Molecular, Electronic, Nonlinear Optical and Spectroscopic Analysis of Heterocyclic 3-Substituted- 4
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Heterocycl. Commun. 2021; 27: 1–16
Research Article Open Access
Murat Beytur* and Ihsan Avinca
Molecular, Electronic, Nonlinear Optical and
Spectroscopic Analysis of Heterocyclic 3-Substituted-
4-(3-methyl-2-thienylmethyleneamino)-
4,5-dihydro-1H-1,2,4-triazol-5-ones:
Experiment and DFT Calculations
https://doi.org/10.1515/hc-2020-0118 the basis set of 6-311G(d,p). The recording of FT-IR fre-
Received June 05, 2020; accepted January 05, 2021. quencies was done for the pertinent compound. The recor-
ded frequencies through DFT/B3LYP and DFT/B3PW91
Abstract: In the present study, 3-p-methoxybenzyl/m-chlo-
methods were compared to experimental values, with a
robenzyl/phenyl-4-(3-methyl-2-thienylmethyleneamino)-
result gained closest to the values of B3LYP. Finally, the
4,5-dihydro-1H-1,2,4-triazol-5-ones were obtained from
Gaussian09W program package in DMSO phase, starting
the reaction between 3-methylthiophene-2-carbaldehyde
from the optimized structure, has been instrumental in
and three different 4-amino-(3-p-methoxybenzyl/m-
calculating the 13C-NMR and 1H-NMR chemical shift values
chlorobenzyl/phenyl)-4,5-dihydro-1H-1,2,4-triazole-
of the GIAO method.
5-ones. In order to compare experimental and theoretical
values, the geometric parameter, electronic, nonlinear
Keywords: 1,2,4-Triazole, Thienyl, DFT calculations,
optical properties, molecular electrostatic potentials and
GIAO-NMR, FT-IR, experimental
spectroscopic properties of 3-substituted-4-(3-methyl-
2-thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-triazol-
5-ones have been simulated. The electronic properties of
the newly synthesized compounds were calculated using Introduction
DFT/B3LYP and DFT/B3PW91 methods revealing parame-
ters such as ionization potential, electron affinity, energy Heterocyclic compounds are considered important classes
gap, electronegativity, molecular hardness, molecular of molecules, and they have been found to be significant
softness, electrophilic index, nucleophilic index and to the structural cores of many natural and synthetic
chemical potential, all obtained from HOMO and LUMO drugs [1]. Synthesis of nitrogen-containing heterocyclic
energies, dipole moments and total energies. UV-visible structures has attracted considerable attention in recent
absorption spectra and the stimulation contributions in years for their benefits in different applications such as
UV-visible transitions were obtained by using TD-DFT/ propellants, explosives, and especially medical fields [2].
B3LYP/6-311G(d,p) and TD-DFT/B3PW91/6-311G(d,p) The 1,2,4-triazole moiety and its derivatives are present
methods in ethanol. The calculated absorption wave- in a variety of therapeutically important agents such as
lengths, oscillator power and excitation energies were ribavirin (antiviral) [3], docetaxel (antineoplastic) [4] and
compared with experimental values. In line with DFT, the rizatriptan (antimigraine) [5]. Heterocyclic derivatives
numbers of molecular vibration were analyzed through containing sulfur possess essential biological properties
too [6, 7]. Antiepileptic drugs including brotizolam [8],
etizolam [9] and tiagabine [10], contain the thiophene
moiety in the active pharmacophore structures.
*Corresponding author: Murat Beytur, Kafkas University, Faculty
Schiff bases containing 1,2,4-triazole in their struc-
of Science and Letters, Department of Chemistry, 36100, Kars,
Turkey; E-mail: muratbeytur83@kafkas.edu.tr
ture have been extensively studied for their applicability
Ihsan Avinca, Kafkas University, Faculty of Science and Letters, in various areas such as biological [11-13], chemical [14,
Department of Chemistry, 36100, Kars, Turkey 15] and pharmaceutical applications [16, 17]. There have
Open Access. © 2021 Beytur and Avinca, published by De Gruyter. This work is licensed under the Creative Commons
Attribution alone 4.0 License.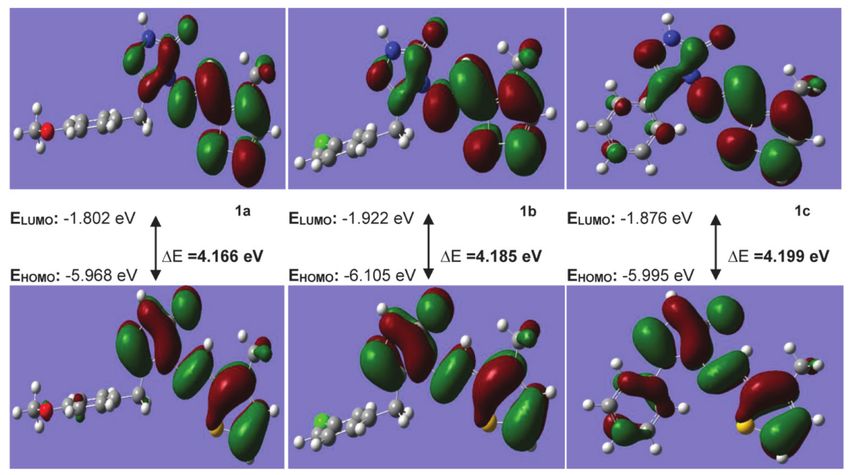
2 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
recently been an increase in studies on Schiff base deri- over polarized functions with B3LYP/DFT and B3PW91/
vatives in relation to corrosion inhibitors [18], optical DFT methods [29, 30].
sensors [19], highly selective polymer membrane electro- The C-C bond lengths of the C3 bonded aryl groups of
des [20], therapeutic properties, highly thermal stability, the different analogues of 1 were compared according to
modern technology (nonlinear optical materials) [21], the Ikizler, based on the optimized structures [31]. Accor-
various coordination complexes, homogenous catalysis ding to the Ikizler, C-C bond lengths in the benzene ring
[21, 22] and biological probes [23]. have been observed as 1.397 Å and C-H bonds as 1.084 Å
Computational chemistry has now reached a stage [31]. The average of C-C bond lengths in the thiophene
whereby new scientific information can be generated to ring in the structure of type S compounds was found to
guide experiments and enable researchers to comprehend be 1.392 Å and 1.390 Å, according to the B3LYP/6-311G(d,p)
and explore the structure and interactions of matter. In and B3PW91/6-311G(d,p) methods, respectively. When the
some areas, it is almost impossible to achieve the targeted theoretical values were compared with values given accor-
results only with laboratory experiments, without compu- ding to the Ikizler [31], value obtained with B3LYP method
tational chemistry and modelling. Physicists and chemists was observed to be closer to the literature [31]. The average
have prior knowledge about the structure of drugs before of C-S bond lengths in the synthesized compounds were
synthesis using a computer, allowing them to determine found to be 1.741 Å according to B3LYP method and 1.730 Å
the desired properties in the drug. Then they may perform according to B3PW91 method (Table 1).
synthesis to generate these properties [24-26]. Density The average C-C bond length values of the C1-linked
functional theory (DFT) methods analyze the structures, benzene ring in the triazole ring in the structure of type
dipole moments, vibration frequencies, nuclear magnetic 1 compounds were found to be 1.392 Å, according to
resonance chemical shifts, optical properties, molecular B3LYP/6-311G(d,p) and B3PW91/6-311G(d,p) methods. It
electrostatic potentials, molecular mechanisms and ther- was observed that the B3LYP method was 1.084 Å, and the
modynamic properties of organic compounds with high B3PW91method was 1.085 Å, when the C-H bond lengths
accuracy. In the present work, Gaussian 09W program is in the benzene ring were examined (Table 2), According
used to determine the most stable locations of each atom to B3LYP method, the obtained value was found to be the
in space. The minimum energy space structure of the most same as that of the literature and the obtained theoretical
optimized compounds was calculated with 6-311G(d,p) data were confirmed against the values according to the
basis set, over polarized functions by B3LYP and B3PW91 Ikizler [31].
methods of DFT. We have analyzed the geometric opti- According to the Ikizler, the experimental C-N length
mization, molecular and electronic properties of the was 1.49 Å and C=N length was 1.27 Å [31]. The results
3-substituted-4-(3-methyl-2-thienylmethyleneamino)- obtained were observed to be 1.368 Å in the B3LYP/6-311G
4,5-dihydro-1H-1,2,4-triazol-5-one compounds and compa- (d, p) method and 1.365 Å, according to the B3PW91/6-311G
red them with studies in the experimental. We have also (d, p) method (Table 3). The average bond lengths obser-
analyzed the spectroscopic properties of molecules both ved with the B3LYP and B3PW91 methods were experimen-
experimentally and theoretically. We have seen that the tally determined to be between the suggested C-N single
theoretical results obtained are highly compatible with bond and C=N double bond lengths. Therefore, it has been
experimental data [27]. observed that the C-NH bond has a partial double bond
property in the 1,2,4-triazole-5-on ring.
