pro- the query sequence and the templates.
tein pairs to be tested, the sequences of all
proteins in the list, the desired cost for false predic-
tions, and two parameters defining the kind of MODPIN: comparative modeling of protein
domains and groups of structural features to complexes
consider. MODPIN is a tool designed to model the structure
of PPIs and derive a range of statistics on the
scores of the interaction [26]. MODPIN uses com-
iFrag: prediction of PPIs based on local parative modeling to obtain an ensemble of confor-
conservation mations of two interacting proteins by using different
templates. The poses are clustered according to
iFrag is a sequence-based predictor of protein– their common interfaces and scored subsequently
protein interfaces [18]. The basis of iFrag prediction using a range state-of-the-art scoring functions
is the presence of similar local regions shared such as ZRANK [33], FoldX [34], Rosetta [35] and
between the two query proteins and other similar the in-house statistic potentials SSP [36], currently
protein-pairs retrieved by a BLAST [22] search of used to analyze docking poses.
each partner. Rather than relying on the presence There are two different ways to execute MODPIN
of conserved motifs (such iLoops above) or protein within Galaxy InteractoMIX. The first is the
domains [23], iFrag relies on the fragments of com- modeling mode where given a list of protein
mon local alignments. The requirements to use sequences in FASTA format and a list of pairs it
iFrag in Galaxy Interactomix are two files: one with returns the all structural models of the list (when
the sequence in FASTA format and the other with this is possible). In the second mode, MODPIN
the IDs of pairs of proteins to predict in text format. analyses the energies of the complexes produced
The sequence coverage, sequence identity and e- and offers a range of statistics comparing the
value of the BLAST search are adjustable distribution of these scores, which may be useful
parameters. for the study of mutations in interfaces.
Tools in Galaxy InteractoMIX: structure-based BADOCK: prediction binding affinity of
tools included in InteractoMIX galaxy complexes
There is a total of eight tools within Galaxy BADOCK [27] is a method that predicts the bind-
InteractoMIX that are structure-based (Table 2). ing energy of protein complexes using the structure
Two of them, M4T [24,25] and MODPIN [26], are of the unbound monomers, i.e. useful when the
actually structure prediction methods based on structure of the protein complex is unknown but
comparative homology for proteins and protein the individual monomers is. BADOCK relies on the
complexes, respectively. BADOCK [27] predicts study of the docking space of proteins as a proxy
the binding affinity of complexes based on unbound for the study of the formation of the protein complex
protein structures. VORFFIP [28] and M-VORFFIP [37]. Because the structure of the protein complex is
[29] are structure-based methods to predict func- not required to estimate the binding affinity with this
tional sites in proteins. V-D2OCK [30] is a data- method, the range of applicability is largely
driven docking method that includes a clustering expanded. BADOCK is based on a scikit-learn
step to reduce the size of the docking space while regression-based predictor [38] trained on the dock-
preserving the conformational richness. Finally, ing affinity benchmark 2 [39]. The use of BADOCK
PCRPi [31] and PiPreD [32] are methods to predict in Galaxy InteractoMIX is straightforward being
critical residues, or hot spots, in protein interface the only requirement for the coordinates of the two
and to model orthosteric peptides to modulate PPIs, proteins of interest. Its sole output is the predicted
respectively. binding affinity, provided in an XML file.
