Le Sindromi Talassemiche - M.Domenica Cappellini MD Professor of Internal Medicine Università di Milano
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

Le Sindromi Talassemiche M.Domenica Cappellini MD Professor of Internal Medicine Università di Milano
Disclosures • Novartis Pharmaceuticals: Consultancy, • Sanofi/Genzyme • Bristol-Myers Squibb (Celgene): Consultancy • Ionis Pharmaceuticals: Consultancy • Vifor: Consultancy • Agios: Consultancy • Novo Nordisk:Consultancy • CRISP: Consultancy
Outline • General overview of thalassemia − How we understand the disease today • Comorbidities in adults • Novel therapies

1925: Cooley description
1880: Cardarelli
1884: Somma 1925: Rietti
1928: Greppi
1935: Miceli
1940–1950:
Caminopetros, Silvestroni, Bianco
Hb abnormalities, hereditary pattern 1949–1960: Pauling: Hb structure
HbS-Mendelian transmission
Ceppellini: HbA2
1960–1970:
Weatheral and Clegg
Hb chain synthesis
1970–1980: transfusional therapy
Iron chelation: deferoxamine
1980–2000:
Prenatal diagnosis (Kan)
Bone marrow transplantation (Lucarelli) 2000… new oral iron chelators
Present:
Gene therapy
New therapies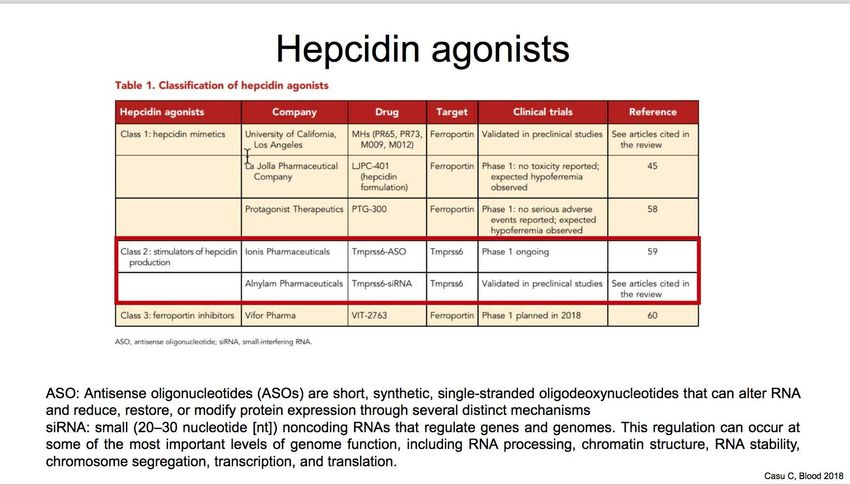
The thalassemias
Group of inherited disorders
Absence or
reduced
synthesis of α thalassemias
α chains of
hemoglobi
n
Absence or
reduced
synthesis of β thalassemias
β chains of
hemoglobi
n
• Reproduced from Muncie HL and Campbell JS. Alpha and Beta Thalassemia. Am
Fam Physician 2009;80:339–344. Copyright 2009 American Academy of Family
Physicians
Epidemiology of thalassemia
β thalassemia is most
common in Mediterranean,
African and Southeast Asian
countries1
α thalassemia occurs
most often in African
and Southeast Asian1
countries
• The annual births of thalassemic disorders is estimated to be nearly 70,0002
• The highest prevalence occurs where malaria was, or still is, endemic2
• Muncie HL and Campbell JS. Am Fam Physician 2009;80:339–344; Review.
• Williams TN and Weatherall DJ. Cold Spring Harb Perspect Med. 2012;2:a011692.
Review.
Evolving global burden due to migration
Predominance in low- or middle-
income countries stretching from sub-
Saharan Africa, through the
Mediterranean region and the Middle
East, to South and Southeast Asia
Recent global population movements
have also led to increasing incidences
in areas of the world previously
relatively unaffected by these
conditions such as Europe and the US
1. Taher AT et al. Lancet. 2018 Jan 13;391(10116):155-167; 2. Weatherall DJ. Blood 2010;115:4331-4336; 3. Weatherall DJ. Blood Rev 2012;26S:S3-S6.