Results and Discussion
Electronic Properties
Geometric Optimization
LUMO (π acceptor) and HOMO (π donor) are successively
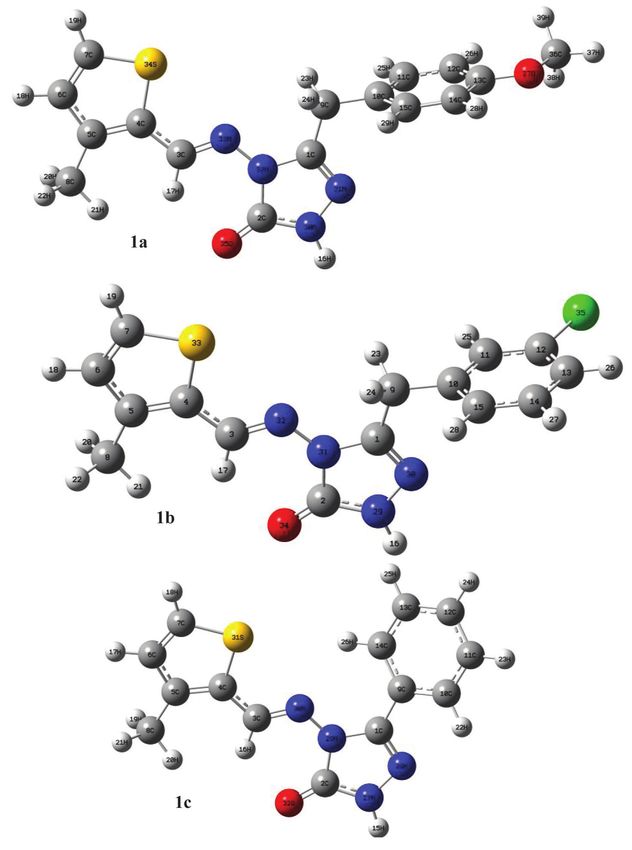
The three-dimensional approximate geometry of the called to be the lowest unoccupied molecular orbital and
3-substituted-4-(3-methyl-2-thienylmethyleneamino)- the highest occupied molecular orbital. ELUMO is the lowest
4,5-dihydro-1H-1,2,4-triazol-5-ones (1) are plotted in energy of unmatched electrons and EHOMO is the highest
Gauss View 5.0 program [28] (Figure 1). Using these geo- energy of matched electrons. HOMO and LUMO can offer
metric structures, Gaussian 09W was used to determine an appropriate qualitative estimate of excitation properties
the most stable positions of each atom in space. The and a molecule’s electron carrying ability [13, 32]. HOMO
minimum energy space structure of the most optimized and LUMO from frontier molecular orbitals play an impor-
compounds was analyzed with the 6-311G (d, p) basis set tant role in determining electrical and optical properties,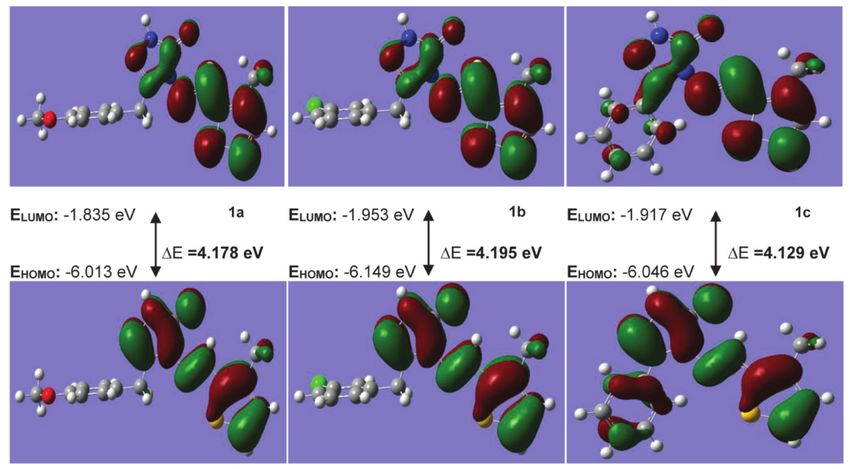
M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 3
Figure 1 The optimized gas-phase molecules at DFT theoretical level using 6-311G(d,p) basis set. 1a: 3-p-methoxybenzyl-4-(3-methyl-2-
thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-triazol-5-one, 1b: 3-m-chlorobenzyl-4-(3-methyl-2-thienylmethyleneamino)-4,5-dihydro-1H-
1,2,4-triazol-5-one, 1c: 3-phenyl-4-(3-methyl-2-thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-triazol-5-one
Table 1 The theoretical C-C and C-S bond lengths of the thiophene group in the structure of 1 type compounds according to DFT/6-311G(d,p)
basis set
Bond Type Compound 1a (Å) Bond Type Compound 1b (Å) Bond Type Compound 1c (Å)
B3LYP B3PW91 B3LYP B3PW91 1c 1c
C4-C5 1.384 1.383 C4-C5 1.384 1.384 C4-C5 1.384 1.384
C5-C6 1.427 1.423 C5-C6 1.427 1.423 C5-C6 1.427 1.423
C6-C7 1.364 1.364 C6-C7 1.364 1.364 C6-C7 1.364 1.364
C4-S34 1.754 1.742 C4-S33 1.754 1.742 C4-S31 1.754 1.742
C7-S34 1.728 1.718 C7-S33 1.728 1.718 C7-S31 1.728 1.718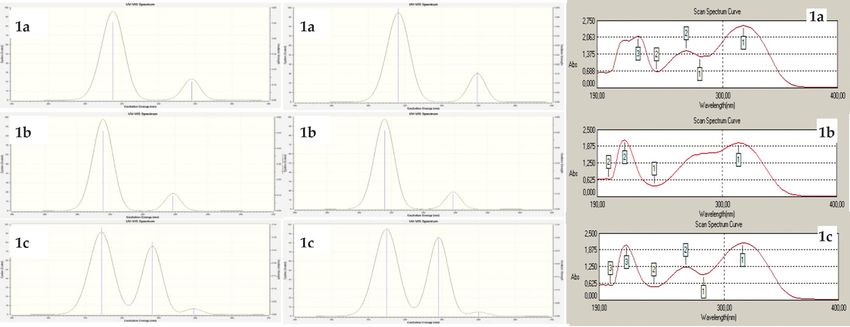
4 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
Table 2 Theoretical C-C and C-H bond lengths in triazole C1 linked benzene ring in the structure of 1 type compounds according to
DFT/6-311G(d,p) basis set
Bond Type Compound 1a (Å) Bond Type Compound 1b (Å) Bond Type Compound 1c (Å)
B3LYP B3PW91 B3LYP B3PW91 1c 1c
C10-C11 1.392 1.390 C10-C11 1.396 1.394 C9-C10 1.401 1.401
C10-C15 1.400 1.398 C10-C15 1.396 1.394 C9-C14 1.399 1.399
C11-C12 1.396 1.393 C11-C12 1.389 1.388 C10-C11 1.387 1.387
C12-C13 1.396 1.394 C12-C13 1.390 1.389 C11-C12 1.393 1.393
C13-C14 1.400 1.398 C13-C14 1.392 1.390 C12-C13 1.390 1.390
C14-C15 1.386 1.398 C14-C15 1.392 1.390 C13-C14 1.390 1.390
C11-H25 1.085 1.086 C11-H25 1.083 1.084 C10-H22 1.084 1.084
C12-H26 1.082 1.083 C12-H26 - - C11-H23 1.085 1.085
C13-H27 - - C13-H26 1.082 1.083 C12-H24 1.085 1.085
C14-H28 1.083 1.084 C14-H27 1.084 1.085 C13-H25 1.085 1.085
C15-H29 1.085 1.086 C15-H28 1.084 1.085 C14-H26 1.081 1.081
Table 3 The theoretical C-N bond lengths of the thiophene group in the structure of 1 type compounds
Bond Type Compound 1a (Å) Bond Type Compound 1b (Å) Bond Type Compound 1c (Å)
B3LYP B3PW91 B3LYP B3PW91 1c 1c
C2-N30 1.368 1.365 C2-N29 1.369 1.365 C2-N27 1.367 1.364
Figure 2 Frontier orbitals (HOMO–LUMO) views, corresponding energies and energy gap of titled compounds (1) according to B3LYP/6-
311G(d,p) method
which are the most important parameters of quantum with positive and negative phases being indicated in red
chemistry. The transitions of selected frontier molecular and green, respectively. The electronic properties of the
orbitals in the gas phase are as shown in Figures 2 and 3, synthesized titled compounds were obtained from the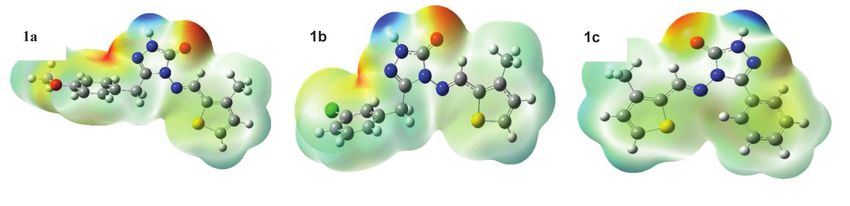
M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 5
Figure 3 Frontier orbitals (HOMO–LUMO) views, corresponding energies and energy gap of titled compounds (1) according to B3PW91/6-
311G(d,p) method
Table 4 The values of electron structure identifiers calculated using 6-311G(d,p) basis set at the B3LYP and B3PW91 theory level of titled
molecules (1) in gas phase
Electronic Properties DFT/B3LYP (eV) DFT/B3PW91 (eV)
1a 1b 1c 1a 1b 1c