5P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
Table 2 List of structure-based tools include in Galaxy InteractoMIX
App Purpose Expected inputs Expected outputs Throughput
BADOCK Estimation of binding Coordinates of both A file in XML format with the predicted A single protein
energy between two proteins in standard binding energy complex per
proteins PDB format round
M4T Comparative Sequence of query Coordinates of structural model of the target A single
modeling of proteins protein in FASTA sequence in standard PDB format sequence per
format round
MODPIN Comparative Sequences of query Coordinates of structural models of No limit on the
modeling of protein proteins in FASTA complexes number of pairs
complexes format Energy-based statistics including several to predict
plots Recommended
A text file defining the not more than
pair of proteins 10 pairs per
sequences to model round
VORFFIP Structure-based Coordinates of the Table with scores in tab-delimited format A single protein
predictor of protein protein of interest in structure per
interfaces standard PDB format Original coordinates where B-factor fields round
are substituted by prediction scores
M-VORFFIP Structure-based Coordinates of the 4 different text files in tab delimited from with A single protein
predictor of functional proteins of interest in prediction for DNA, RNA, protein and structure per
sites standard PDB format peptide sites round
4 different files including the original
coordinates where B-factor fields are
substituted by prediction scores (DNA,
RNA, protein and peptide)
V-D2OCK Biased protein– Coordinates of A tar-compressed file containing A single pair of
protein docking proteins to dock in coordinates docking poses. protein
standard PDB format Text file with tab-delimited docking scores structures per
round
PCPRi Structure-based Coordinate of protein A tab-delimited text file with prediction A single protein
predictor of hot spot complex of interest in scores. structure
residues in protein standard PDB format A tab-delimited text file with the atomic complex per
interfaces interaction at the interface. round
A file with remediated coordinates where B-
factor fields are substituted by prediction
scores
PiPreD Structure-based Coordinate of protein A tar-compressed file containing A single protein
modeling of complex of interest in coordinates of modeled orthosteric peptides structure
orthosteric peptides standard PDB format complex per
round
VORFFIP AND M-VORFFIP: prediction of V-D2OCK: prediction of complexes using
functional sites in proteins biased docking
VORFFIP and M-VORFFIP [28,29] are methods Protein docking can be used to derive structural
that predict functional sites in proteins. While models of protein complexes, and this the
VORFFIP predicts protein interfaces, M- functionality of V-D2OCK [30] on the platform. V-
VORFFIP also predicts peptide-, DNA-, and D2OCK is a data-driven protein-docking algorithm.
RNA-binding sites. VORFFIP is also used by V- On the first stage, V-D2COK invokes VORFFIP to
D2OCK, another of the tools present in Galaxy delineate the binding sites in unbound proteins;
InteractoMIX, as the predicted interface(s) is information that is subsequently used to drive the
(are) used to guide and bias the protein docking docking exploration. Resulting docking decoys are
(see next section). The use of both tools within then clustered to reduce the size of the docking
the platform is very simple, as only the coordi- space while preserving conformational diversity.
nates of the protein of interest are needed. The Several scoring functions are available to users to
coordinates can be downloaded from the PDB rank the docking models. Within Galaxy Inter-
databank or local repository, but also obtained actoMIX, users need to upload the coordinates for
with M4T or MODPIN, thus highlighting the inte- the proteins to dock and define clustering tolerance
gration offered by the platform (Figure 1). and the scoring function for ranking. The structural
6P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
models derived from M4T can be also used as provides an online repository to upload several
inputs (Figure 1). workflows.
Beside tailored and user-made, we have
PCRPi: prediction of critical residues in developed seven ready-to-use workflows each
interfaces addressing a set of analyses that otherwise would
need to be done manually by running individual
Not all residues of protein interfaces contribute tools and combining their respective outputs.
equally to the binding energy of a complex, where While the use of individual tools is open, in order
some of them, i.e. hot-spot residues, have the to access the workflow users will need to create
largest contribution [40]. The identification of hot an account in the platform. To do so, users will
spots or critical residues in interfaces is a relevant have to register on the email address provided in
and critical question particularly in drug discovery the main webpage. No personal data are required
and biotechnological applications dealing with PPIs. besides an email address where details on the
PCRPi [31,41] uses a machine-learning classifier username and password will be emailed.
integrating a range of features (conservation, struc- Registered users will have both longer running
tural determinant and energy-based terms) to pre- times and larger disk space quotas in line with the
dict hot spots in protein interfaces. Users need to usual requirement workflows as compare to
provide the coordinates of the protein complex, individual tools. Moreover, users will be able to
the chain identifier of the protein of interest and access the historical record on the platform, such
the type of Bayesian network to execute PCRPi. previous results that are not kept for more than a
As of other tools in the platform, the coordinates week in the case of individual tools.
