INTRODUCTION TO NEXT GENERATION SEQUENCING
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
ECOLE DE BIOINFORMATIQUE
INITIATION AU TRAITEMENT DES DONNÉES DE GÉNOMIQUE OBTENUES PAR SÉQUENÇAGE À HAUT DÉBIT
05-10 OCTOBRE 2014 - STATION BIOLOGIQUE - ROSCOFF
INTRODUCTION TO
NEXT GENERATION SEQUENCING
Claude Thermes
Genome analysis
Centre de Génétique Moléculaire
Gif-sur-Yvette
06/10/2014
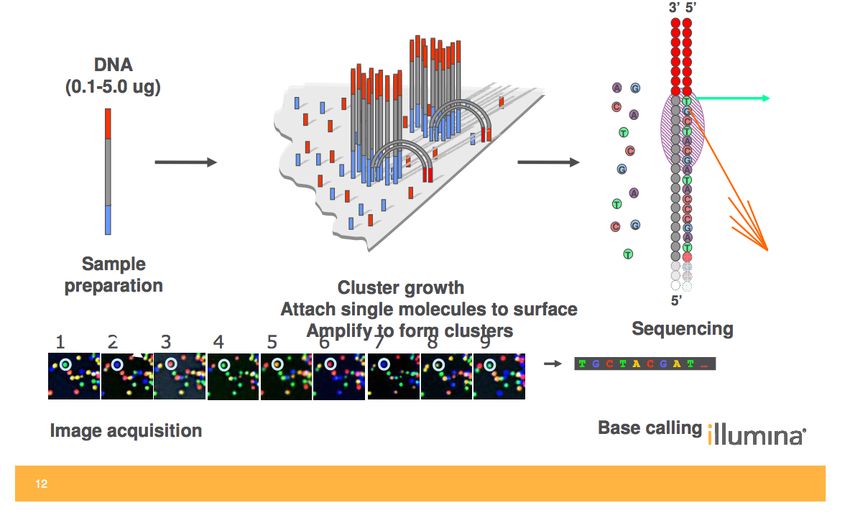
Step 1: sample preparation
Step 2: sequencing (Illumina)
Step 3: data analysis
(with permission of ABIMS)
Situation in 2009
1-5 µg genomic DNA
Genome
sequencing
10 ng DNA 10 µg total RNA
10 µg total RNA
Adapted from Science 306:636-640, 2004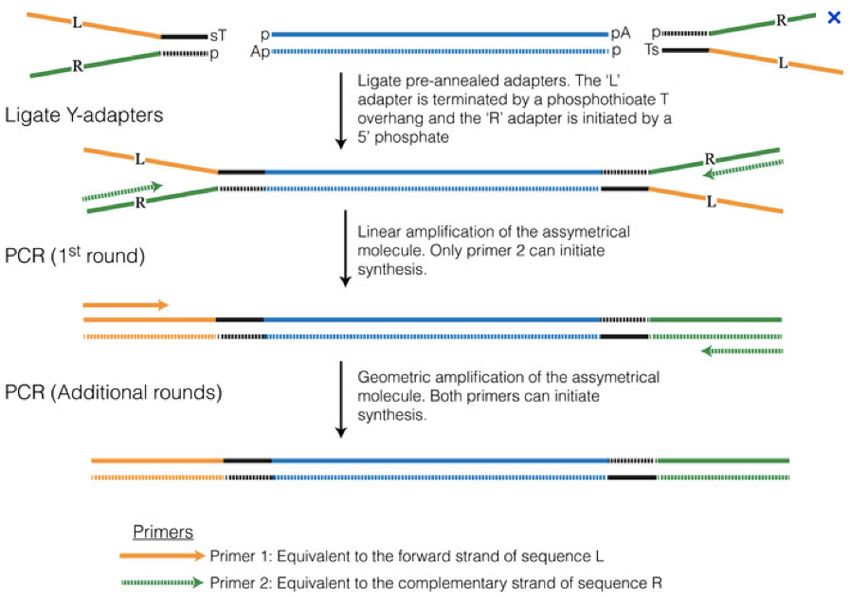
Situation today
1-5 µg genomic DNA
50 ng
Genome
sequencing
10 ng DNA 10 µg total RNA
1-2 ng 1 µg
10 µg total RNA
1ng
Adapted from Science 306:636-640, 2004
Libraries from DNA samples
DNA-seq Libraries
Illumina TruSeq technology
Genomic DNA
Sonication
Size selection
Adaptors ligation
PCR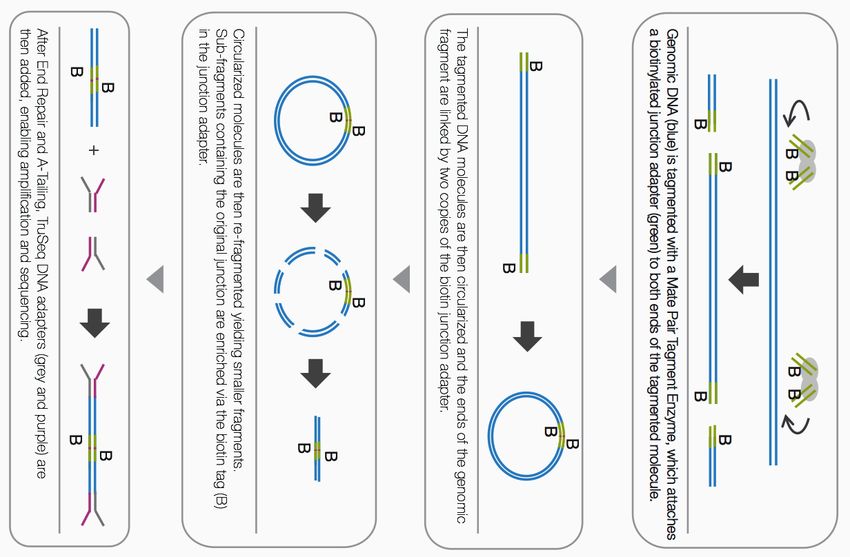
DNA-seq Libraries
Illumina TruSeq technology
Genomic DNA
Sonication
Size selection
?
Adaptors ligation
PCR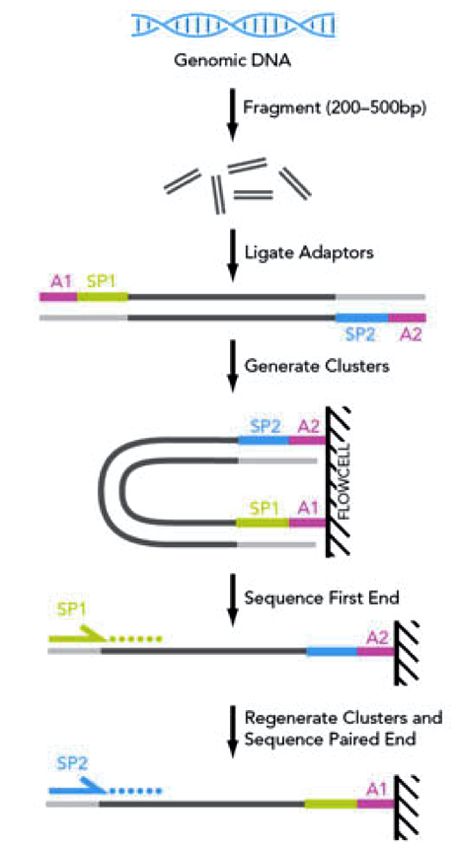
Ligate Y-adaptors
PCR Primer 1: complementary to R
Primer 2: equivalent to R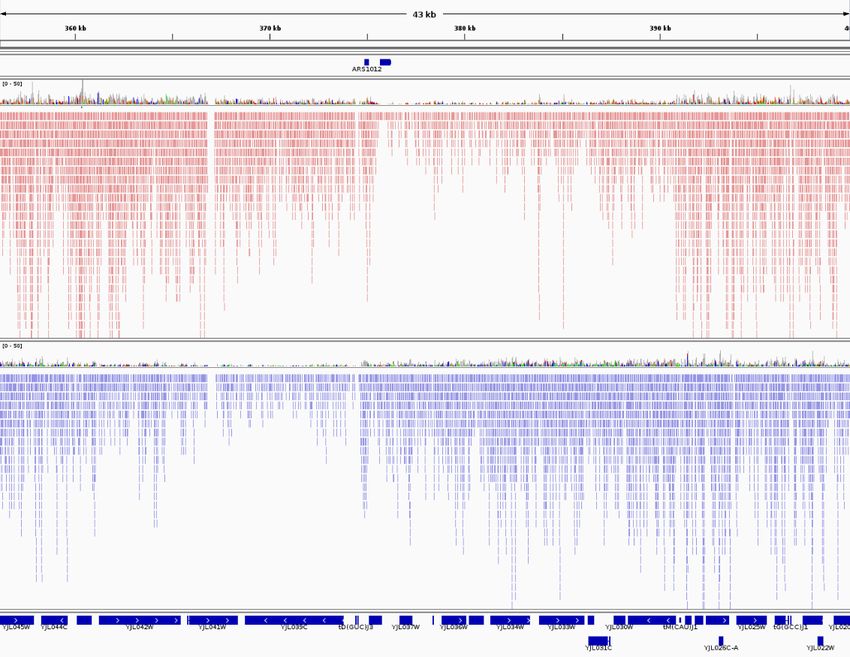
DNA-seq Libraries
Nextera “tagmentation”
Transposomes / Tagment Enzyme
Tagment Enzyme fragments
DNA and attaches junction
adapters (blue and green) to
both ends of the tagmented
molecule
Tagmentation
Dual barcode
approach
up to 96 indexed
samples
rapid
(
2
hours)
and
requires
small
quan33es
(50
ng)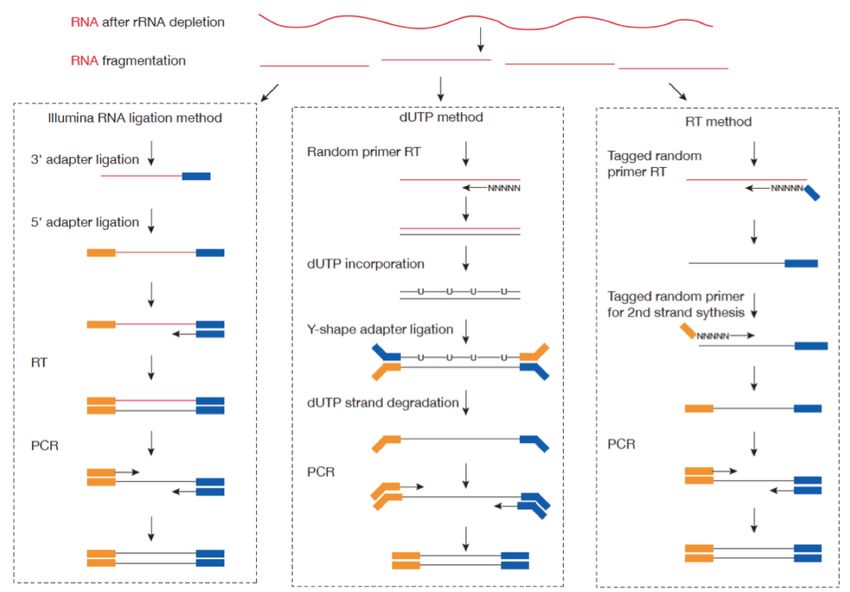
Paired end sequencing 1rst read 2d read
Comparison
of
single
read
versus
paired
end
sequencing
Single
read
density
?
?
?
Paired
end
density
Single
read
density
?
?
?
Paired
end
density
Paired
end
density
Paired
end
sequencing
:
•
improves
genome
assembly
•
but
requires
a
good
control
of
DNA
fragmenta3on