I; Ionization Potential 5.968 6.105 5.995 6.013 6.149 6.046
A; Electron Affinity 1.802 1.922 1.876 1.835 1.953 1.917
ΔE; Energy Gap 4.166 4.182 4.119 4.178 4.195 4.129
χ; Electronegativity 3.885 4.014 3.935 3.924 4.051 3.982
η; Molecular Hardness 2.083 2.091 2.059 2.089 2.098 2.065
Ѕ; Molecular Softness 0.480 0.478 0.486 0.479 0.477 0.484
µ; Chemical Potential −3.885 −4.014 −3.935 −3.924 −4.051 −3.982
ω; Electrophilic Index 3.624 3.852 3.760 3.685 3.911 3.839
ɛ; Nucleophilic Index −0.297 −0.308 −0.298 −0.301 −0.312 −0.302
calculated HOMO and LUMO energies, by using B3LYP/6- The electron distribution is quite variable and pola-
311G(d,p) and B3PW91/6-311G(d,p) methods [18]. The rization is low, especially when the LUMO-HOMO gap is
results obtained are given in Table 4. small. The electron distribution within the molecule is
less variable and polarization is low when the energy gap
Ionization Potential I = –EHOMO (1) [33,34] is large. The molecules examined contain substrates such
Electron Affinity A = –ELUMO (2) [33,34] as p-methoxybenzyl (1a), m-chlorobenzyl (1b) and phenyl
Energy Gap ∆E = (ELUMO – EHOMO) (3) [35] (1c) moieties bound to C1 in the 1,2,4-triazole-5-on ring.
Electronegativity χ = (I + A) / 2 (4) [36] When the donor and acceptor substituents examined the
Molecular Hardness ɳ = (I – A) / 2 (5) [37] effect on structures, LUMO/HOMO energies differences
Molecular Softness Ѕ = 1/ɳ (6) [38] of 1a, 1b and 1c molecules are calculated as 4.166/4.178,
Chemical Potential µ = –χ (7) [39] 4.182/4.195 and 4.199/4.129 eV according to DFT/B3LYP
Electrophilic Index ω = µ 2/2ɳ (8) [40] and DFT/B3PW91, respectively (Table 4). It was found
Nucleophilic Index ɛ=µ.ɳ (9) [41] that the greater the energy gap of the molecule, the higher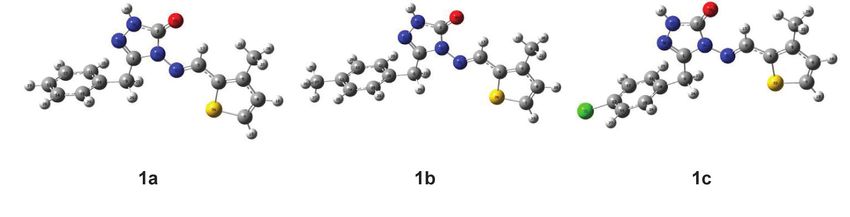
6 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
the intramolecular charge density. The energy gap in the Isotropic polarization calculation equation
studied molecules is 1b > 1a > 1c in the B3PW91/6-311G(d,p)
set. Therefore, the 1b molecule with an electron-donating α xx + α yy + α zz
substituent in the ring has the highest energy gap, within α0 =
the substituents explored. 3
( ) + (α ) + (α ) +6
2 2 2
∆α = 2 α − α − α zz − α xx 2
xx yy yy zz xx
Nonlinear optical Features
Polarizability and hyperpolarizability provide useful The average calculation equation availability
information for frequency changing, optical modu- hyperpolarized
lation, optical switching and optical logic for tech-
nologies evolving in areas such as non-linear optical β0 = β x2 + β y2 + β z2
(NLO) activity, communication, signal processing,
and optical interconnection [42]. Organic materi-
β x = β xxx + β xyy + β xzz
als are expected to have relatively strong NLO pro-
perties, due to the delocalized electrons in the π->π*
β y = β yyy + β xxy + β yzz
orbitals [43].
The first hyperpolarizability (β0) of the Schiff base
β z = β zzz + β xxz + β yyz
molecular systems under consideration is calcula-
ted using the DFT method based on the finite-field
(β ) ( ) ( )
2 2 2
approach. The first hyperpolarizability is a third-grade β= xxx
+ β xyy + β xzz + β yyy + β yzz + β yxx + β zzz + β zxx + β zyy
tensor that can be defined by a 3×3×3 matrix. 27 compo-
nents of the 3D matrix can be reduced to 10 components
The NLO properties of the molecules were calculated
due to Kleinman symmetry. The components of β are
with the above equations using the basis sets B3LYP/6-
defined as coefficients in the expansion of energy in the
311G (d, p) and B3PW91/6-311G (d, p). Total static dipole
external electric field in the Taylor series energy. When
moment, polarizability and first order hyperpolarizability
the electric field is weak and homogeneous, expansion
are given in Table 5. The data obtained were compared with
occurs.
the reported values of similar derivatives reported by Binil
Where E0 is the energy of the free molecule, F i is the
et al. [45]. The related compounds were compared to urea,
area in origin; μi, μij, βijk and γijkl are components of the
referenced as a NLO material (urea: 0.3728 × 10-30 esu),
dipole moment, polarizability, first hyperpolarization and
according to Adant et al. [46]. The calculated hyperpo-
second hyperpolarizability.
larizability of 1 analogues appears to be approximately
10 times higher than the urea value, a noted significant
1 1 increase.