can be uploaded or re-used from structural models The workflows currently available in in Galaxy
derived by MODPIN, allowing for the discovery of InteractoMIX include those to retrieve information
critical regions in protein interfaces or to understand on explicit pathways and eventually model the
the potential impact of mutations. structure of associated protein complexes;
combined prediction of protein interfaces by
PiPreD: modeling of orthosteric peptides sequence-, structure-based or combined;
PiPreD [32] is a structure- and knowledge-based modeling of protein complexes either by
approach to model the conformation of peptides tar- comparative modeling or docking; structural
geting protein interfaces. The modulation of PPIs is modeling of protein complexes linked to diseases
a very active field where peptides are posed to play including analyses of protein interfaces and
an important role. Given its nature, PiPreD is a fast mutations (with their potential effects); and
method that ensures a systematic and comprehen- eventually the de novo modeling of orthosteric
sive exploration of the entire interface in an unbi- peptides to target a particular PPI. Each of the
ased manner from the point of view of the workflows in Galaxy InteractoMIX is fully
conformation of peptides. PiPreD requires the coor- documented including a tutorial with a step-by-
dinates of a protein complex and specifying the step explanation and test data. These tutorials can
chain IDs of the target (i.e. the specific protein to be accessed through the main web page on the
be targeted by peptides) and mirror protein(s) (i.e. galaxy platform.
other protein(s) of the complex whose native ele- As an example of a workflow, we present a
ments are used to drive the modeling of peptides). combined structure-based analysis of protein
As in other cases, the coordinates of the protein complexes. This workflow utilizes eight tools
complexes can be uploaded or re-used from other included in Galaxy InteractoMIX platform
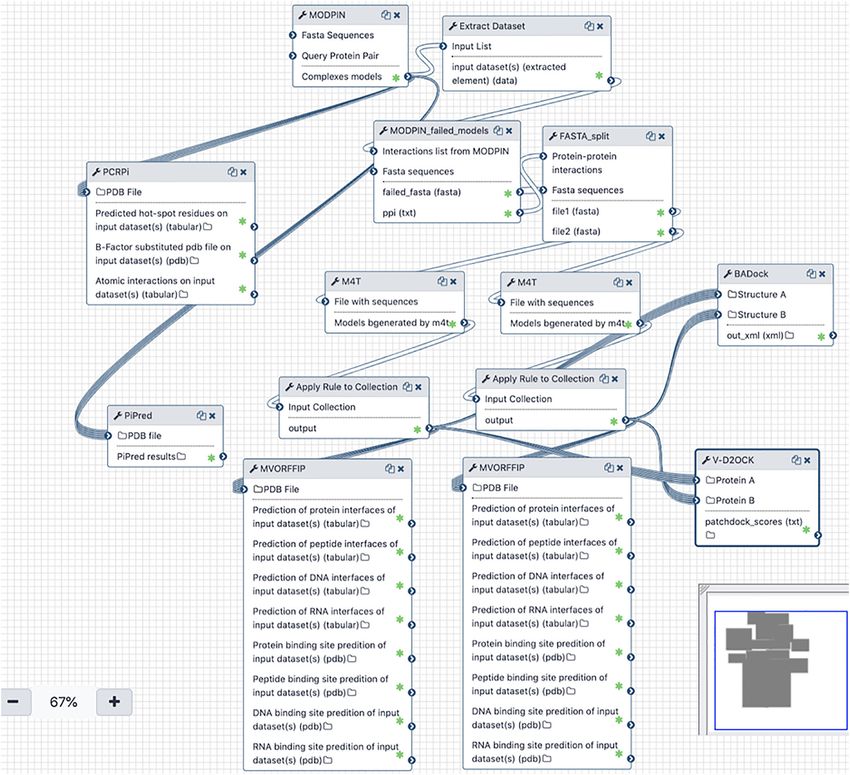
tools, e.g. V-D2OCK or MODPIN (Figure 1). (Figure 2). The workflow starts with a list of
protein pairs and their sequences in FASTA
format. On the first step, it attempts to model the
InteractoMIX galaxy provides ready-to-use
structure of the complexes based on homology
workflows for complex analyses
modeling using MODPIN. For the successful
An obvious advantage of integrating the different complexes, i.e. with at least one suitable template
tools described above in the Galaxy framework is to model the structure of the complex; the
the option of developing computational workflows workflow submits the complexes to both PCRPi
by linking together the tools of the platform. and PiPreD. The former will assess the hot spots
Galaxy provides a simple graphical interface that in protein interfaces, while the latter will model
allows users to combine the different tools using a peptides to target such interfaces.