(purifying
gels/columns)
•
3me
consuming
and
requires
large
quan33es
(1-‐5
µg)
BUT :
Paired end fragments are too short for assembling large genomes with many
repeated elements
mate pair libraries“Classical”
Illumina
mate
pair
library
several kilobases
Problems
:
• low
coverage
• few
fragments,
over-‐amplified
A new method : Nextera Mate Pair
Tagment Enzyme fragments
DNA and attaches a biotinylated
junction adapter (green) to both
ends of the tagmented molecule
circularization
Fragmentation enrichment via the biotin tag
adapters ligation at both endsA new method : Nextera Mate Pair
Tagment Enzyme fragments
DNA and attaches a biotinylated
junction adapter (green) to both
ends of the tagmented molecule
circularization
Fragmentation enrichment via the biotin tag
rapid
(
few
hours)
and
requires
small
quan33es
(50
ng)
adapters ligation at both endsQuelques remarques
Protocole Illumina Truseq Nextera
Ligations d’adaptateurs Tagmentation
Matériel de départ Fragments d’ADN (dble brin)
Génomique ou ChIP ADN génomique, 50 ng (grands génomes)
1-1000ng
• Peu sensible à qualité du matériel Très rapide (4h)
Avantages
• Très versatile, contrôle précis de la taille (purif sur gel )
• Protocole préféré si on veut des tailles homogènes, ou
grandes pour du paired end 2x250
• Fonctionne également sans PCR si quantité de
matériel suffisante (>100ng)
• Très sensible à qualité de l’ADN de départ
(intégrité, pureté)
• Difficile de contrôler la taille des inserts qui
inconvénients • Protocole long : 1-2 journées sont trop petits pour paired end 2X250
• dimères possibles, fragmentation nécessaire • PCR obligatoire
Remarques • Très adaptable, on peut ajuster le nombre de • Possibilité de double tag (96 index)
cycles PCR à la quantité de matériel de départ • Non miltiplexable avec Truseq (primers
• Si petites quantités : utiliser des billes différents de Truseq)
• la taille des fragments de départ déterminera la