There are many types of
thalassemia
α thalassemias1 β thalassemias2
α+ thalassemia trait β thalassemia minor
α0 thalassemia trait β thalassemia intermedia
Hb Constant Spring β thalassemia major
Hb H β thalassemia with
Hb Barts hydrops fetalis associated Hb variants:
Hb H/Hb Constant Spring Hb C/β thalassemia
Hb E/β thalassemia
Hb S/β thalassemia
• Muncie HL and Campbell JS. Am Fam Physician 2009;80:339–334. Review.
• Galanello R and Origa R. Orphanet J Rare Dis 2010;5:11. Review.
How we view the disease today: Transfusion requirement is
commonly used to distinguish phenotypes
Non-transfusion-dependent thalassemia
§ β-thalassaemia intermedia
§ Mild/moderate HbE/β-thalassemia NTD
§ HbH disease (α-thalassemia intermedia) T
Transfusions Occasional transfusions Intermittent transfusions required Regular, lifelong
seldom required required (e.g. surgery, (e.g. poor growth and development, transfusions
pregnancy, infection) specific morbidities) required for survival
Transfusions not required
§ α-thalassemia trait
§ β-thalassemia minor TDT Transfusion-dependent thalassemia
§ β-thalassemia major
§ Severe HbE/β-thalassemia
§ Hb Barts hydrops (α-thalassemia major)
Musallam KM et al. Haematologica 2013;98:833-844.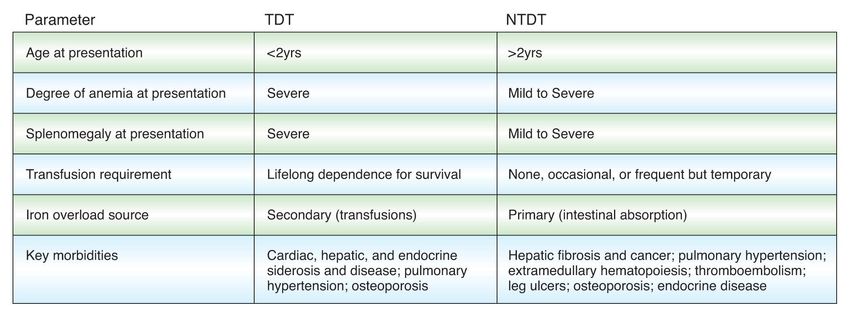
PATHOPHYSIOLOGY OF THALASSEMIAS
Excess free Formation of heme
α-globin chains Denaturation and hemichromes
Degradation
§Chronic anaemia & haemolysis Iron-mediated toxicity
§Ineffective erythropoiesis Membrane
Ineffective
§Iron overloaderythropoiesis
Hemolysis binding of
IgG and C3 Removal of
damaged red cells
Increased
erythropoietin Reduced tissue Anaemia Splenomegaly
synthesis oxygenation
Skeletal Erythroid
deformities, Increased
marrow Iron overload
osteopenia expansion Iron absorption
Olivieri NF, et al. N Engl J Med. 1999;341:99-109.How we view the disease today: Transfusion requirement is
commonly used to distinguish phenotypes
Non-transfusion-dependent thalassemia
§ β-thalassaemia intermedia
§ Mild/moderate HbE/β-thalassemia NTD
§ HbH disease (α-thalassemia intermedia) T
Transfusions Occasional transfusions Intermittent transfusions required Regular, lifelong
seldom required required (e.g. surgery, (e.g. poor growth and development, transfusions
pregnancy, infection) specific morbidities) required for survival
Transfusions not required
§ α-thalassemia trait
§ β-thalassemia minor TDT Transfusion-dependent thalassemia
§ β-thalassemia major
§ Severe HbE/β-thalassemia
§ Hb Barts hydrops (α-thalassemia major)
Musallam KM et al. Haematologica 2013;98:833-844.TDT vs. NTDT Management of Non-Transfusion-Dependent Thalassemia: A Practical Guide Taher A Cappellini MD Drugs. 2014 Sep 26.