“drag-and-drop” approach. The tools can be linked For the remaining unmodeled interactions, the
by either reusing the same inputs or by using the workflow attempts to model the individual,
output of one as input for another, which may be unbound, structures using M4T. A range of tools
used as downstream analysis. Once a pipeline is will use these structures as inputs. VORFFIP and
created within Galaxy InteractoMIX, this can be M-VORFFIP are used to predict the interfaces and
reused, shared or modified by the user at any functional sites at the proteins. Paired structures
time. Moreover, the Galaxy Project as a whole are used as inputs to V-D2OCK, to run a data-
7P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
Figure 2. Schematic representation of one for the workflows implemented in Galaxy InteractoMIX. The different
tools and associations between them are shown as presented in the Galaxy workflow engine. Each box represents a
tool with a number of inputs and outputs that can be connected for other tools and/or parsed.
driven docking and obtain structural models of the Materials and Methods
interaction, and to BADock, for predicting the
binding affinity of the complex based on the Galaxy platform
unbound monomers.
Overall the workflow will deliver the structure of Galaxy InteractoMIX instance is running on a
protein complexes obtained by either comparative Linux operating system using the January 2020
modeling or protein docking, together with several Galaxy Release (v 20.01) and an external MySQL
downstream analyses such as the prediction of database. The featured tools were integrated
functional sites, peptides and estimation of binding using Python 2.7.5 and bash. The different tools
affinities. This example illustrates the possibilities are implemented in different languages including
and versatility of the Galaxy InteractoMIX platform Python 2.7.5, Perl, C++ and C. An OVA image of
and how the implementation on the Galaxy a virtual machine running the Galaxy InteractoMIX
framework facilitates such complex analyses. platform is available upon request to the authors.
8P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
Databases and accessory bioinformatics tools DECLARATION OF COMPETING INTEREST
Several tools rely on external databases. BIANA None declared.
uses data from IntAct [1], BioGRID [2], DIP [3], HIP-
PIE [4], InBio_Map [5], ConsensusPathDB [6], I2D
[7]and STRING [42]. GUILDIFY uses PPI networks
from six species (human, mouse, yeast, worm, fly,
Acknowledgments
and plant) that are computed from the consensus
BIANA databases above. These databases are The authors received support from the following: the
updated every time new consensus interactomes Spanish Ministry of Sci (MINECO; BIO2017-85329-R,
are derived. MODPIN and M4T rely on the PDB RYC2015-17519, BIO2017-83591-R); the Marı́a de
[43], 3DiD [44], and BLAST preformatted databases Maeztu Programme for Units of Excellence in R&D
(NR and PDBaa) that are updated on a regular (award AEI CEX2018-000792-M), IMI-JU under grants
basis. The BLAST preformatted PDBaa set is also agreements no. 116030 (TransQST) resources of
used by PCPRi to derive sequence profiles. iLoops which are composed of financial contribution from the
make use of the ArchDB database as well as the EU-FP7 (FP7/2007-2013) and EFPIA companies in
definition of SCOP [45] and PFAM [46] databases. kind contribution. J.P.I. is supported by the grant
Finally, PiPreD relies on the iMotifsDB that is pre- MSCAfellow@MUNI (CZ.02.2.69/0.0/16_027/0008360).