taille finale des fragmentsSome examples of libraries prepared from DNA samples
Hi-C Re-sequencing
Long-range Indels, SNP, CNV
interactions
De novo
Exome Rad-seq sequencing
sequencing
DNA replication
origins
Adapted from Science 306:636-640, 2004Re-sequencing : identification of SNP, indels
“Mutations” specific to forward strand“Mutations” due to mono-directional sequence effect
Nakamura et al. NAR (2011)
Partial blockage of DNA synthesis“Dephasing” due to partial blockage of DNA synthesis
“Dephasing” due to partial blockage of DNA synthesis
“Mutations” due to bi-directional sequence effect
Libraries from RNA samples
RNA-seq Libraries
Quelques remarques
Tous les protocoles sont directionnels
Protocole TruSeq small RNA ScriptSeq TotalScript
(Illumina) (Epicentre) (Epicentre, Nextera)
Matériel de départ ARN déplété ou polyA ARN déplété ou polyA ARN total (ou polyA)
25-100 ng 0,5 - 50 ng 1-5ng
ARN NON DEGRADÉ
(tagmentation)
Principe fragmentation RT par random priming RT par oligo dT
Ligation sur ARN PCR PCR++
RT & PCR
• Petites quantités RNA-seq possible même
Avantages Taille des fragments bien contrôlée
• Possible même si dégradé (FFPE) si très petites quantités
Adapté pour paired end 2X250
• Rapide, automatisable d’ARN total
inconvénients • Aberrations si trop petites quantités • Sensible à contamination par gDNA • L’ARN doit être peu
• 2-3 jours de manip • Fragmentation non contrôlée (200-800nt) dégradé
• non automatisable • Semble donner pas mal de duplicats • Non adapté pour paired
quand les quantités sont dans la gamme end 2X250
basse
Remarques Non multiplexable avec
TruSeq (index Nextera)Comparison
of
two
RNA
fragmenta3on
protocols
:
SOLiD
(Transcriptome
Analysis
kit)
:
RNase
III
fragmenta.on
and
Illumina
(Direc3onal
mRNA-‐Seq
kit)
:
Zinc
fragmenta.on
SOLiDTM Whole Transcriptome Analysis Kit: RNase III fragmentation
RiboMinus
RNA
5’
3’
RNaseIII
N
NNNNNN
fragmented
RNA
Reverse
transcrip6on
Hybridiza6on
with
adapters,
liga6on
Size
selec6on
PCR