Clinical distinction between NTDT and TDT
TDT NTDT
Silent cerebral ischemia
Hypothyroidism
Hypoparathyroidism Pulmonary Hypertension
Right-sided heart failure
Cardiac siderosis
Left-sided Heart failure Extramedullary
hematopoietic pseudotumors
Hepatic failure
Viral hepatitis
Malignancy Hepatic fibrosis,
cirrhosis, and cancer
Diabetes mellitus
Renal Dysfunction
Gallstones
Hypogonadism
Osteoporosis Splenomegaly
Osteoporosis
Venous thrombosis
Leg ulcers
1. Musallam KM et al. Haematologica.2013;98:833-844; 2. Taher AT, Cappellini MD. Drugs 2014;74:1719-1729.Thalassemia major survival in
the early sixty
Ø In 1965 no patient treated at the Italian
Thalassemia Centres reached the age
of 13 years
Ø Between 1960 and 1976 patients
followed at Cornell Medical Center had
a median survival of 17.1 yearsMedian TDT patient age has increased during the last 3
decades
Age distribution of β thalassemia patients
over several surveys North
30 America Italy/Greece
1973 (n=243)
25
1985 (n=303)
Percent of patients
20 1993 (n=443) 1993 (n=271)
2002 (n=319) 2003 (n=170)
15
10
5
0
0–5 6–10 11–15 16–20 21–25 26–30 31–35 36–40 41–45 46–50 51–55
Age (years)
Vichinsky E P et al. Pediatrics 2005;116:e818-e825. Cross-sectional study; n=781.Reason behind the change of survival 1960s: Regular transfusion to maintain mean hemoglobin in the normal range 1964: introduction of the first iron chelator 1984: first bone marrow transplant was initiated 1995/2005: new oral iron chelators 1999: T2* cardiovascular magnetic resonance (CMR) technique which became Today: Multidisciplinary approach
UK: Progress in thalassemia management has
improved survival
Regular DFO therapy T2* CMR
transfusion became DFP approval DFX approval
1960s 1980s 1999 introduced 1999 2006
in Europe
became the standard in Europe
in the UK
norm practice
50
45
40 Unknown
Number of deaths
35 Other
Malignancy
30
Iron overload
25 Infection
20 BMT complication
15 Anemia
10
5
0
3*
9
9
4
4
9
4
9
4
4
9
98
99
99
97
98
96
96
97
95
95
00
–1
–1
–1
–1
–1
–1
–1
–1
–1
–1
–2
90
95
75
80
85
60
65
70
50
55
00
19
19
19
19
19
19
19
19
19
19
20
*The number of deaths in the 2000–2003 interval represents deaths during 4 years; in all the other groups, the number of deaths is over 5 years.
• BMT, bone marrow transplantation; CMR, cardiovascular Modell B et al. J Cardiovasc Magn Reson 2008;10:4
magnetic resonance. Case-control study; n=1089.It also changed the profile of causes for morbidity and mortality
Italy2
United Kingdom1
Causes of mortality
100
90
80 Hepatitis C
complications
70 Other/unknown
Malignancy
60 Infection
Patients (%)
50 BMT complication
Anaemia
40 Iron overload
30
20
10
03
59
69
79
89
99
0
20
19
19
19
19
19
–
–
–
–
–
–
80
90
00
50
60
70
19
19
19
19
19
20
01 rt
–2 ho
3
99 Co
19 hit
W
LH
C
U
1. Modell B et al. J Cardiovasc Magn Reson 2008;10:42; 2. Borgna-Pignatti C et al. Haematologica 2004;89:1187-1193..Evolution of thalassemia patient’s survival over the past decades 1. Farmakis D et al. Eur J Haematol. 2020 Jul;105(1):16-23.
