Data Analysis for Ion Torrent Sequencing
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
IFU022
v140202
Research Use Only
Instructions For Use Part III
Data Analysis
for Ion Torrent™ Sequencing
MANUFACTURER:
Multiplicom N.V.
Galileilaan 18
2845 Niel
Belgium
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 1 of 9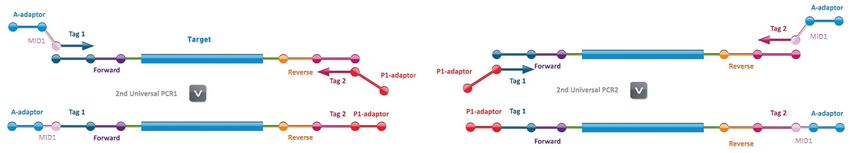
INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
TABLE OF CONTENTS
1. KITS AND INTENDED USE ................................................................................................................. 3
2. PRINCIPLE OF THE METHOD............................................................................................................. 3
3. MATERIALS AND EQUIPMENT REQUIRED BUT NOT PROVIDED ........................................................ 5
4. FILES PROVIDED .............................................................................................................................. 5
5. GENERAL CONSIDERATIONS ............................................................................................................ 5
5.1. DATA FILES .......................................................................................................................................5
5.2. STRUCTURE OF THE SEQUENCING READS ........................................................................................ 5
5.3. DEMULTIPLEXING OF THE SEQUENCING READS .............................................................................. 5
5.4. TRIMMING OF THE SEQUENCING READS ......................................................................................... 6
5.5. ALIGNMENT TO THE REFERENCE SEQUENCE ................................................................................... 6
5.6. VARIANT CALLING ............................................................................................................................ 6
5.6.1. MINIMAL COVERAGE ........................................................................................................... 6
5.6.2. QUALITY SCORES .................................................................................................................. 7
5.7. CNV ANALYSIS ..................................................................................................................................7
6. SPECIFIC INSTRUCTIONS .................................................................................................................. 8
6.1. TORRENT SUITETM SOFTWARE.......................................................................................................... 8
6.2. DROPGEN INSTRUCTIONS ................................................................................................................ 8
7. LIST OF ABBREVIATIONS .................................................................................................................. 9
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 2 of 9
INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
1. KITS AND INTENDED USE
The combined use of Multiplicom’s MASTR (Multiplex Amplification of Specific Targets for
Resequencing) kits with one or more of Multiplicom’s molecular identifier (MID) kit(s) or Short Read
Amplification kit enables the preparation of libraries for sequencing the gene(s) of interest using
massively parallel sequencing (MPS) instruments. A list of available MASTR assays and Complementary
MASTR products can be found on Multiplicom’s website (http://www.multiplicom.com), under
Products section.
These MASTR assays are for Research Use Only, unless otherwise stated, enabling the identification or
confirmation of the presence or absence of mutations and/or copy number variations (CNV) in target
regions.
2. PRINCIPLE OF THE METHOD
Multiplicom’s MASTR assays enable multiplex PCR amplification of all required target regions of the
gene(s) of interest in a limited number of PCR reactions. The recommended amount of DNA for each
multiplex PCR reaction is between 20 and 50 ng of purified genomic DNA for the germline MASTRs and
somatic MASTRs for DNA derived from fresh‐frozen tissue (FFT), or a minimum of 20 ng for DNA derived
from FFPE (formalin‐fixed paraffin‐embedded) material for somatic MASTRs. Next, the resulting
amplicons are barcoded, pooled and sequenced using a MPS instrument according to the
manufacturer’s instructions. The resulting sequence read pairs are subsequently analyzed to identify
variant positions compared with the reference sequence of the targeted gene(s). Comparing those
variants with public and/or private databases and analyzing the predicted change on the protein level
will allow the identification of mutations associated with health and disease. Moreover, a number of
MASTR assays enable CNV analysis directly from MPS data.