amplifica6on
Illumina directional mRNA-Seq Library: Zinc fragmentation
RiboMinus
RNA
5’
3’
Zinc
N
NNNNNN
fragmented
RNA
Reverse
transcrip6on
Hybridiza6on
with
adapters,
liga6on
Size
selec6on
PCR
amplifica6on
Sequencing Illumina (Zinc) and Solid (Rnase III) libraries
intron
YBR078W
Zinc
Same number of reads
RNase
III
Examples of libraries from RNA samples
miRNA-seq
Ribo-seq Long non-coding RNAs
Identification
mRNA 5’ ends
of
Pol II
CLIP-seq
NET-seq
FRT-seqNET-seq : Native Elongating Transcript sequencing
Churchman and Weissman, 2011
• sequencing of 3’ ends of nascent RNAs still associated with RNA polymerase
• distribution of transcribing polymerases along the genome in a strand specific manner
• allows studies of transcription termination
Pol II Pol II Cells in desired condition
Pol II
RNA polymerase II immunoprecipitation
Pol II Pol II
Recovery of nascent transcripts
Associated with the polymerase
RNA-seq and mapping on the genomeFRT-seq:
amplification-free, strand-specific transcriptome sequencing
Mamanova et al. Nature Methods (2010)
• The reverse transcription reaction takes place on the flowcell
• No PCR amplification, so PCR biases and duplicates are avoided
• Because the template is poly(A)+ RNA rather than cDNA, the resulting RT on the flowcell
sequences are necessarily strand-specific
• The method is compatible with paired- or single-end sequencing
Cluster
generationSome problems
Libraries
prepared
from
very
small
amounts
of
DNA
or
RNA
(Sequencing of very small amounts of genome fragments (
New
direc3ons
with
single-‐cell
sequencing
• FLUIDIGM C1™ System : allows measurement of gene expression in 96 single-cells
• MALBAC “Multiple Annealing and Looping-based Amplification Cycles”
Allows sequencing the genome of a unique cell (Zong C. et al. Science, 2012)
• Many other systems are in development :
• larger cell numbers,
• single-cell ChIP-seq, etc.You can also read

















