This resulted in improvement in life expectancy and evolving concerns when patients advance in age Taher AT, Cappellini MD. Blood 2018;132:1781–1791
Summary 1 • The survival of patients with TDT (b-thalassaemia major) has significantly increased all over the world providing that they receive regular transfusions in order to prevent sequelae resulting from anaemia, including growth retardation • Complications due to iron overload, mainly cardiac failure, are decreased providing that patients are properly chalated, but still there are differences • Bone marrow transplantation had a significant impact in several countries • Prevention programs have been implemented or are under evaluation in developing countries
TDT guidelines • 6 years worth of new data to solidify evidence-based recommendations • Change of scope of management with emergence of more novel approaches and therapeutics • Awareness to the need to focus on quality of life and other supportive care
Outline • General overview of thalassemia – How we understand the disease today • Comorbidities in adults • Novel therapies
Malignancy as an emerging concern in Adult patients with
Thalassemia
Hodroj M et al. Blood Rev. 2019 Sep;37:100585Epidemiology of cancer in thalassemia
Thalassaemia and risk of cancer: a
population-based cohort study
Malignancies in Patients With β-
Thalassemia Major and β-
Taiwan
Thalassemia Intermedia: A
(2014)
Multicenter Study in Iran (4
Thalassemia Centers)
Objective: To investigate the incidence
and risk of cancer in 2, 655
Iran
thalassemia patients
(2009)
Results: The overall incidence of
• 11 cases of malignancy among
cancer was 1.52 times higher in the
4,630 patients
thalassemia cohort than in the
comparison cohort.
• Of the 11 cases, 5 had
lymphoma, 5 had leukemia, and 1
Patients with thalassemia had a
patient had a non-hematological
considerably higher risk of
malignancy.
hematological malignancy and
abdominal cancer
Karimi M et al. Pediatr Blood Cancer 2.009;.53:10. Chung WS et al. J Epidemiol Community Health 2015;69:1066-10670. 64-1067Additional cofactors may further substantiate risk of hepatic
injury and cancer from iron overload
Cofactors which accelerate Alcohol HCV Obesity and
iron overload in the liver Insulin resistance
and subsequent injury: Steatosis
Oxidative stress/Lipid peroxidation
• Infection (eg, HCV)
• Alcoholic liver disease Accelerated liver iron uptake
• Obesity and insulin Hepatocyte Kupffer cell Stellate cell
resistance Cytokines
• Ineffective (Inflammation)
erythropoiesis
Apoptosis Proliferation Carcinogenesis Fibrosis
Alcoholic cirrhosis, hepatitis C and insulin resistance may increase steatosis
and oxidative stress, which accelerate liver iron uptake and increase risk of
liver fibrosis or HCC
Kohgo Y et al. World J Gastroenterol 2007;13:4699–4706.Evidence of solid malignancies Hodroj M et al. Blood Rev. 2019 Sep;37:100585.
Hematologic Malignancies in thalassemia
While the increased prevalence of
solid cancers, most notably
hepatocellular carcinoma
secondary to hepatitis C and iron
overload, has been noted, the
occurrence of hematologic
malignancies has been
proposed to be even higher
Hodroj M et al. Blood Rev. 2019 Sep;37:100585. Zanella S et al. Ann N Y Acad Sci 2016;1368:140–148.Evidence on Hematologic malignancies Hodroj M et al. Blood Rev. 2019 Sep;37:100585.
Possible mechanisms of hematologic malignancies in thalassemia Halawi R et al. Am J Hematol 2017;92:414–416.
Insights on mechanisms
IRON OVERLOAD AND TRANSFUSION THERAPY
• Iron overload aggravates genomic instability.