MASTR assays serve as front‐end amplification for sequence analysis on all commercially available
bench top MPS instruments. The technology is based on “target amplification”. The principle of the
MASTR assays relies on two key technologies: multiplex PCR amplification and Massively Parallel
Sequencing (the detection method).
In the first step, all target regions of the gene of interest are amplified in separate multiplex PCR
amplification reactions (number of multiplex reactions is defined per MASTR assay) per individual, using
a hot‐start DNA polymerase (Figure 1). The resulting amplicons of each multiplex are diluted 2,000 fold.
Figure 1. First step: multiplex PCR
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 3 of 9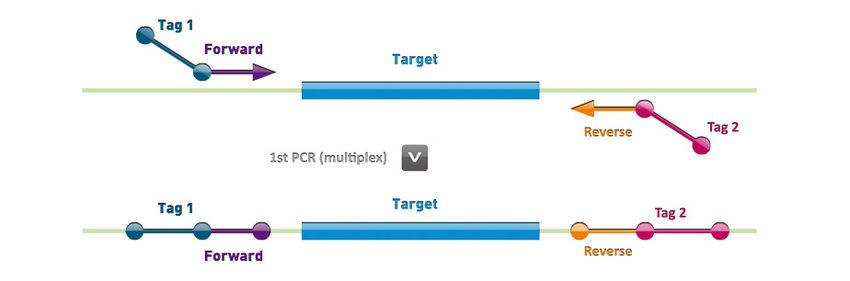
INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
For detailed workflow of this first step, please refer to the Instructions for Use Part I
Multiplex PCR with amplicon specific primers: MASTR assays (IFU016).
In the second step, a second round of PCR is performed enabling tagging of all the amplicons to
incorporate MID and A and P1 adaptors required for Ion Torrent Sequencing (Figure 2).
Figure 2. Second step: Universal PCR (example for Ion Torrent systems)
The resulting tagged amplicons are mixed per individual applying a predefined assay‐specific mixing
scheme. Each amplicon library is subsequently purified from small residual DNA fragments and the DNA
concentration determined.
For the detailed workflow of the second Universal PCR and subsequent mixing, purification and
pooling steps please refer to the IFU Part II MID for Ion PGMTM System (IFU241 or IFU242).
Next, these purified and individually tagged amplicon libraries are pooled equimolar, resulting in an
amplicon pool or sequencing sample, which is then further processed with the Ion PGMTM Template
OT2 400 Kit resulting in a template that is sequenced on an MPS Instrument according to the
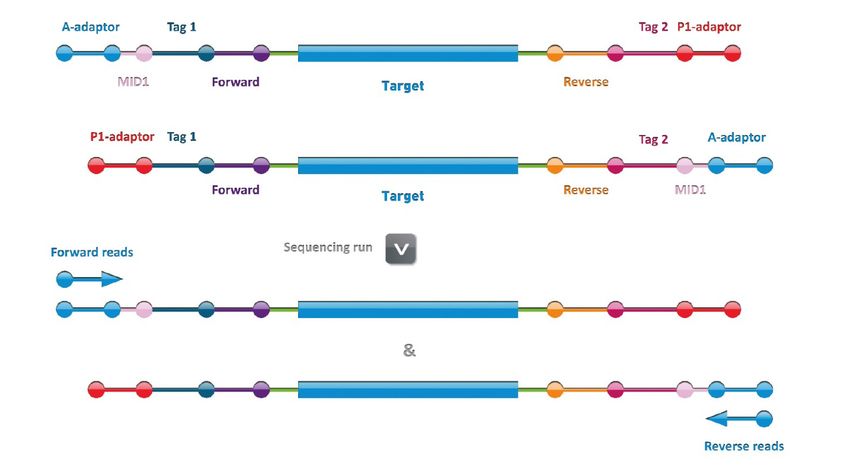
manufacturer’s instructions. The positions of the Ion Torrent sequencing primers are indicated in
Figure 3.