computed using the biounit complexes from the
PDB databank [43]. Received 29 May 2020;
In term of accessory bioinformatics tools, there Accepted 16 September 2020;
are several methods in Galaxy InteractoMIX that Available online 23 September 2020
use BLAST [22] to perform sequence searches
and this include BIPS, iFrag, iLoops, MODPIN, Keywords:
M4T, and PCRPi. MODPIN and M4T both use Mod- interactomics;
eller [47] for comparative modeling as well as network analyses;
PROSA2 [48] (M4T only), Zrank [33], FoldX [34] integration;
and InterfaceAnalyzer [49] to score structural mod- Galaxy platform;
els. M4T also uses CD-HIT [50] and ClustalW [51]. workflows
VORFFIP and M-VORFFIP require QHULL [52],
PSAIA [53] and NACCESS [54]. V-D2OCK requires
PatchDock [55], as does BADOCK, and GroMACS Abbreviations:
[56] to perform the clustering of docking poses. PPI, protein–protein interaction; RF, random forest
BADOCK also utilizes FiberDOCK [57] to refine
docking poses. PCRPi makes use of CSU [58], References
al2co [59], 3DCO [60] and FoldX [34] to compute
the features for the Bayesian network classifier. [1]. Kerrien, S., Aranda, B., Breuza, L., Bridge, A., Broackes-
Finally, PiPreD requires DSSP [61] and Scwrl4.0 Carter, F., Chen, C., et al., (2012). The IntAct molecular
[62]. interaction database in 2012. Nucleic Acids Res., 40,
D841–D846.
[2]. Stark, C., Breitkreutz, B.J., Chatr-Aryamontri, A.,
Boucher, L., Oughtred, R., Livstone, M.S., et al., (2011).
CRediT authorship contribution The BioGRID Interaction Database: 2011 update. Nucleic
statement Acids Res., 39, D698–D704.
[3]. Xenarios, I., Salwinski, L., Duan, X.J., Higney, P., Kim, S.
Patricia Mirela-Bota: Software, Methodology, M., Eisenberg, D., (2002). DIP, the database of interacting
Resources, Validation, Data curation, proteins: a research tool for studying cellular networks of
Investigation. Joaquim Aguirre-Plans: protein interactions. Nucleic Acids Res., 30, 303–305.
[4]. Salwinski, L., Miller, C.S., Smith, A.J., Pettit, F.K., Bowie,
Methodology, Resources, Software, Visualization,
J.U., Eisenberg, D., (2004). The database of interacting
Investigation. Alberto Meseguer: Resources,
proteins: 2004 update. Nucleic Acids Res., 32, D449–
Validation. Cristiano Galletti: Resources, Writing
D451.
- review & editing. Joan Segura: Resources, [5]. Li, T., Wernersson, R., Hansen, R.B., Horn, H., Mercer, J.,
Writing - review & editing. Joan Planas-Iglesias: Slodkowicz, G., et al., (2017). A scored human protein–
Resources, Writing - review & editing. Javi protein interaction network to catalyze genomic
Garcia-Garcia: Software, Resources. Emre interpretation. Nat. Methods, 14, 61–64.
Guney: Resources, Writing - review & editing. [6]. Herwig, R., Hardt, C., Lienhard, M., Kamburov, A., (2016).
Baldo Oliva: Resources, Writing - review & Analyzing and interpreting genome data at the network
editing, Funding acquisition, Supervision. Narcis level with ConsensusPathDB. Nat. Protoc., 11, 1889–
Fernandez-Fuentes: Conceptualization, 1907.
Resources, Writing - original draft, Writing - review [7]. Brown, K.R., Jurisica, I., (2007). Unequal evolutionary
& editing, Funding acquisition, Supervision, conservation of human protein interactions in interologous
Project administration. networks. Genome Biol., 8, R95.
9P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
[8]. Venkatesan, K., Rual, J.F., Vazquez, A., Stelzl, U., PSI-BLAST: a new generation of protein database search
Lemmens, I., Hirozane-Kishikawa, T., et al., (2009). An programs. Nucleic Acids Res., 25, 3389.
empirical framework for binary interactome mapping. Nat. [23]. Sprinzak, E., Margalit, H., (2001). Correlated sequence-
Methods, 6, 83–90. signatures as markers of protein–protein interaction. J.
