Anti-SARS Drug Discovery and Development
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
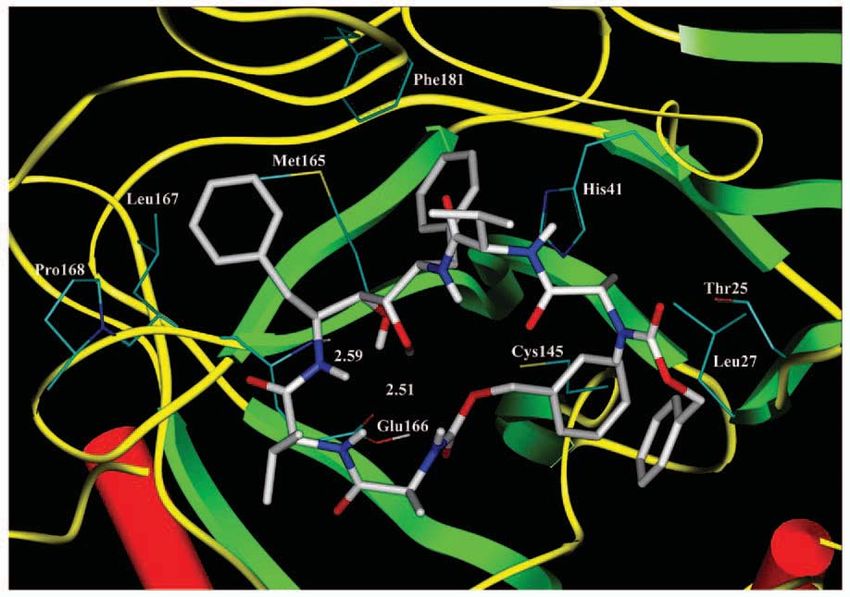
Fig 1. The life cycle of SARS
coronavirus.
Anti-SARS Drug Discovery and
Development
Severe Acute Respiratory Syndrome (SARS) is caused by a novel coronavirus
originally found in Guangdong, China in 2002, creating a global medical and eco-
nomical crisis. Due to the unknown nature of this newly emerged disease and the
lack of effective treatments, the initial rapid spread of the disease resulted in more
than 700 deaths around the world (1, 2). Figures
1 and 2 show the life cycle and replication of
SARS coronavirus.
At the early stage of the pandemic, we
organized an anti-SRAS drug discovery pro-
gram to screen about ten thousand compounds
and natural products that were collected from
many research laboratories. The anti-SARS
compound screening was initiated in a P-4 labo-
ratory at Institute of Preventive Medicine,
National Defense University, Taipei, Taiwan
using the prevention of SARS-mediated cyto-
toxicity as the criterion for the selection of anti-
SARS compounds. Figure 3 summarizes the
protocol used for our anti-SARS screening (3).
The positive compounds (or hits) identified
were subjected to a serial validation studies
including virology studies (infection induced Fig 2. SARS coronavirus replication.
51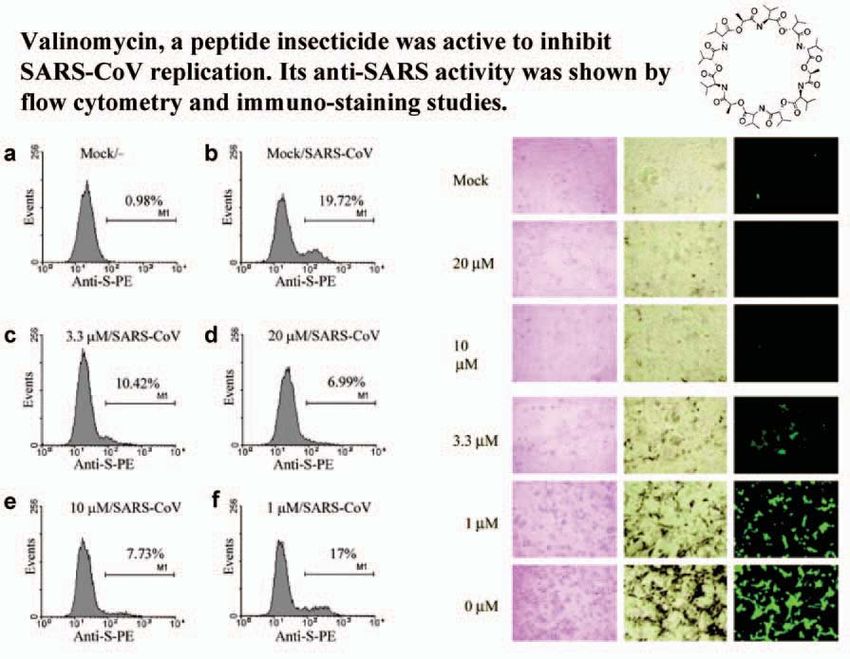
Fig 3. Anti-SARS Screening Flow Chart.
cytopathic effects, viral titer determinations by end-point titration and plaque
count), and various techniques to determine viral antigen levels produced in the
presence of increasing concentrations of test compounds. For compounds that might
be inhibitory to viral enzymes, such as the 3CL-protease (3CLpro; the main pro-
tease), enzymatic assays were conducted.