• Iron overload triggers an immune regulatory imbalance which may promote cancer
development
• Blood transfusions are associated with the transmission of oncogenic viruses that may trigger
the development of hematologic malignancies (eg EBV, CMV, HTLV-1 vs lymphomas)
• A host of abnormalities involving the immune systems have been described in patients with
thalassemia on transfusions and their role in cancer development merits investigation
Pullarkat V et al. Adv Hematol 2010;2010:12; Walker EM et al. Ann Clin Lab Sci 2000;30:354–365; Switzer WM et al. AIDSRes Hum Retroviruses 2013;29:1006–1009; Suarez
F et al. Blood 2006;107:3034–3044; Refaai MA et a. Expert Rev Hematol 2013;6:653–663.Hypercoagulability and vascular disease in NTDT
↑
Transfusion Splenectomy Iron overload
↑ ↑ ↑
↓
Pathologic RBCs Endothelial injury Platelet abnormalities
§ ↑ Thrombin generation § Expression of adhesion § Thrombocytosis
(phosphatidyl serine exposure) molecules and tissue factor § Chronic activation
§ ↑ Rigidity, deformability, and § ↑ Adhesion and aggregation
aggregation
↑ Circulating
microparticles
↓ Antithrombin III Endocrine & hepatic
↓ Protein C dysfunction
↓ Protein S
?↑ Atherosclerosis
THROMBOSIS
Musallam KM, et al. Haematologica. 2013;98:833-4.Patient stratification according to splenectomy and TEE status:
OPTIMAL CARE
• Three groups of patients identified
− Group I, splenectomized patients with a documented TEE (n = 73)
− Group II, age- and sex-matched splenectomized patients without TEE (n = 73)
− Group III, age- and sex-matched non-splenectomized patients without TEE (n = 73)
Type of thromboembolic event in
n (%)
splenectomized TI patients (Group I)
DVT 46 (63.0)
PE* 13 (17.8)
STP 12 (16.4)
PVT 11 (15.1)
Stroke 4 (5.5)
*All patients who had PE had confirmed DVT.
TEE = thromboembolic events
Taher AT et al. J Thromb Haemost 2010;8:2152-2158.OPTIMAL CARE study: multivariate analysis on risk factors
for thrombosis in splenectomised patients
Parameter Group OR 95% CI p value
NRBC count ≥ 300 x 106/L Group III 1.00 Referent
Group II 5.35 2.31–12.35
< 0.001
Group I 11.11 3.85–32.26
Platelet count ≥ 500 x 109/L Group III 1.00 Referent
Group II 8.70 3.14–23.81
Group I patients had significantly higher NRBC,
Group I 76.92 22.22–250.00
< 0.001
platelets, and PHT occurrence, and were mostly
PHT Group III 1.00 Referent
Group II non-transfused
4.00 0.99–16.13
0.020
Group I 7.30 1.60–33.33
Transfusion naivety Group III 1.00 Referent
Group II 1.67 0.82–3.38 0.001
Group I 3.64 1.82–7.30
NRBC = nucleated red blood cell; PHT = pulmonary hypertension; OR = adjusted odds ratio; CI = confidence interval. Taher AT, et al. J Thromb Haemost. 2010;8:2152-8.Development of a thalassemia-related thrombosis risk scoring system Taher A et al. Am J Hematol. 2019 Aug;94(8):E207-E209. .
Mechanisms of renal disease in β-thalassaemia Musallam KM et al. J Am Soc Nephrol 2012;23:1299–1302.
Pulmonary Hypertension in β-Thalassemia § Pulmonary hypertension in β-Thalassemia is characterized by precapillary pulmonary hypertension in the absence of left-sided heart disease, lung disease, or chronic thromboembolism. § Exact mechanism remains unknown § In newer classification, it would belong to group 5 pulmonary hypertension associated with chronic hemolytic anemia with unclear/multifactorial mechanism
There exists a higher prevalence noted in NTDT than
TDT patients
Suggested Mechanisms include:
– Hypercoagulability with thrombosis suggested to play a major role
– Hemolysis and anemia halting the arginine-NO pathway disallowing dilation
– Over expression of endothelin inducing vasoconstriction
• Tricuspid-valve regurgitant jet velocity (TRV) exceeding 2.5-2.8, with
confirmation by right heart catheterization, as TRV tends to overestimate