[9]. Aloy, P., Russell, R.B., (2004). Ten thousand interactions Mol. Biol., 311, 681–692.
for the molecular biologist. Nat. Biotechnol., 22, 1317– [24]. Fernandez-Fuentes, N., Madrid-Aliste, C.J., Rai, B.K.,
1321. Fajardo, J.E., Fiser, A., (2007). M4T: a comparative
[10]. Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, protein structure modeling server. Nucleic Acids Res., 35,
D., Cech, M., et al., (2018). The Galaxy platform for W363–W368.
accessible, reproducible and collaborative biomedical [25]. Fernandez-Fuentes, N., Rai, B.K., Madrid-Aliste, C.J.,
analyses: 2018 update. Nucleic Acids Res., 46, W537. Fajardo, J.E., Fiser, A., (2007). Comparative protein
W44. structure modeling by combining multiple templates and
[11]. Poglayen, D., Marin-Lopez, M.A., Bonet, J., Fornes, O., optimizing sequence-to-structure alignments.
Garcia-Garcia, J., Planas-Iglesias, J., et al., (2016). Bioinformatics., 23, 2558–2565.
InteractoMIX: a suite of computational tools to exploit [26]. Meseguer, A., Dominguez, L., Bota, P.M., Aguirre-Plans,
interactomes in biological and clinical research. Biochem. J., Bonet, J., Fernandez-Fuentes, N., et al., (2020). Using
Soc. Trans., 44, 917–924. collections of structural models to predict changes of
[12]. Garcia-Garcia, J., Guney, E., Aragues, R., Planas- binding affinity caused by mutations in protein–protein
Iglesias, J., Oliva, B., (2010). Biana: a software interactions. Protein Sci., 29 (10), 2112–2130.
framework for compiling biological interactions and [27]. Marin-Lopez, M.A., Planas-Iglesias, J., Aguirre-Plans, J.,
analyzing networks. BMC Bioinformatics., 11, 56. Bonet, J., Garcia-Garcia, J., Fernandez-Fuentes, N.,
[13]. Guney, E., Garcia-Garcia, J., Oliva, B., (2014). et al., (2018). On the mechanisms of protein
GUILDify: a web server for phenotypic characterization interactions: predicting their affinity from unbound
of genes through biological data integration and tertiary structures. Bioinformatics., 34, 592–598.
network-based prioritization algorithms. Bioinformatics., [28]. Segura, J., Jones, P.F., Fernandez-Fuentes, N., (2011).
30, 1789–1790. Improving the prediction of protein binding sites by
[14]. Aguirre-Plans, J., Pinero, J., Sanz, F., Furlong, L.I., combining heterogeneous data and Voronoi diagrams.
Fernandez-Fuentes, N., Oliva, B., et al., (2019). BMC Bioinformatics., 12, 352.
GUILDify v2.0: a tool to identify molecular networks [29]. Segura, J., Jones, P.F., Fernandez-Fuentes, N., (2012).
underlying human diseases, their comorbidities and their A holistic in silico approach to predict functional sites in
druggable targets. J. Mol. Biol., 431, 2477–2484. protein structures. Bioinformatics., 28, 1845–1850.
[15]. Planas-Iglesias, J., Marin-Lopez, M.A., Bonet, J., Garcia- [30]. Segura, J., Marin-Lopez, M.A., Jones, P.F., Oliva, B.,
Garcia, J., Oliva, B., (2013). iLoops: a protein–protein Fernandez-Fuentes, N., (2015). VORFFIP-driven dock:
interaction prediction server based on structural features. V-D2OCK, a fast and accurate protein docking strategy.
Bioinformatics, 29, 2360–2362. PLoS One, 10, e0118107
[16]. Planas-Iglesias, J., Bonet, J., Garcia-Garcia, J., Marin- [31]. Assi, S.A., Tanaka, T., Rabbitts, T.H., Fernandez-
Lopez, M.A., Feliu, E., Oliva, B., (2013). Understanding Fuentes, N., (2010). PCRPi: Presaging Critical Residues
protein–protein interactions using local structural features. in Protein interfaces, a new computational tool to chart hot
J. Mol. Biol., 425, 1210–1224. spots in protein interfaces. Nucleic Acids Res., 38, e86
[17]. Garcia-Garcia, J., Schleker, S., Klein-Seetharaman, J., [32]. Oliva, B., Fernandez-Fuentes, N., (2015). Knowledge-
Oliva, B., (2013). BIPS: BIANA Interolog Prediction based modeling of peptides at protein interfaces: PiPreD.