E = E0 −∑ µ i F i − ∑α ij
FiF j − ∑β ijk
FiF j Fk
2 ij
6 ijk
1
− ∑γ F F F F +.....
i j k l
24 ijkl
ijkl Molecular Electrostatic Potential Analysis
Molecular electrostatic potential (MEP), which is related
Total static dipole moments (μtot), average polariz-
to electron density proves to be useful in understanding
ability (α0), anisotropy (α) and average first hyperpola-
the regions of electrophilic and nucleophilic reactions
rizability values of polarizations (β) were determined
[47]. Electrostatic potential is also well suited to analyzing
according to Zhang et al., using the X, Y and Z com-
processes based on the “recognition” of one molecule
ponents [44]. Total static dipole moment calculation
by another, such as drug-receptor and enzyme-substrate
equation;
interactions [48]. Molecular electrostatic potentials were
calculated in optimized geometry with the B3LYP and
µtot = µ x2 + µ y2 + µ z2 B3PW91 methods and the basis set of 6-311G (d,p) to esti-
mate the reactive regions of electrophilic and nucleophilicM. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 7
Table 5 Calculated dipole moment, polarizability and hyperpolarizability values of the related molecules (1)
B3LYP B3PW91
1a 1b 1c 1a 1b 1c
μx Debye 0.2104 2.6327 −0.0528 0.2104 2.6410 −1.2607
μy Debye −3.2757 −0.8705 −2.3138 −3.2757 −0.8729 −1.2222
μz Debye 0.2652 1.5044 0.0204 0.2652 1.4898 0.1637
μToplam Debye 3.2932 3.1547 2.3145 3.2932 3.1553 1.7635
αxx a.u. 55,075 51,663 44,389 54,763 51,276 44,187
αyy a.u. 31,360 29,893 36,370 31,150 29,754 36,148
αzz a.u. 21,722 23,220 14,105 21,611 23,128 14,061
Α x10−24 esu 36,052 34,925 31,621 35,842 34,719 31,465
∆α x10−24 esu 29,730 25,764 27,177 29,560 25,489 27,019
βx a.u. −1966,378 −507,524 −2294,653 1909,348 −643,629 2567,096
βy a.u. −3380,453 −3236,769 −3887,853 −3411,620 −3205,343 −4024,102
βz a.u. 722,926 1927,783 −86,438 −705,287 2030,639 95,285
Β x10−30 esu 3,977 3.801 4.515 3.973 3.849 4.774
E a.u. −1233.33 −1233.33 −1233.33 −1232.95 −1232.95 −1232.95
Β value For Urea: 0.3728 x10-30 esu
Figure 4 Molecular electrostatic potentials of 1 type compounds according to B3PW91 method
attacks for the studied molecules. Different values of the 6-311G(d,p) polarized set based on optimized structure.
electrostatic potential on the surface are indicated by dif- The calculated absorption wavelengths (λ), oscillator
ferent colors. Potential increases are listed as red < orange power (f) and excitation energies are shown in Table 6 in
< yellow < green < blue. On the molecular electrostatic the ethanol solvent phase. The stronger the donor charac-
potential, negative regions (red and yellow) are associated ter of the substitution in the molecules, the more electrons
with electrophilic reactivity, and positive regions (blue) pushed into the molecule and the greater the λmax. These
are associated with nucleophilic reactivity (Figure 4) values may change slightly due to the effect of a given
[49]. It appears that the negative charge covers the carbo- solvent. The role of the substrate and solvent effect acts
nyl group and the positive region is above the remaining on the UV spectrum too.
groups. The highest electronegativity is located in the car- Three absorption bands were seen in the theoreti-
bonyl group, the most reactive parts of the molecules are cally obtained electronic spectrum of the synthesized
therefore elsewhere (1). compounds (1a-c) in ethanol (Figure 5). The calculated
absorption wavelengths were determined to be close to
experimental values (Table 6).
UV-vis Spectral Analysis The absorption bands below 300 nm belong to the
π->π* transitions in the benzene ring and azomethine
UV-vis absorption spectra of analogues of 1 were obtai- group. Absorption bands between 300-400 nm are due
ned in ethanol (Figure 5). Calculations were obtained to n->π* transitions of the imine group [50]. Gauss-
with TD-DFT/B3LYP and TD-DFT/B3PW91 methods and Sum3.0 program was used to determine the stimulation8 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
Figure 5 Theoretically generated (DFT/B3LYP, DFT/B3PW91 and experimental) UV-vis spectra graphics of 1 type compounds, respectively
Table 6 Experimental and theoretical (DFT/B3LYP and DFT/B3PW91) UV-vis values, transition types and the main transition contribution of
S molecules
λ (nm) Excitation Energy (eV) Oscillator Power (f) The Main Transition Contribution
Exp./B3LYP/B3PW91 B3LYP/B3PW91 B3LYP/B3PW91 B3LYP/B3PW91
1a 314.00/324.82/323.58 3.8170/3.8317 0.5047/0.4970 H->L (96%)/H->L (96%)/
284.00/318.39/316.43 3.8941/3.9182 0.0011/0.0001 H-1->L (99%)/H-1->L (99%)
226.00/281.96/280.58 4.3972/4.4188 0.1208/0.1590 H-2->L (77%)/H-2->L (80%)
1b 314.00/324.21/322.80 3.8242/3.8408 0.5136/0.5060 H->L (96%)/H->L (96%)/
274.00/281.47/279.99 4.4049/4.4281 0.0966/0.1317 H-1->L (71%)/H-1->L (75%)
214.00/275.58/276.47 4.4991/4.4845 0.0029/0.0007 H-4->L (58%), H-2->L (39%)/
H-4->L (64%), H-2->L (34%)/
1c 318.00/331.14/330.09 3.7442/3.7561 0.3395/0.3337 H->L (94%)/H->L (94%)/
276.00/303.43/301.66 4.0861/4.1101 0.2813/0.3013 H-1->L (91%)/H-1->L (90%)
226.00/280.90/279.88 4.4139/4.4299 0.0240/0.0163 H->L+1 (89%)/H-4->L (35%),
H->L+1 (40%)
Table 7 Comparison of theoretical data and experimental data obtained according to DFT/B3LYP/6-311G(d,p) and DFT/B3PW91/6-311G(d,p)
methods of 1 type compounds
NH C=O N=C CH C=C
1a Experimental 3161 1702 1590 3063-2928 1537, 1447
B3LYP 3646 1782 1632,1622 3209-2971 1644, 1606-1485, 1440-1341
B3PW91 3520 1733 1584,1575 3093-2862 1594, 1556-1429
1b Experimental 3180 1700 1579 3097-2925 1538, 1444
B3LYP 3645 1785 1621 3209-3000 1626-1604, 1511-1432
B3PW91 3519 1735 1574 3093-2893 1577-1554, 1455-1389
1c Experimental 3160 1695 1575 3052-2917 1541, 1442
B3LYP 3641 1783 1622 3210-2999 1613-1515, 1419-1394
B3PW91 3515 1734 1575 3090-2892 1564-1460
contributions in UV-visible transitions (Table 7) [51]. (H->L) (96%/96%) was determined as n->π* transitions
According to B3LYP/B3PW91 for TD-DFT calculations, for at 324.82/323.58 nm and the main transition contribution
1a, the main transition contribution from HOMO to LUMO from HOMO-1 to LUMO (H-1->L) (99%/99%) was determinedM. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 9 as n->π* transitions at 318.39/316.43 nm. In addition, Infrared Spectral Analysis the main transition contribution from HOMO-2 to LUMO (H-2->L) (77%/80%) was determined at 281.96/280.58 nm Derivatives of 1 were calculated by B3LYP and B3PW91 and are observed in the benzene ring and in the azome- methods, and 6-311G(d,p) polarized set of vibration fre- thine group π>π* transitions. Secondly for 1b, the main quencies in gas phase to generate infra-red spectral infor- transition contribution from H->L (96%/96%) was deter- mation. There are 3N–6 free vibrational motions, therefore mined as n->π* transitions at 324.21/322.80 nm and In the synthesized compounds are of planar and nonlinear addition, the main transition contribution from H-1->L structure. 1a-c consist of 39, 35 and 32 atoms respectively, (77%/80%) was determined at 281.47/279.99 nm. The and have 111, 99 and 90 normal modes of fundamental main transition contribution from H-4->L (58%/64%) vibrations, respectively. The calculated FT-IR spectra were and H-2->L (39%/34%) were determined as π->π* transi- obtained from B3LYP and B3PW91 levels with 6-311G(d,p) tions at 318.39/316.43 nm. It is observed that these π>π* set (Figure 6). Negative frequency was not found in the transitions are in the benzene ring and in the azomethine data obtained from the optimized structure. The vibratio- group. Finally, the 1c molecule, the main transition con- nal frequencies obtained by Gaussian 09W are multiplied tribution from H->L (94%/94%) was determined as n->π* by 0.9516 for the B3LYP/6-311G(d,p) method and 0.9905 transitions at 331.14/330.09 nm and the main transition for the B3PW9/6-311G(d,p) method [29]. Veda4f program contribution from H-1->L (91%/90%) was determined as was used to determine the vibrational types obtained by n->π* transitions at 303.43/301.66 nm. In addition, the both methods [52]. The experimental IR spectral values main transition contribution from H->L+1 and (89%/40%) were compared with the theoretical IR spectral values and was determined at 280.90/279.88 nm are observed in some functional group regions were analyzed experimen- the benzene ring and in the azomethine group π>π* tally and theoretically. The obtained data were made com- transitions. patible with experimental data. Figure 6 Theoretically generated (B3LYP and B3PW91) IR spectrums of 1 type compounds, respectively