PHT prevalence
• Generally causes significant morbidity with decreased functional capacity
and life-threatening right ventricular dysfunction
Chueamuangphan N, et al. J Med Assoc Thai 2012;95(1):16-21.Risk of Pulmonary Hypertension in NTDT increases with
high LIC, advancing age, and splenectomy
SF
LIC 5,000
40 TCG and SF 100 All patients
Probability of PHT (%)
TCG and LIC
90
LIC (mg Fe/g dry wt)
r = 0.514 Non-splenectomised
4,000
30
p = 0.01 80 Splenectomised
70
SF (µg/L)
3,000 60
20 50
2,000 40
10
30
r = 0.097 1,000 20
p = 0.513 10
0 0 0
20 40 60 80 18 28 38 48 58 68 78
Tricuspid gradient (mmHg) Age (years)
PHT (defined as PASP ≥ 30 mmHg) present
in 38.5% PHT prevalence in thalassaemia was 2.1%
Significantly correlated with LIC (TI 4.8%, TM 1.1%)
Not correlated with age, Hb level, and SF level
PASP, pulmonary artery systolic pressure;
1. Derchi G, et al. Circulation. 2014;129:338-45. 2. Isma’eel H, et al. Am J Cardiol. 2008;102:363-7. TCG, tricuspid gradient.Approach of pulmonary HTN in patients with β-Thalassemia Taher AT et al. Blood. 2018 Oct 25;132(17):1781-1791
Outline • General overview of thalassemia −How we understand the disease today • Comorbidities in adults • Novel therapies
Pathophysiology of Thalassemia
Cappellini MD,Motta I.Hematology 2017
Taher A., Weatherall DJ,Cappellini MD Lancet 2017Gene therapy trials
Gene Vector Location Protocol # Sponsor Condition Conditioning Intervention Phase Start
date
βA-T87Q- HPV569 France LG001 study bluebird bio β-TM and severe Myeloablative Transplantation of I/II Sept
globin (formerly SCD conditioning HSCs transduced ex 2006
Genetix vivo with a LV
Pharmaceutical
s)
βA-T87Q- BB305 France NCT02151526 bluebird bio β-TM and severe Myeloablative Transplantation of I/II July 2013
globin (HGB-205 study) SCD conditioning HSCs transduced ex
vivo with a LV
βA-T87Q- BB305 USA, NCT01745120 bluebird bio β-Thalassemia Myeloablative Transplantation of I/II Aug
globin Thailand, (HGB-204 study) major conditioning HSCs transduced ex 2013
Australia vivo with a LV
βA-T87Q- BB305 USA NCT02140554 bluebird bio Severe sickle cell Myeloablative Transplantation of I Aug 2014
globin (HGB-206 study) disease conditioning HSCs transduced ex
vivo with a LV
Negre O et al. Hum Gene Ther February 2016;27:148-165.Gene therapy trials
Gene Vector Location Protocol # Sponsor Condition Conditioning Intervention Phase Start
date
β-globin TNS9.3.55 USA NCT01639690 Memorial Sloan β-Thalassemia Partial Transplantation of I July 2012
Kettering Cancer major cytoreduction (Bu HSCs transduced ex
Center 8 mg/kg) for 3 vivo with a LV
patients,
myeloablative
conditioning (Bu
14 mg/kg) for 1
patient
β-globin GLOBE Italy NCT02453477 IRCCS San β-Thalassemia Myeloablative Transplantation of I/II May
Raffaele major conditioning HSCs transduced ex 2015
vivo with a LV
(intrabone injection)
γ-globin sGbG USA NCT02186418 Children's Severe sickle cell Unknown Transplantation of I/II July 2014
Hospital Medical disease HSCs transduced ex
Center, Cincinnati vivo with a LV
βAS3- Lenti/βAS3 USA NCT02247843 University of Severe sickle cell Unknown Transplantation of I Aug
globin -FB California, disease HSCs transduced ex 2014
(T87Q, Children's vivo with a LV
G16D, Hospital, Los
E22A) Angeles
Negre O et al. Hum Gene Ther February 2016;27:148-165.New therapeutic targets in β-thalassaemia
• HSCT
• Gene therapy • α-chain
• HbF induction synthesis reduction
Haemolysis
• Minihepcidins
• JAK2 inhibitors • Hepcidin analogues
• TMPRSS inhibitors
• Sotatercept
• Luspatercept
Luspatercept is FDA and EU approved for adult patients with transfusion dependent anaemia.
Cappellini MD, Motta I. Hematology. 2017;1:278-83. Taher AT, et al. Lancet. 2017;391:155-67.