Server. A tool for protein–protein interaction inference. Bioinformatics., 31, 1405–1410.
Nucleic Acids Res., 40, W147–W151. [33]. Pierce, B., Weng, Z., (2007). ZRANK: reranking protein
[18]. Garcia-Garcia, J., Valls-Comamala, V., Guney, E., docking predictions with an optimized energy function.
Andreu, D., Munoz, F.J., Fernandez-Fuentes, N., et al., Proteins., 67, 1078–1086.
(2017). iFrag: a protein–protein interface prediction server [34]. Schymkowitz, J., Borg, J., Stricher, F., Nys, R.,
based on sequence fragments. J. Mol. Biol., 429, 382– Rousseau, F., Serrano, L., (2005). The FoldX web
389. server: an online force field. Nucleic Acids Res., 33,
[19]. Bonet, J., Planas-Iglesias, J., Garcia-Garcia, J., Marin- W382–W388.
Lopez, M.A., Fernandez-Fuentes, N., Oliva, B., (2014). [35]. Leaver-Fay, A., Tyka, M., Lewis, S.M., OF, Lange,
ArchDB 2014: structural classification of loops in proteins. Thompson, J., Jacak, R., et al., (2011). ROSETTA3: an
Nucleic Acids Res., 42, D315–D319. object-oriented software suite for the simulation and
[20]. Blohm, P., Frishman, G., Smialowski, P., Goebels, F., design of macromolecules. Methods Enzymol., 487,
Wachinger, B., Ruepp, A., et al., (2014). Negatome 2.0: a 545–574.
database of non-interacting proteins derived by literature [36]. Feliu, E., Aloy, P., Oliva, B., (2011). On the analysis of
mining, manual annotation and protein structure analysis. protein–protein interactions via knowledge-based
Nucleic Acids Res., 42, D396–D400. potentials for the prediction of protein–protein docking.
[21]. Bonet, J., Segura, J., Planas-Iglesias, J., Oliva, B., Protein Sci., 20, 529–541.
Fernandez-Fuentes, N., (2014). Frag’r’Us: knowledge- [37]. Wass, M.N., Fuentes, G., Pons, C., Pazos, F., Valencia,
based sampling of protein backbone conformations for de A., (2011). Towards the prediction of protein interaction
novo structure-based protein design. Bioinformatics., 30, partners using physical docking. Mol. Syst. Biol., 7, 469.
1935–1936. [38]. Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V.,
[22]. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Thirion, B., Grisel, O., et al., (2011). Scikit-learn: machine
Zhang, Z., Miller, W., et al., (1997). Gapped BLAST and learning in Python. J. Mach. Learn. Res., 12, 2825–2830.
10P. Mirela-Bota, J. Aguirre-Plans, A. Meseguer, et al. Journal of Molecular Biology 433 (2021) 166656
[39]. Vreven, T., Moal, I.H., Vangone, A., Pierce, B.G., [51]. Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson,
Kastritis, P.L., Torchala, M., et al., (2015). Updates to T.J., Higgins, D.G., et al., (2003). Multiple sequence
the integrated protein–protein interaction benchmarks: alignment with the Clustal series of programs. Nucleic
docking benchmark version 5 and affinity benchmark Acids Res., 31, 3497.
version 2. J. Mol. Biol., 427, 3031–3041. [52]. Barber, C.B., Dobkin, D.P., Huhdanpaa, H., (1996). The
[40]. Clackson, T., Wells, J.A., (1995). A hot spot of binding Quickhull algorithm for convex hulls. ACM Trans. Math.
energy in a hormone–receptor interface. Science., 267, Softw., 22, 469–483.