10 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
Table 8 Experimentally and theoretically 13C and 1H-NMR (B3LYP/(DMSO) and B3PW91/(DMSO)) chemical shift values of 1a molecule
according to TMS standard (δ/ppm)
13
C-NMR Experimental B3LYP/ B3PW91/ 1
H-NMR Experimental B3LYP/ B3PW91/
6311(d,p) 6311(d,p) 6311(d,p) 6311(d,p)
1C 146.18 153.61 147.92 16H 11.93 7.23 7.32
2C 151.14 153.89 148.79 17H 9.84 10.44 10.66
3C 147.33 146.60 142.41 18H 7.02 6.97 7.15
4C 131.77 142.18 136.07 19H 7.71 7.49 7.63
5C 142.87 150.71 145.57 20H 2.32 2.25 2.37
6C 129.98 134.16 130.38 21H 2.32 2.29 2.42
7C 131.22 138.57 133.48 22H 2.32 2.34 2.48
8C 13.72 13.73 10.74 23H 3.89 3.86 3.98
9C 30.28 34.61 30.48 24H 3.89 3.89 4.04
10C 127.44 130.43 125.16 25H 7.24 7.43 7.61
11C 129.93 135.84 131.93 26H 6.85 6.82 7.01
12C 113.84 111.41 107.63 28H 6.85 7.38 7.55
13C 158.11 165.06 159.42 29H 7.24 3.64 3.71
14C 113.84 120.40 116.44 H37 3.7 3.64 3.71
15C 129.93 135.55 131.58 H38 3.7 4.02 4.11
36C 55.01 54.55 50.65 H39 3.7 7.23 7.32
Table 9 Experimentally and theoretically 13C and 1H-NMR (B3LYP/(DMSO) and B3PW91/(DMSO)) chemical shift values of 1b molecule
according to TMS standard (δ/ppm)
13
C-NMR Experimental B3LYP/ B3PW91/ 1
H-NMR Experimental B3LYP/ B3PW91/
6311(d,p) 6311(d,p) 6311(d,p) 6311(d,p)
1C 145.33 152.66 147.00 16H 12.01 7.25 7.33
2C 151.14 153.74 148.65 17H 9.85 10.42 10.63
3C 147.51 148.85 142.65 18H 7.02 6.97 7.16
4C 132.95 142.03 135.94 19H 7.72 7.49 7.64
5C 143.00 150.99 145.84 20H 2.32 2.25 2.39
6C 128.93 134.16 130.39 21H 2.32 2.28 2.39
7C 131.23 138.72 133.63 22H 2.32 2.34 2.48
8C 13.73 13.75 10.78 23H 4.00 3.88 4.03
9C 30.78 35.11 31.07 24H 4.00 3.94 4.07
10C 131.65 142.52 137.36 25H 7.34 7.35 7.52
11C 130.21 134.72 130.72 26H 7.41 7.36 7.54
12C 127.62 145.85 139.79 27H 7.26 7.49 7.67
13C 138.02 131.44 127.49 28H 7.34 7.37 7.54
14C 127.62 133.76 129.77
15C 130.06 133.37 129.25
The corresponding heterocyclic 1,2,4-triazole com- cm−1 for B3LYP/6-311G(d,p) method and 1733-1735 cm−1 for
pounds have signals corresponding to N-H stretching B3PW91/6-311G(d,p) method were obtained. As shown in
vibrations. While NH stretching vibrations are observed Table 7, the peaks of the imine group in the Schiff base ring
in the range of 3160-3184 cm−1 in the experimental data, are observed in the experimental in the range of 1575-1590
theoretical signals were obtained using the B3LYP/6- cm−1 [27], whereas the calculated values in the range of 1621-
311G(d,p) method in the range of 3641-3646 cm−1 and for 1632 cm−1 for the B3LYP/6-311G(d,p) method and 1574-1584
the B3PW91/6-311G(d,p) method in the range of 3515-3520 cm−1 for the B3PW91/6-311G(d,p) method were obtained.
cm−1. The carbonyl peaks in the 1,2,4-triazol-5-one ring Experimental data [27] were found to be more compatible
were observed the range of 1695-1702 cm−1 in the experi- with the data obtained from B3PW91 when comparing vib-
mental data, whereas the theoretical ranges of 1782-1785 rational frequencies obtained by both methods.M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 11
Table 10 Experimentally and theoretically 13C and 1H-NMR (B3LYP/(DMSO) and B3PW91/(DMSO)) chemical shift values of 1c molecule
according to TMS standard (δ/ppm)
C-NMR
13
Experimental B3LYP/ B3PW91/ 1
H-NMR Experimental B3LYP/ B3PW91/
6311(d,p) 6311(d,p) 6311(d,p) 6311(d,p)
1C 144.11 150.07 144.20 15H 12.40 7.66 7.77
2C 151.55 154.30 149.21 16H 9.82 10.67 10.89
3C 150.23 147.21 143.07 17H 7.05 6.98 7.17
4C 131.60 142.17 136.01 18H 7.71 7.50 7.65
5C 143.33 150.97 145.94 19H 2.38 2.26 2.38
6C 130.27 134.20 130.44 20H 2.38 2.32 2.45
7C 131.28 138.88 133.82 21H 2.38 2.39 2.53
8C 13.77 13.76 10.77 22H 7.92 8.17 8.37
9C 126.66 131.72 126.67 23H 7.51 7.69 7.87
10C 128.39 131.71 127.68 24H 7.52 7.70 7.88
11C 127.74 132.46 128.56 25H 7.53 7.73 7.91
12C 130.01 134.40 130.42 26H 7.94 8.48 8.80
13C 127.74 132.02 128.12
14C 128.39 133.59 129.24
Figure 7 Comparison of experimental data with theoretical 13C-NMR and 1H-NMR chemical shift values obtained by B3LYP (DMSO) and
B3PW91 (DMSO) methods of 1 type compounds, respectively
NMR Spectral Analysis C-NMR and 1H-NMR chemical shift values were cal-
13
culated by regression analysis via analysing experimen-
The isotropic chemical shift analysis allowed us to iden- tal data using the least squares method. The obtained R2
tify relative ionic species, and to calculate reliable mag- values were found to be nearly 1, especially for 13C-NMR
netic properties in nuclear magnetic resonance (NMR) data (Figure 7).
spectroscopy, providing accurate predictions of molecular It is well known that aromatic carbon atoms give
geometries [53-55]. In the study, 13C-NMR and 1H-NMR che- NMR signals in the range of 100-150 ppm. However, in
mical shift values of 1a-c were obtained from optimized coordination with electronegative atoms, these NMR
structures with minimum energy. Chemical shift values of signals resulting from aromatic carbon atoms shift
1a-c were obtained by using optimized structures, obtai- to higher values [57, 58]. Experimentally and theore-
ned from of B3LYP and B3PW91 methods, by using Gauge- tically generated 13C-NMR and 1H-NMR isotropic shift
Independent Atomic Orbital (GIAO) NMR using 6-311G(d,p) values were compared and a linear correlation was
basis set in a DMSO solvent phase (Tables 8-10) [56]. observed (Figure 6). Theoretical chemical shifts of12 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
3-substituted-4-(3-methyl-2-thienylmethyleneamino)- transitions. It was found that the calculated absorption
4,5-dihydro-1H-1,2,4-triazol-5-one compounds were opti- wavelengths closely matched those of the experimentally-
mized to the most stable structure using the B3LYP and derived values. Vibrational frequencies were calculated
B3PW91 methods using the 6-311G(d,p) basis set. It has too from the optimized structures and it was determined
been found out that 13C-NMR chemical shift values are the experimental FT-IR spectral values compared favoura-
highly compatible between the GIAO-NMR approach bly with the theoretical values. NMR chemical shift values
and experimental data [27]. In the 1H-NMR chemical shift of the titled compounds were obtained, using B3LYP and
values, it was determined that the R2 value was lower than B3PW91 methods, by using the GIAO NMR approach using
expected, since the N-H proton in the 1,2,4-triazole-5-on 6-311G(d,p) basis set in DMSO. It was found that 13C-NMR
ring has an acidic value [59]. chemical shift values were highly comparable between
GIAO-NMR data and experimental data. For 1H NMR it was
determined that the correlation was lower than expected,
Conclusions since the N-H proton in the 1,2,4-triazole-5-on ring has an
acidic value in the 1H-NMR chemical shift values.