Figure 3. Third step: Sequencing run.
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 4 of 9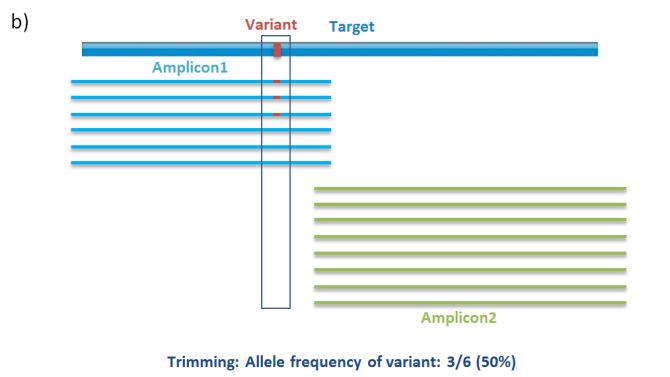
INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
3. MATERIALS AND EQUIPMENT REQUIRED BUT NOT PROVIDED
Equipment Recommendations/Comments
Analysis software for read counts and
Several software packages are commercially available.
variant calling of the MPS data
4. FILES PROVIDED
Table 1. Explanation of files supplied for data analysis
File description Type and content
MID sequences* General .pdf file listing the sequences of the MIDs present in the MID
(IFU333) for Ion PGMTM System kits: for demultiplexing of reads (Section 5.3)
MASTR‐specific .txt file listing the primers used for the amplification of
PCR specific primers
the different amplicons: for sequence trimming (Section 5.4)
MASTR‐specific .txt file listing the amplicon positions in Homo sapiens
BED‐file hg19 (MASTR‐specific primers are trimmed off): target info for data
analysis in general format (Section 5.5)
All files listed above can be downloaded from http://www.multiplicom.com/keycode All documents
mentioned above can be downloaded from http://www.multiplicom.com/keycode using the KEY‐
CODE printed on the box label of the specific MASTR kit (or MID for Ion PGMTM System kit*).
5. GENERAL CONSIDERATIONS
5.1. Data files
For Ion Torrent sequencing, the Torrent SuiteTM Software generates for each MID an SFF (Standard
Flowgram Format) file or a FASTQ file containing all filter passed sequencing reads generated during the
run.
5.2. Structure of the sequencing reads
The structure of the sequencing reads is depicted in Figure 3: the reads start with the MID, followed by
the universal tag sequence (Tag1 or Tag2), the PCR specific primer (Forward or Reverse) and the
amplified region. Depending on the size of the amplified region and the length of the read, this
sequence of the amplified region is further followed by the other PCR specific primer, universal tag and
P1‐adaptor.
5.3. Demultiplexing of the sequencing reads
The MID sequences at the beginning and/or at the end of the reads are used to demultiplex the
sequencing reads: to attribute the reads to one of the analysed samples or a no‐match residual
category.
Depending on the software tool used, the default being the Torrent SuiteTM Software the number of
allowed mismatches between the observed MID sequence and the expected MID sequences is an input
parameter for the demultiplexing step. We advise to allow maximally 2 (tolerant) mismatches. Reducing
the allowable mismatches reduces the risk for barcode misassignment; however, the number of reads
assigned to a barcode will be reduced concomittantly.
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 5 of 9INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
5.4. Trimming of the sequencing reads
The PCR specific primer part in the sequencing reads is by definition equal to the genomic reference
sequence and thus independent of the individual sample that is sequenced. As depicted in Figure 4,
when 2 amplicons overlap, failure to trim the PCR primer sequences from the reads can result in
skewed variant allele frequencies. Since virtually all MASTR assays contain overlapping amplicons,
primer trimming is a mandatory step in the data analysis.
The sequences of PCR primers (Figure 4a – Forward2 and Reverse2) should be removed from those
reads generated directly with them (Figure 4a – Amplicon2 reads), and should not be removed from
reads generated with other PCR primers (ie, from overlapping amplicons; Figure 4a – Amplicon1 reads).