One of the active anti-SARS compounds was later identified as Valinomycin, a
peptide insecticide. Some of the studies conducted to confirm Valinomycin anti-
Fig 4. Anti-SARS activity of Valinomycin as shown in flow cytometry and immuno-staining studies.
52
SARS activity are summarized in Figure 4. We developed a protocol, using flow Fig 5. Anti-SARS com-
pounds identified by
cytometry, to evaluate the SARS spike protein levels that were detectable only in screening that are also
approved drugs or under
SARS-infected cells (Figure 4, a and b). In the presence of increasing amounts of
clinical studies.
Valinomycin, the amounts of the spike protein were reduced significantly. Similarly,
increased Valinomycin doses resulted in proportional increases in anti-SARS activi-
ties as observed by the lack of cytotoxicity, and the reduction of the associated viral
antigens as measured by immuno-staining and immuno-fluorescence studies (see
Figure 4 right panel).
Among the ten thousand small molecule compounds and natural products
screened in this program, fifteen active anti-SARS compounds were identified. Two
compounds, Reserpine and Aescin are approved drugs. Reserpine is used primarily
as a peripheral anti-hypertensive and sedative. Aescin is used for chronic venous
insufficiency and hemorrhoids. In addition, three other compounds went through
clinical studies. These compounds could be developed as anti-SARS drugs in a rela-
tively short time, if needed
(Figure 5). For the discovery
of SARS specific drug leads,
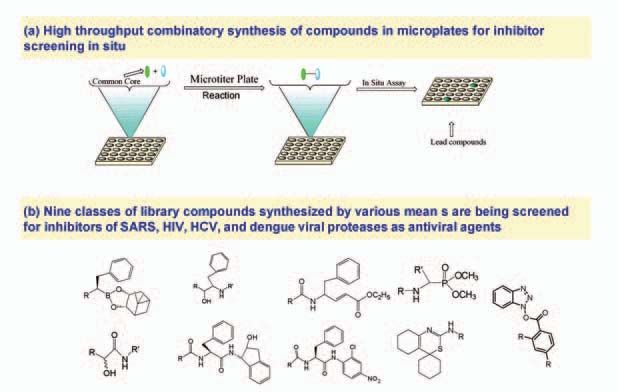
we designed and synthesized
greater than 5,000 compounds
to target the SARS main pro-
tease (or 3CL pro ) (4). To
achieve high throughput syn-
thesis of protease inhibitor
compounds, a new synthetic
approach was developed for
parallel synthesis of com-
pounds in 96-microwell plates
Fig 6. Synthesis in microtiter plates for screening in situ, and the structure diversity of the protease
followed by screening in situ. inhibitors identified.
53
The yields of compounds thus
synthesized were high enough to
allow in situ screening of these
compounds without purification
(Figure 6a). The combination of
the parallel compound synthesis
and in situ screening resulted in
shorter turn around times to iden-
tify potent inhibitors (5). Using
this and other synthetic schemes,
we synthesized a diversified pro-
tease inhibitor library containing
nine classes of chemical skele-
tons as shown in Figure 6b.
Table 1. Perspectives
The protease inhibitors syn-
after the anti-SARS drug thesized were evaluated by enzymatic screening using recombinant 3CLpro and a
discovery effort.