The 3-substituted-4-(3-methyl-2-thienylmethyleneamino)-
4,5-dihydro-1H-1,2,4-triazol-5-ones used in the study were
optimized with DFT methods and polarized functions Materials and Methods
using the Gaussian 09W program, and the minimum
energy, most stable placements and space structure of each Experimental Method
atom in the compounds were determined. Based on the
optimized structures, the C‒C, C-H and C-S bond lengths In the study, 3-p-methoxybenzyl-4-(3-methyl-2-thieny
of the S-type compounds were compared with the data lmethyleneamino)-4,5-dihydro-1H-1,2,4-triazol-5-one (1a),
in the literature according to the DFT/B3LYP/6-311G(d,p) 3-m-chlorobenzyl-4-(3-methyl-2-thienylmethyleneamino)-
method. The obtained values were found to match those 4,5-dihydro-1H-1,2,4-triazol-5-one (1b) and 3-phenyl-4-
reported in the literature. The electronic properties of the (3-methyl-2-thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-
synthesized compounds obtained from HOMO and LUMO triazol-5-one (1c) were obtained from reaction between
energies were theoretically calculated. The molecules 3-methylthiophene-2-carbaldehyde and three different
examined contained substrates such as p-methoxybenzyl 4-amino-3-p-methoxybenzyl/m-chlorobenzyl/phenyl-
(1a), m-chlorobenzyl (1b) and phenyl (1c). When the donor 4,5-dihydro-1H-1,2,4-triazole-5-ones [27] (Scheme 1).
and acceptor substituents examined the effect on structu-
res, it was found that 1b with its electron-donating ring
substituent had a high energy gap. 1c was found to be the General procedure for the synthesis of 1 type compounds
molecule with the highest intra-molecular charge density.
Molecular electrostatic potentials were calculated in opti- 3-methylthiophene-2-carboxialdehyde A (0.01 mol)
mized geometry to estimate the reactive regions of electro- was dissolved in acetic acid (15 mL) and reacted
philic and nucleophilic attacks for the studied molecules. with the corresponding compounds T (0.01 mol) to
NLO properties of molecules were calculated such as total 3-p-methoxybenzyl/3-m-chlorobenzyl/3-phenyl-4-(3-
static dipole moment, polarizability and first order hyper- methyl-2-thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-
polarizability. The data obtained were compared with the triazol-5-ones (1a-c) and was refluxed for 1.5 hour. Then,
reported values of similar derivatives in the literature and the solution evaporated at 50-55 °C in vacuo. The residue
it was observed that they have provided better results. The was crystallized several times in ethanol and pure 1a-c
calculated hyperpolarizability of molecules appears to be compounds were obtained as white crystals.
significantly higher than the urea value, so we can con- 3-p-Methoxybenzyl- 4-( 3-methyl-2-
clude that the theoretically studied molecules are attrac- thienylmethyleneamino)-4,5-dihydro-1H-1,2,4-tri-
tive for their potential value given their NLO properties. azol-5-one (1a) Yield (white solid) 94%; IR (υ, cm−1):
UV-visible absorption spectra of 1a-c were investigated 3161 (NH), 1702 (C=O), 1590 (C=N), 850 (1,4-disubstituted
experimentally and theoretically in ethanol. The role of benzenoid ring); 1H-NMR (400 MHz, DMSO-d6): δ 11.93 (s,
the substrate and the effect of solvent on the UV spect- 1H, NH), 9.84 (s, 1H, N=CH), 7.71 (d, 1H, ArH; J=5.20 Hz),
rum were considered and the GaussSum3.0 program was 7.24 (d, 2H, ArH; J=8.80 Hz), 7.02 (d, 1H, ArH; J=5.20 Hz),
used to determine the stimulation contributions in UV-vis 6.85 (d, 2H, ArH; J=8.80 Hz), 3.89 (s, 2H, CH2Ph), 3.70 (s,M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 13
Scheme 1 Synthesis route of 1 type compounds
Figure 8 The optimized molecular structure of 3-benzyl/p-methylbenzy/p-chlorobenzyl-4-(3-methyl-2-thienylmethyleneamino)-4,5-dihydro-
1H-1,2,4-triazol-5-one (1) with DFT/B3LYP/ 6-311G(d,p) level
3H, PhOCH3), 2.32 (s, 3H, CH3); 13C-NMR (100 MHz, DMSO- Calculation Methods
d6): δ 151.14 (triazole-C2), 147.33 (N=CH), 146.18 (triazole-
C1), 158.11, 142.87, 131.77, 131.22, 129.98, 129.93 (2C), 127.44, Approximate geometry of three dimensions in Denning-
113.84 (2C) (Ar-C), 55.01 (OCH3), 30.28 (CH2Ph), 13.72 (CH3); ton et al., the gas phase and basis state molecules were
mp 207 °C (dec). recorded and drawn in GaussView5.0 molecular imaging
3-m-Chlorobenzyl-4-(3-methyl-ı2- software (Figure 8) [28]. The initial geometries of the mole-
thienylmethyleneamino)-4,5-dihydro-1H-1, 2,4- cules were obtained in GaussView 5.0 package software
triazol-5-one (1b) Yield (white solid) 74%; IR (cm−1): 3180 and transferred to Gaussian 09W software as input data
(NH), 1700 (C=O), 1579 (C=N), 788 and 622 (1,3-disubsti- [29, 30]. Many parameters such as geometric, spectrosco-
tuted benzenoid ring); 1H-NMR (400 MHz, DMSO-d6): pic, electronic and thermodynamic properties of molecu-
δ 12.01 (s, 1H, NH), 9.85 (s, 1H, N=CH), 7.72 (d, 1H, ArH; les to be examined from the optimized structure can be
J=5.20 Hz), 7.41 (s, 1H, ArH), 7.26-7.34 (m, 3H, ArH), 7.02 analysed. The basis or excited states of compounds or
(d, 1H, ArH; J=4.80 Hz), 4.00 (s, 2H, CH2Ph), 2.32 (s, 3H, atoms can be used in theoretical calculation processes [29,
CH3); 13C-NMR (100 MHz, DMSO-d6): δ 151.14 (triazole-C2), 30, 60]. All calculations were made on computers located
147.51 (N=CH), 145.33 (triazole-C1), 143.00, 138.02, 132.95, in Chemistry Department of Kafkas University Science
131.65, 131.23, 130.21, 130.06, 128.93, 127.62, 126.78 (Ar-C), Faculty.
30.78 (CH2Ph), 13.73 (CH3); mp 165 °C (dec). The ab-initio method is based on the analysis of the
3-Phenyl-4-(3-methyl-2-thienylmethyleneamino)- Schrödinger wave equation without experimental values
4,5-dihydro-1H-1,2,4-triazol-5-one (1c) Yield (white [61]. It seems that the solution of my Schrödinger wave
solid) 97%; IR (υ, cm−1): 3160 (NH), 1695 (C=O), 1575 (C=N), equation is possible with a single electron hydrogen
766 and 686 (monosubstituted benzenoid ring); 1H-NMR atom. However, mathematical approaches such as DFT
(400 MHz, DMSO-d6): δ 12.40 (s, 1H, NH), 9.82 (s, 1H, (density function theory) are used as it has been challen-
N=CH), 7.94-7.92 (m, 2H, Ar-H), 7.71 (d, 1H, ArH; J=5.20 Hz), ging to analyse in multi-electron structures. In an attempt
7.52 (t, 3H, ArH; J=6.40 Hz), 7.05 (d, 1H, ArH; J=4.80 Hz), to determine the electronic properties of the structures
2.38 (s, 3H, CH3); 13C-NMR (100 MHz, DMSO-d6): δ 151.55 better, the DFT method was used which takes into account
(triazole-C2), 150.23 (N=CH), 144.11 (triazol-C1), 143.33, the electron density and generates the desired data on this
131.60, 131.28, 130.27, 130.01, 128.39 (2C), 127.74 (2C), 126.66 electron density. In addition, the B3LYP hybrid function
(Ar-C), 13.77 (CH3); mp 202 °C (dec). in the Gaussian 09W software has been applied suitable14 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
for workstation capacity and polarized 6-311G (d,p) basis [3] Cai S, Li QS, Borchardt RT, Kuczera K, Schowen RL. The
set [62]. antiviral drug ribavirin is a selective inhibitor of S-adenosyl-L-
homocysteine hydrolase from Trypanosoma cruzi. Bioorg Med
In this study, we were optimized using DFT/B3LYP
Chem. 2007 Dec;15(23):7281–7.
and DFT/B3PW91 methods In order to find the minimum [4] Rao BM, Chakraborty A, Srinivasu MK, Devi ML, Kumar PR,
energy and the most stable structure of the synthesized Chandrasekhar KB, et al. A stability-indicating HPLC
molecules, the bond lengths of the related compounds assay method for docetaxel. J Pharm Biomed Anal. 2006
were determined from the optimized geometric structure May;41(2):676–81.
with minimum energy found. Vibration frequencies were [5] Ashish C, Ravi V, Rachana I, Thrimoorthy P. Intranasal spray
formulation containing rizatriptan benzoate for the treatment
calculated from the optimized structure of the molecules.
of migraine. Int J Pharm. 2019;5173(19):30747–51.