51Luspatercept
• Luspatercept is an investigational first-in-class erythroid maturation agent that
neutralizes select TGF-β superfamily ligands to inhibit aberrant Smad2/3 signaling and
enhance late-stage erythropoiesis1,2
Luspatercept
ActRIIB / IgG1 Fc recombinant
fusion protein
Modified TGF-β superfamily
extracellular ligand
ActRIIB
domain of P
ActRIIB Cytoplasm Smad2/3
Complex
Nucleus
Human
IgG1 Fc
domain
Erythroid maturation
1. Attie KM, et al. Am J Hematol. 2014;89:766-770.
• ActRIIB, human activin receptor type IIB; IgG1 Fc, immunoglobulin G1 fragment crystallizable; 2. Suragani RN, et al. Nat Med. 2014;20:408-414.BELIEVE Trial
A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study
β-thalassemiaa
Current study
patients statusc
≥ 18 years, requiring
Luspaterceptb
regular RBC
1 mg/kg s.c. every 21 days + BSC
transfusions
Randomized
(n = 224)
(defined as: Open- Post-
unblinding
2:1
6–20 RBC units in the 24 treatment
label
Study
May be titrated up to 1.25 mg/kg
weeks prior to
(up to 5 follow-up
randomization with no ≥ Placebob
35-day transfusion-free years) (3 years)
s.c. every 21 days + BSC
period during that time)
(n = 112)
(N = 336)
12-week period 12-week period Double-blind period Crossover permitted
historical screening / run-in (48 weeks)
transfusions transfusions
a
β-thalassemia or hemoglobin E / β-thalassemia (β-thalassemia with mutation and / or multiplication of α-globin was allowed. b RBC transfusions and iron chelation therapy to maintain each patient’s baseline hemoglobin level. c The trial is fully enrolled and patients continue to receive treatment or follow-up.
BSC, best supportive care; RBC, red blood cell; s.c., subcutaneously.
The BELIEVE Trial studied adult patients.BELIEVE Trial
Study endpoints
Primary endpoint:
• ≥ 33% reduction from baseline in RBC transfusion burden (with a reduction of ≥ 2 RBC units)
during weeks 13–24
Key secondary endpoints:
• ≥ 33% reduction from baseline in RBC transfusion burden during weeks 37–48
• ≥ 50% reduction from baseline in RBC transfusion burden during weeks 13–24
• ≥ 50% reduction from baseline in RBC transfusion burden during weeks 37–48
• Mean change from baseline in RBC transfusion burden during weeks 13–24
Additional endpoint:
• ≥ 33% or ≥ 50% reduction from baseline in RBC transfusion burden during any
12 weeks or 24 weeks on study
The BELIEVE Trial studied adult patients.BELIEVE Trial
Primary endpoint MET: Rate of Erythroid Response
A significantly greater proportion of luspatercept-treated patients achieved a ≥ 33% reduction from baseline in
transfusion burden during weeks 13 to 24
Luspatercept
30
Placebo
P < 0.0001
(OR 5.79, 95% CI 2.24–14.97)
25
Transfusion Burden Reduction (%)
20
Patients Achieving ≥ 33%
15
21.4%
(n = 48)
10
5 4.5%
(n = 5)
0
Luspatercept Placebo
(n = 224) (n = 112)
CI, confidence interval; OR, odds ratio.