This discrimination can be made based on the fact that the sequences of the PCR primers are flanked by
the universal tags (Tag1, AAGACTCGGCAGCATCTCCA, or Tag2, GCGATCGTCACTGTTCTCCA), while the
same sequences in the overlapping amplicons are not.
Figure 4. PCR Primer trimming. a) Illustration before PCR primer trimming: alignment of Amplicon1 and
Amplicon2 reads with Forward and Reverse primers. b) Illustration after PCR primer trimming.
Remark: During design, great care was taken to select primer binding sites avoiding regions with
variants. In addition, a periodic review is performed to identify newly reported variants in those regions
and to test their impact on amplification. It can however not be excluded that a variant in a binding site
of a primer may be present in a sample, which may lead to the amplification of only one of the alleles,
masking the presence of a clinically relevant mutation in the amplicon. If such a case is suspected,
calculation of the dosage quotient of each amplicon can be used for confirmation (as desctibed in
Section 5.7). For further support, contact customer services at customerservice@multiplicom.com.
5.5. Alignment to the reference sequence
The sequence reads can be aligned to the targeted regions or to the entire human genomic sequence.
To facilitate the transfer of assay specific information to the different analysis software packages, a BED
file with the trimmed amplicon positions on hg19 is available for download at our website.
5.6. Variant calling
Different parameters can be analyzed to discriminate true positive variants from false positive or
background signals. Below, you find a non‐exhaustive list of parameters whose effect on the sensitivity
and specificity of variant calling might be evaluated:
5.6.1. Minimal coverage
The coverage, or number of aligned reads, at the site of the variant has to reach a given threshold for
confident variant detection. The minimal coverage recommended by Multiplicom for MASTRs in
combination with an Ion PGM System is 100 reads for each position at the region of interest (50 reads
per allele) for SNV analysis and 300 reads per amplicon for CNV analysis. It is advised that target regions
that do not reach this minimal coverage are eliminated from the list of analysed target regions in the
final variant calling report.
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 6 of 9INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
In case of an amplicon library derived from a tumor tissue sample (FFPE or FFT) deeper sequencing
might be needed to obtain the required minimal coverage of 50 reads per affected allele. Examples are
when the sample contains clonal populations of tumor cells and/or has a lower percentage of tumor
cells. In these cases the minimal numbers of reads should be recalculated accordingly (eg, 2‐fold higher
to identify positions with a variant allele frequency (VAF) of 25%, or for a sample with 50% tumor tissue
content).
5.6.2. Quality scores
The quality of the aligned bases at the position of the potential variant has an effect on the confidence
in the variant call. This quality is generally influenced by the position in the read (the overall quality
decreases along the reads) and the genomic context (eg, homopolymer stretches have a negative
impact on the quality of the following bases). This leads to two derived parameters:
Presence in forward and reverse reads
Since the quality decreases along the reads and forward and reverse reads start at opposite
positions on an amplicon, the quality of the forward reads is highest where the quality of the
reverse reads is the lowest (and vice versa). If all target positions are covered by both forward and
reverse reads, the presence of a variant in both forward and reverse reads is a good predictor for a
true positive variant call.
Changes in/around homopolymeric stretches
In view of the inherent difficulties of the Ion Torrent sequencing technology to call the actual
length of homopolymer stretches, special care has to be taken when calling variants in or flanking a
homopolymeric stretch. Based on our experience, homopolymeric stretches with a length of 4 bp
or more require special care.
Remark: for specific MASTR assays, we offer a complementary homopolymer (HP) kit. For an
overview of all available HP kits, please refer to the Products section on Multiplicom’s website
(http://www.multiplicom.com).
5.7. CNV analysis
CNV analysis is possible for a selected number of MASTR assays. These MASTR assays contain a
separate set of control amplicons for each plex (located on chromosomes different from the target
genes), which are amplified, tagged and sequenced in parallel with the targeted region. Only MASTRs
listing such control amplicons on their GS Reference Pattern are suited for CNV analysis.