Veda4f program [44] was used to determine the vibration [6] Molvi KI, Vasu KK, Yerande SG, Sudarsanam V, Haque N.
types of the calculated IR frequencies by using computer- Syntheses of new tetrasubstituted thiophenes as
aided Gaussian 09W package program. The theoretically novel anti-inflammatory agents. Eur J Med Chem. 2007
calculated vibration frequency values are multiplied by Aug;42(8):1049–58.
[7] Shukla R, Mohan TP, Vishalakshi B, Chopra D. Synthesis,
appropriate scale factors, and they are compared with
crystal structure and theoretical analysis of intermolecular
the experimental values [27]. Theoretical IR spectra interactions in two biologically active derivatives of 1, 2,
were drawn according to DFT/B3LYP and DFT/B3PW91 4-triazoles. J Mol Struct. 2017;1134:426–34.
methods. Chemical shift values of 1H-NMR and 13C-NMR [8] Noguchi H, Kitazumi K, Mori M, Shiba T. Electroencepha-
were calculated according to GIAO method using opti- lographic properties of zaleplon, a non-benzodiazepine
mized structure. The theoretically obtained chemical shift sedative/hypnotic, in rats. J Pharmacol Sci. 2004
Mar;94(3):246–51.
values were compared with the experimental values and it
[9] Polívka Z, Holubek J, Svatek E, Metys J, Protiva M. Potential
was observed that they were compatible. In addition, the hypnotics and anxiolytics: synthesis of 2-bromo-
3-substituted-4-(3-methyl-2-thienylmethyleneamino)-4,5- 4-(2-chlorophenyl)-9-[4-(2-methoxyethyl)piperazino]-
dihydro-1H-1,2,4-triazole-5-ones calculated HOMO-LUMO 6H-thieno[3,2,4-triazolo[4,3-a]-1,4-diazepine and of
energies, energy differences and Electronic parameters some related compounds. Collect Czech Chem Commun.
1984;49(3):621–36.
such as I; Ionization potential, A; electron affinity, ΔE;
[10] Arroyo S, Salas-Puig J, Grupo Español de Investigación sobre
Energy Gap, χ; electronegativity, S; molecular softness, ω; Tiagabina. Estudio abierto con tiagabina en epilepsia parcial.
Electrophilic Index, IP; Nucleophilic Index Pi, Chemical [An open study of tiagabine in partial epilepsy]. Rev Neurol.
Potential derived from HOMO-LUMO energies and Total 2001 Jun;32(11):1041–6.
Energy. [11] Slivka M, Korol N, Pantyo V, Baumer V, Lendel V. Regio- and
stereoselective synthesis of [1,3]thiazolo[3,2-b][1,2,4]
triazol-7-ium salts via electrophilic heterocyclization of
Research funding: The author(s) received no financial
3-S-propargylthio-4Н-1,2,4-triazoles and their antimicrobial
support for the research, authorship, and/or publication activity. Heterocycl Commun. 2017;23(2):109–13.
of this article. [12] Aktaş-Yokuş Ö, Yüksek H, Manap S, Aytemiz F, Alkan M,
Beytur M, et al. In-vitro biological activity of some new
Conflict of interest: Authors state no conflict of interest. 1,2,4-triazole derivatives with their potentiometric titrations.
Bulg Chem Commun. 2017;49(1):98–106.
[13] Yüksek H, Göksu B, Manap S, Beytur M, Gürsoy Kol Ö.
Data Availability Statement: All data generated or ana- Synthesis of some new 4-[2-(2-methylbenzoxy)-
lyzed during this study are included in this published benzylidenamino]-4,5-dihydro-1H-1,2,4-triazol-5-one
article. derivatives with their antioxidant properties. Int J Chem.
2018;22(2):1–29.
[14] Körödi F, Szabo Z. Szabo. Z. Fused 1,2,4-trıazole
References
heterocycles. III. Syntheses and structures of novel [1,2,4]
trıazolo[1,3]thıazınoquınolınes. Heterocycl Commun.
1995;1(4):297–306.
[1] Nithyabalaji R, Krishnan H, Sribalan R. Synthesis, [15] Bahçeci Ş, Yıldırım N, Gürsoy-Kol Ö, Manap S, Beytur M,
molecular structure and multiple biological activities Yüksek H. Synthesis, characterization and antioxidant
of N-(3-methoxyphenyl)-3-(pyridin-4-yl)-1H-pyrazole-5- properties of new 3-alkyl (aryl)-4-(3-hydroxy-4-methoxy-
carboxamide. J Mol Struct. 2019;1186:1–10. benzylidenamino)-4,5-dihydro-1H-1,2,4-triazol-5-ones.
[2] Dadiboyena S, Valente EJ, Hamme AT 2nd. A novel synthesis Rasayan J Chem. 2016;9(3):494-501.
of 1,3,5-trisubstituted pyrazoles through a spiro-pyrazoline [16] Zolezzi S, Spodine E, Decinti A. Electrochemical studies of
intermediate via a tandem 1,3-dipolar cycloaddition/ copper(II) complexes with Schiff-base ligands. Polyhedron.
elimination. Tetrahedron Lett. 2009 Jan;50(3):291–4. 2002;21(1):55–9.M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted 15
[17] Ambike V, Adsule S, Ahmed F, Wang Z, Afrasiabi Z, Sinn E, et al. [34] Sastri VS, Perumareddi JR. Molecular orbital theoretical
Copper conjugates of nimesulide Schiff bases targeting VEGF, studies of some organic corrosion inhibitors. Corrosion.
COX and Bcl-2 in pancreatic cancer cells. J Inorg Biochem. 2007 1997;53(8):617–22.
Oct;101(10):1517–24. [35] Jesudason EP, Sridhar SK, Malar EJ, Shanmugapandiyan P,
[18] Beytur M, Turhan Irak Z, Manap S, Yüksek H. Synthesis, charac- Inayathullah M, Arul V, et al. Synthesis, pharmacological
terization and theoretical determination of corrosion inhibitor screening, quantum chemical and in vitro permeability studies
activities of some new 4,5-dihydro-1H-1,2,4-Triazol-5-one of N-Mannich bases of benzimidazoles through bovine cornea.
derivatives. Heliyon. 2019 Jun;5(6):e01809. Eur J Med Chem. 2009 May;44(5):2307–12.
[19] Beytur M, Kardaş F, Akyıldırım O, Özkan A, Bankoğlu B, Yüksek [36] Masoud MS, Ali AE, Shaker MA, Elasala GS. Synthesis,
H, et al. A highly selective and sensitive voltammetric sensor computational, spectroscopic, thermal and antimicrobial
with molecularly imprinted polymer based silver@gold activity studies on some metal-urate complexes. Spectrochim
nanoparticles/ionic liquid modified glassy carbon electrode Acta A Mol Biomol Spectrosc. 2012 May;90:93–108.
for determination of ceftizoxime. J Mol Liq. 2018;251:212–7. [37] Gökce H, Bahçeli S. A study on quantum chemical calculations
[20] Al Zoubi W, Al Mohanna N. Membrane sensors based on Schiff of 3-, 4-nitrobenzaldehyde oximes. Spectrochim Acta A Mol
bases as chelating ionophores—a review. Spectrochim Acta A Biomol Spectrosc. 2011 Sep;79(5):1783–93.