fluorescence substrate (6). This was followed by measurements to determine the
inhibition constants of these inhibitors. The structural and activity relationship of the
active inhibitors were selected so as to generate inhibitors that were more potent in
the next round of structure design and development. Most of the active inhibitors
were subjected to anti-SARS tests as described above. A combination of all of these
studies resulted in the identification of several potent 3CLpro inhibitors with Ki val-
ues at nM levels (7, 8). A few of them were also found active as potent anti-SARS
agents (Figure 7). These compounds are therefore drug development candidates for
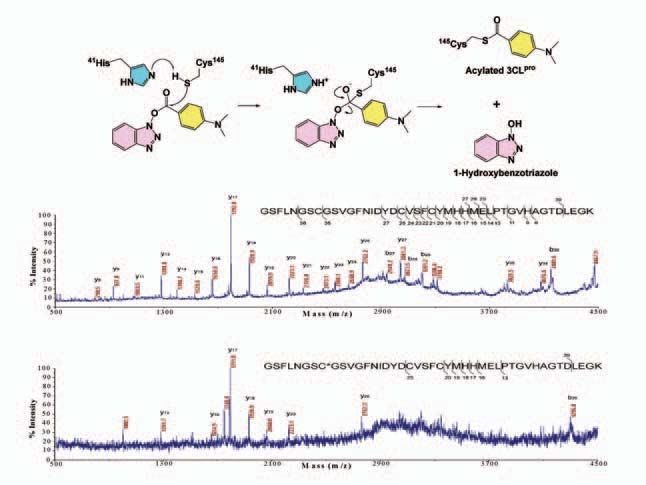
specific anti-SARS therapies. To further understand the mechanism of inhibition, a
benzotriazole ester candidate was investigated using mass spectrometry and kinetic
Fig 7. Structures and inhibition constants of potent inhibitors against SARS-CoV 3CLpro and the SARS virus.
54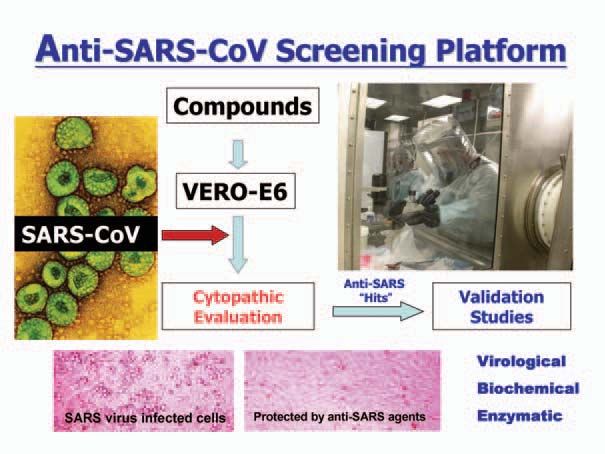
analysis and was shown to
form a covalent S-acyl
enzyme intermediate with
3CL pro (Figure 8). Since
the covalently modified
3CL pro protease is no
longer active, the life cycle of
SARS coronavirus is thus
blocked.
We are now exploring
the potential of these lead
inhibitors for other viral
proteases, such as HIV,
HCV, and dengue viruses.
A recently developed screening
protocol for HIV protease was used to test these compounds. Preliminary results indicated Fig 8. A new class of stable
benzotriazole esters as
that some new HIV protease inhibitors are also inhibitors of these viruses. We are SARS-CoV 3CLpro inhibitors.
These inhibitors act as mech-
particularly interested in developing TL-3, an antiviral for SARS, HIV and FIV
anism-based inactivators.
(Feline Immunodeficiency Virus), as a drug candidate for SARS and HIV. Figure 9
shows how TL-3 interacts with the SARS protease using computer-based molecular
modeling. We have successfully scaled up TL-3 synthesis, and started various phar-
macology studies. In the initial four toxicity studies, TL-3 has been found to be free
of toxic properties. A follow-up GLP toxicity study on mice also showed that at the
highest administered dose of 200 mg/kg, TL-3 was non-toxic. At the end of preclini-
cal studies, we intend to transfer the intellectual property to a private sector to apply
an IND for TL-3 to start its clinical development. In addition to the preparation for
TL-3 drug development, we are working to develop protocols to study antiviral drug
candidates in animal disease models and pharmacology using P3 facilities. At this
time, a murine model for SARS infection has been established. An improved model
using ferrets that produce disease symptoms as well as allow SARS infections is cur-
rently under development. The facility built in the Institute of Preventive Medicine is
equipped to conduct these studies. In addition, preparations to study virally infected
primates are also under way. Table 1 summarizes our current and future work after
the anti-SARS drug discovery study. The immediate threat of SARS is over; never-
theless, we intend to continue the anti-SARS program, partly to guard for its possible
return. More importantly, we need to transform what we learned from the SARS
experience to other emerging viral diseases that almost certainly will come to threat-
en in the future.