Remark:
Excel template sheets are available upon request (at customerservice@multiplicom.com) for the
specific MASTR assays enabling CNV analysis. To use these sheets, the read counts (number of reads)
of all amplicons in all samples should be extracted from the sequencing data.
For CNV analysis using MPS data, read count comparison between target and control amplicons is
performed to calculate the Dosage Quotient (DQ) as described:
Read count of the amplicon of interest is divided by the sum of read counts of control amplicons of
that plex (in other words: normalize on sum of control amplicons) = “normalized read count”
The average of the normalized read counts of that amplicon for all samples is calculated =
“reference normalized read count”
The “normalized read count” is divided by the “reference normalized read count” = DQ
When the DQ ≥ 1.3, the corresponding genomic fragment is considered to be present in 3 copies
(duplication of one allele); when the DQ ≤ 0.7, the genomic fragment is considered to be present in
only 1 copy (deletion of one allele).
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 7 of 9INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
Remarks:
(1) CNV analysis calculations always need to be made “within a plex”.
(2) For the proper calculation of the “reference normalized read count” (in the calculation of the DQ
as described above), the set of samples should meet the following requirements:
o When using a set of known samples as references (no CNVs), the libraries of these samples
should be constructed together with the unknown samples.
o When using the other unknown samples of your run as references, only a 40% of samples from
the total set is allowed to have a CNV.
(3) Since polymorphisms in primer sites may lead to amplification of only one of the alleles, resulting
in a false positive DQ ≤ 0.5, a detected CNV is only considered to be valid when 2 adjacent
amplicons show a significantly altered DQ and/or when confirmed by an independent method.
(4) Compared to variant analysis deeper sequencing is required for CNV analysis.
For the precise list of amplicons that will be amplified using a certain PCR Mix,
refer to the MASTR‐specific GS Reference Pattern, which can be obtained from
http://www.multiplicom.com/keycode using the KEY‐CODE
printed on the box label of the used MASTR kit.
6. SPECIFIC INSTRUCTIONS
Data analysis can be performed using a variety of analysis software packages. Below we provide some
specific instructions for the use of the Torrent SuiteTM software of Life Technologies (Section 6.1), and
the dropGen application of the Integrated Clinical NGS Dry Lab Service of Sophia Genetics 6.2).
6.1. Torrent SuiteTM software
Life Technologies advises to align the generated sequences using the Torrent Suite Software and
analyse the generated BAM‐files with the Torrent Variant Caller. One step in this process is the
definition of the target regions. For this, the BED‐file mentioned in Table 1 should be used. More
detailed information on these software solutions can be found on the Ion Community website
(http://ioncommunity.lifetechnologies.com).
6.2. dropGen instructions
The dropGen application should be used according to manufacturer’s instructions.
To access and use Sophia Genetics' service, laboratories shall request the creation of an account
on the dropGen application by contacting Sophia Genetics directly:
http://www.sophiagenetics.com/contact.php.
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 8 of 9INSTRUCTIONS FOR USE Ion Torrent™ Data Analysis
7. LIST OF ABBREVIATIONS
CNV: Copy Number Variant
DNA: Deoxyribonucleic acid
FFPE: formalin‐fixed paraffin‐embedded
IFU: Instructions For Use
MASTR: Multiplex Amplification of Specific Target for Resequencing
MID: Molecular Identifiers
MPS: Massively Parallel Sequencing
PCR: Polymerase Chain Reaction
Plex: Set of MASTR derived amplicons
ROI: Region of Interest
SFF: Standard Flowgram Format
TTC: Tumor Tissue Content
VAF: Variant Allele Frequency
www.multiplicom.com © 2014 Multiplicom NV, all rights reserved.
Revision date: August 21, 2014 Page 9 of 9You can also read