Mol Biomol Spectrosc. 2014 Nov;132:854–70. [38] Arivazhagan M, Subhasini VP. Quantum chemical studies on
[21] Di Bella S, Oliveri IP, Colombo A, Dragonetti C, Righetto S, structure of 2-amino-5-nitropyrimidine. Spectrochim Acta A
Roberto D. An unprecedented switching of the second-order Mol Biomol Spectrosc. 2012 Jun;91:402–10.
nonlinear optical response in aggregate bis(salicylaldiminato) [39] Mebi CA. DFT study on structure, electronic properties, and
zinc(II) Schiff-base complexes. Dalton Trans. 2012 reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. J Chem Sci.
Jun;41(23):7013–6. 2011;123(5):727–31.
[22] Kumar S, Dhar DN, Saxena PN. Applications of metal [40] Kiyooka S, Kaneno D, Fujiyama R. Parr’s index to describe
complexes of Schiff bases-A review. J Sci Ind Res (India). both electrophilicity and nucleophilicity. Tetrahedron Lett.
2009;68(3):181–7. 2013;54(4):339–42.
[23] Hosny NM, Hussien MA, Radwan FM, Nawar N. Synthesis, [41] Pearson RG. Absolute electronegativity and hardness:
spectral characterization and DNA binding of Schiff-base metal application to inorganic chemistry. Inorg Chem.
complexes derived from 2-amino-3-hydroxyprobanoic acid and 1988;27(4):734–40.
acetylacetone. Spectrochim Acta A Mol Biomol Spectrosc. 2014 [42] Geskin VM, Lambert C, Brédas JL. Origin of high second- and
Nov;132:121–9. third-order nonlinear optical response in ammonio/borato
[24] Gümüş S, Türker M. Substituent effect on the aromaticity of diphenylpolyene zwitterions: the remarkable role of polarized
1,3-azole systems. Heterocycl Commun. 2012;18(1):12–6. aromatic groups. J Am Chem Soc. 2003 Dec;125(50):15651–8.
[25] Lienard P, Gavartin J, Boccardi G, Meunier M. Predicting drug [43] Rajeshirke M, Sekar N. NLO properties of ester containing
substances autoxidation. Pharm Res. 2015 Jan;32(1):300–10. fluorescent carbazole based styryl dyes – Consolidated
[26] Rai NS, Kalluraya B, Lingappa B, Shenoy S, Puranic VG. spectroscopic and DFT approach. Opt Mater. 2018;76:191–09.
Convenient access to 1,3,4-trisubstituted pyrazoles carrying [44] Zhang CR, Chen HS, Wang GH. Changping A stable-manifold-
5-nitrothiophene moiety via 1,3-dipolar cycloaddition of based method for chaos control and synchronization. Chem
sydnones with acetylenic ketones and their antimicrobial Res Chin Univ. 2004;5:947–54.
evaluation. Eur J Med Chem. 2008 Aug;43(8):1715–20. [45] Binil PS, Mary YS, Varghese HT, Panicker CY, Anoop MR,
[27] Gürsoy-Kol Ö, Yüksek H, İslamoğlu F. Synthesis and Manojkumar TK. Infrared and Raman spectroscopic
in vitro antioxidant activities of novel 4-(3-methyl-2- analyses and theoretical computation of 4-butyl-
thienylmethyleneamino)-4-5-dihydro-1H-1,2,4-triazol-5-one 1-(4-hydroxyphenyl)-2-phenyl-3,5-pyrazolidinedione.
derivatives with their acidic properties. J Chem Soc Pak. Spectrochim Acta A Mol Biomol Spectrosc. 2012 Aug;94:101–9.
2013;35(4):1179–90. [46] Adant C, Dupuis M, Bredas JL. Ab initio study of the nonlinear
[28] Dennington R, Keith T. Millam J. GaussView. Version 5.0. optical properties of urea: electron correlation and dispersion
Shawnee Mission: Semichem Inc.; 2009. effects. Int J Quantum Chem. 1995;56(S29):497–507.
[29] Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, [47] Luque FJ, Lopez JM, Orozco M. Perspective on electrostatic
Mennucci B, et al. Gaussian 09. Version A.02. Wallingford: interactions of a solute with a continuum. a direct utilization
Gaussian Inc.; 2009. of ab initio molecular potentials for the prevision of solvent
[30] Foresman JB, Frisch A. Exploring Chemistry with electronic effects. Theor Chem Acc. 2000;103(3-4):343–5.
structure methods. Pittsburgh: Gaussian Inc., 1996. [48] Li Y, Liu Y, Wang H, Xiong X, Wei P, Li F. Synthesis, crystal
[31] İkizler AA. Organik Kimyaya Giriş: Dördüncü Baskı. Trabzon: structure, vibration spectral, and DFT studies of 4-aminoan-
KTÜ Basımevi; 1996. tipyrine and its derivatives. Molecules. 2013 Jan;18(1):877–93.
[32] Anand S, Muthusamy A. Synthesis, characterization, [49] Moro S, Bacilieri M, Ferrari C, Spalluto G. Autocorrelation of
electrochemical, electrical, thermal and ESIPT behaviour of molecular electrostatic potential surface properties combined
oligobenzimidazoles of certain substituted benzimidazole with partial least squares analysis as alternative attractive tool
carboxylic acids and their diode applications. J Mol Struct. to generate ligand-based 3D-QSARs. Curr Drug Discov Technol.
2019;1177:78–89. 2005 Mar;2(1):13–21.
[33] Koopmans T. Über die zuordnung von wellenfunktionen [50] O’Boyle NM, Tenderholt AL, Langner KM. cclib: a library for
und eigenwerten zu den einzelnen elektronen eines atoms. package-independent computational chemistry algorithms.
Physica. 1934;1(1-6):104–13. J Comput Chem. 2008 Apr;29(5):839–45.16 M. Beytur and I. Avinca: Spectroscopic Analysis of Heterocyclic 3-Substituted
[51] Zhenming D, Heping S, Yufang L, Diansheng L, Bo L. [56] Wolinski K, Hinton JF, Pulay P. Efficient implementation of the
Experimental and theoretical study of 10-methoxy-2- gauge-independent atomic orbital method for NMR chemical
phenylbenzo[h]quinoline. Spectrochim Acta A Mol Biomol shift calculations. J Am Chem Soc. 1990;112(23):8251–60.
Spectrosc. 2011 Mar;78(3):1143–8. [57] Pihlaja K, Kleinpeter E. Carbon-13NMR Chemical Shifts in
[52] Jamróz MH. Vibrational Energy Distribution Analysis VEDA 4 Structural and Sterochemical Analysis: VCH Publishers.
program, Warsaw, 2004-10 [cited 2021 Jan 21]. Available from: Deerfield: Beach; 1994.
https://smmg.pl/software/veda. [58] Kalinowski HO, Berger S, Braun S. Carbon-13 NMR
[53] Rani AU, Sundaraganesan N, Kurt M, Cinar M, Karabacak Spectroscopy. Chichester: John Wiley & Sons; 1988.
M. FT-IR, FT-Raman, NMR spectra and DFT calculations on [59] Bahçeci Ş, Yüksek H, Ocak Z, Köksal C, Özdemir M. Synthesis
4-chloro-N-methylaniline. Spectrochim Acta A Mol Biomol and non-aqueous medium titrations of some new 4,5-dihydro-
Spectrosc. 2010 May;75(5):1523–9. 1H-1,2,4-triazol-5-one derivatives. Acta Chim Slov. 2002;49:
[54] Subramanian N, Sundaraganesan N, Jayabharathi J. 783–94.
Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR, [60] Turhan Irak Z, Gümüş S. Heterotricyclic compounds via click
UV) studies and first-order molecular hyperpolarizabilities of reaction: A computational study. Noble Int J Sci Res. 2017;1(7):80–9.
1,2-bis(3-methoxy-4-hydroxybenzylidene)hydrazine by density [61] Jensen F. Introduction to Computational Chemistry. John
functional method. Spectrochim Acta A Mol Biomol Spectrosc. Wiley&Sons Ltd; 1999.
2010 Jul;76(2):259–69. [62] Becke AD. Density-functional thermochemistry. IV. A new
[55] Wade LG. Organic Chemistry. New Jersey: Pearson Prentice dynamical correlation functional and implications for exact-
Hall; 2006. exchange mixing. J Chem Phys. 1996;104(3):1040–6.You can also read

















