383–386. [53]. Mihel, J., Sikif á, M., Tomif á, S., Jeren, B., Vlahovicek, K.,
[41]. Segura Mora, J., Assi, S.A., Fernandez-Fuentes, N., (2008). PSAIA—protein structure and interaction
(2010). Presaging critical residues in protein interfaces- analyzer. BMC Struct. Biol., 8, 21.
web server (PCRPi-W): a web server to chart hot spots in [54]. Hubbard, T.J.P., Blundell, T.L., (1987). Comparison of
protein interfaces. PLoS One, 5, e12352 solvent-inaccessible cores of homologous proteins:
[42]. Szklarczyk, D., Gable, A.L., Lyon, D., Junge, A., Wyder, definitions useful for protein modelling. Protein Eng., 1,
S., Huerta-Cepas, J., et al., (2019). STRING v11: protein– 159–171.
protein association networks with increased coverage, [55]. Schneidman-Duhovny, D., Inbar, Y., Nussinov, R.,
supporting functional discovery in genome-wide Wolfson, H.J., (2005). PatchDock and SymmDock:
experimental datasets. Nucleic Acids Res., 47, D607. servers for rigid and symmetric docking. Nucleic Acids
D13. Res., 33, W363–W367.
[43]. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., [56]. Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof,
Bhat, T.N., Weissig, H., et al., (2000). The Protein Data G., Mark, A.E., Berendsen, H.J., (2005). GROMACS:
Bank. Nucleic Acids Res., 28, 235–242. fast, flexible, and free. J. Comput. Chem., 26, 1701–
[44]. Stein, A., Ceol, A., Aloy, P., (2011). 3did: identification 1718.
and classification of domain-based interactions of known [57]. Mashiach, E., Nussinov, R., Wolfson, H.J., (2010).
three-dimensional structure. Nucleic Acids Res., 39, FiberDock: flexible induced-fit backbone refinement in
D718–D723. molecular docking. Proteins., 78, 1503–1519.
[45]. Conte, L.L., Brenner, S.E., Hubbard, T.J.P., Chothia, C., [58]. Sobolev, V., Sorokine, A., Prilusky, J., Abola, E.E.,
Murzin, A., (2002). SCOP database in 2002: refinements Edelman, M., (1999). Automated analysis of
accommodate structural genomics. Nucleic Acids Res., interatomic contacts in proteins. Bioinformatics., 15,
30, 264–267. 327–332.
[46]. Finn, R.D., Coggill, P., Eberhardt, R.Y., Eddy, S.R., [59]. Pei, J., Grishin, N.V., (2001). AL2CO: calculation of
Mistry, J., Mitchell, A.L., et al., (2016). The Pfam protein positional conservation in a protein sequence alignment.
families database: towards a more sustainable future. Bioinformatics., 17, 700–712.
Nucleic Acids Res., 44, D279–D285. [60]. Landgraf, R., Xenarios, I., Eisenberg, D., (2001). Three-
[47]. Webb, B., Sali, A., (2016). Comparative protein structure dimensional cluster analysis identifies interfaces and
modeling using MODELLER. Curr. Protoc. Protein Sci., functional residue clusters in proteins. J. Mol. Biol., 307,
86, 2 9 1–2 9 37. 1487.
[48]. Sippl, M.J., (1993). Recognition of errors in three- [61]. Kabsch, W., Sander, C., (1983). Dictionary of protein
dimensional structures of proteins. Proteins., 17, 355– secondary structure: pattern recognition of hydrogen-
362. bonded and geometrical features. Biopolymers., 22,
[49]. Stranges, P.B., Kuhlman, B., (2013). A comparison of 2577–2637.
successful and failed protein interface designs highlights [62]. Krivov, G.G., Shapovalov, M.V., Dunbrack Jr., R.L.,
the challenges of designing buried hydrogen bonds. (2009). Improved prediction of protein side-chain
Protein Sci., 22, 74–82. conformations with SCWRL4. Proteins., 77, 778–795.
[50]. Li, W., Jaroszewski, L., Godzik, A., (2001). Clustering of
highly homologous sequences to reduce the size of large
protein databases. Bioinformatics, 17, 282.
11You can also read

















