Chi-Huey Wong
Genomics Research Center, Academia Sinica
Proceedings of the National Academy of Sciences 101, 10012-10017 (2004)
55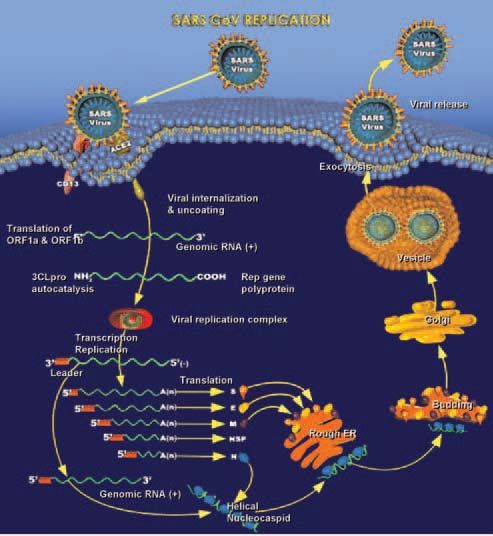
Fig 9. The molecular
model of 3CLpro-TL3
complex.
References
1. Ksiazek, T. G.; Erdman, D.; Goldsmith, C. S.; Zaki, S. R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.;
Comer, J. A.; Lim, W.; Rollin, P. E.; Dowell, S. F.; Ling, A. E.; Humphrey, C. D.; Shieh, W. J.;
Guarner, J.; Paddock, C. D.; Rota, P.; Fields, B.; DeRisi, J.; Yang, J. Y.; Cox, N.; Hughes, J. M.;
LeDuc, J. W.; Bellini, W. J.; Anderson, L. J., A novel coronavirus associated with severe acute respira-
tory syndrome. N Engl J Med 2003, 348, (20), 1953-66.
2. Chinese, S. M. E. C., Molecular evolution of the SARS coronavirus during the course of the SARS
epidemic in China. Science 2004, 303, (5664), 1666-9.
3. Wu, C. Y.; Jan, J. T.; Ma, S. H.; Kuo, C. J.; Juan, H. F.; Cheng, Y. S.; Hsu, H. H.; Huang, H. C.; Wu,
D.; Brik, A.; Liang, F. S.; Liu, R. S.; Fang, J. M.; Chen, S. T.; Liang, P. H.; Wong, C. H., Small mole-
cules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci U S A 2004,
101, (27), 10012-7.
4. Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J. R.; Hilgenfeld, R., Coronavirus main proteinase
(3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, (5626), 1763-7.
5. Chang, C. F.; Ho, C. W.; Wu, C. Y.; Chao, T. A.; Wong, C. H.; Lin, C. H., Discovery of picomolar slow
tight-binding inhibitors of alpha-fucosidase. Chem Biol 2004, 11, (9), 1301-6.
6. Kuo, C. J.; Chi, Y. H.; Hsu, J. T.; Liang, P. H., Characterization of SARS main protease and inhibitor
assay using a fluorogenic substrate. Biochem Biophys Res Commun 2004, 318, (4), 862-7.
7. Shie, J. J.; Fang, J. M.; Kuo, C. J.; Kuo, T. H.; Liang, P. H.; Huang, H. J.; Yang, W. B.; Lin, C. H.;
Chen, J. L.; Wu, Y. T.; Wong, C. H., Discovery of potent anilide inhibitors against the severe acute res-
piratory syndrome 3CL protease. J Med Chem 2005, 48, (13), 4469-73.
8. Shie, J. J.; Fang, J. M.; Kuo, T. H.; Kuo, C. J.; Liang, P. H.; Huang, H. J.; Wu, Y. T.; Jan, J. T.; Cheng,
Y. S.; Wong, C. H., Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimet-
ic alpha,beta-unsaturated esters. Bioorg Med Chem 2005, 13, (17), 5240-52.
56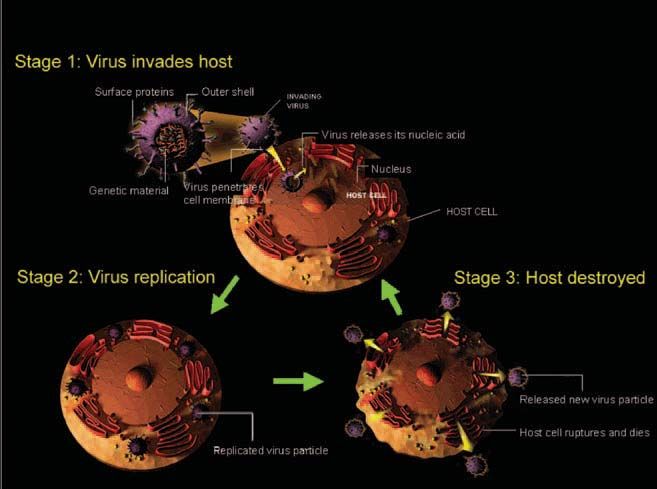
You can also read

















