The BELIEVE Trial studied adult patients.BELIEVE Trial
Primary endpoint: Subgroup analysis favors luspatercept
Luspatercept Placebo
Sub-groups OR (95% CI) P value
n/N (%) n/N (%)
Overall 48/224 (21.4) 5/112 (4.5) 5.79 (2.24, 14.97) < 0.0001
Region: North America & Europe 23/100 (23.0) 1/51(2.0) 14.94 (1.95, 114.12) 0.0009
Region: Middle East & North Africa 11/52 (21.2) 2/26 (7.7) 3.22 (0.66, 15.77) 0.1351
Region: Asia–Pacific 14/72 (19.4) 2/35 (5.7) 3.98 (0.85, 18.62) 0.0629
Age: ≤ 32 years 22/129 (17.1) 4/63 (6.3) 3.00 (0.98, 9.20) 0.0476
Age: > 32 years 26/95 (27.4) 1/49 (2.0) 17.50 (2.27, 134.98) 0.0004
Splenectomy: Yes 31/129 (24.0) 2/65 (3.1) 9.72 (2.22, 42.53) 0.0003
Splenectomy: No 17/95 (17.9) 3/47 (6.4) 2.94 (0.81, 10.69) 0.0918
Sex: Female 35/132 (26.5) 4/63 (6.3) 5.33 (1.80, 15.80) 0.0011
Sex: Male 13/92 (14.1) 1/49 (2.0) 8.05 (1.01, 64.16) 0.0218
β-thalassemia Gene: β0/β0 9/68 (13.2) 2/35 (5.7) 2.54 (0.48, 13.51) 0.2708
β-thalassemia Gene: Non-β0/β0 39/155 (25.2) 3/77 (3.9) 8.35 (2.47, 28.23) < 0.0001
Baseline Transfusion Burden: ≤ 6 units/12 weeks 27/112 (24.1) 3/56 (5.4) 5.61 (1.60, 19.65) 0.0033
Baseline Transfusion Burden: > 6 units/12 weeks 21/112 (18.8) 2/56 (3.6) 6.16 (1.38, 27.44) 0.0082
Baseline Hemoglobin: < 9 g/dL 22/87 (25.3) 4/51 (7.8) 3.78 (1.25, 11.42) 0.0128
Baseline Hemoglobin: ≥ 9 g/dL 26/137 (19.0) 1/61 (1.6) 14.17 (1.85, 108.79) 0.0012
Baseline Liver Iron: ≤ 3 mg/g dry weight 12/70 (17.1) 1/37 (2.7) 7.18 (0.88, 58.63) 0.0335
Baseline Liver Iron: > 3 to ≤ 7 mg/g dry weight 13/51 (25.5) 0/30 (0) Infinity 0.0053
Baseline Liver Iron: > 7 to ≤ 15 mg/g dry weight 10/38 (26.3) 1/19 (5.3) 5.41 (0.67, 43.34) 0.0741
Baseline Liver Iron: > 15 mg/g dry weight 13/65 (20.0) 3/26 (11.5) 1.79 (0.47, 6.78) 0.3831
0.1 1 10 100
Favors placebo Favors luspatercept
The BELIEVE Trial studied adult patients.BELIEVE Trial
All key secondary endpoints MET: Rates of Erythroid Response
A significantly greater proportion of luspatercept-treated patients achieved clinically meaningful reductions in
transfusion burden of ≥ 33% and ≥ 50%
30
Luspatercept
P< 0.0001a
25
Patients Achieving Transfusion Burden
Placebo
20
P = 0.0017c
Reduction (%)
15
P = 0.0303b
10 19,6
5 10,3
7,6
1,8
3,6 0,9
0
≥ 33% ≥ 50% ≥ 50%
(from week 37 to 48) (from week 13 to 24) (from week 37 to 48)
• The least squares mean change in transfusion burden from baseline to weeks 13–24 (luspatercept versus placebo) was −1.35
RBC units/12 weeks (95% CI −1.77 to −0.93; P < 0.0001)
a
OR 6.44, 95% CI 2.27–18.26. b OR 4.55, 95% CI 1.03–20.11. c OR 11.92, 95% CI 1.65–86.29.
The BELIEVE Trial studied adult patients.BELIEVE: Reduction in RBC Transfusion Burden During
Any 12-Wk and 24-Wk Interval
Any 12-Wk Interval Any 24-Wk Interval
100 P < .0001 100 Luspatercept
(OR: 5.69;
Placebo
Patients Achieving Transfusion Burden
Patients Achieving Transfusion Burden
80 95% CI: 3.46-9.35) 80
P < .0001 P < .0001
(OR: 9.95; (OR: 25.02;
95% CI: 7.76-80.71) P < .0001
Reduction (%)
60
Reduction (%)
95% CI: 4.44-22.33) 60
(OR: 20.37;
95% CI: 2.86-144.94)
40 40
70,5
20 40,2 20 41,1 16,5
29,5
6,3 2,7 0,9
0 0
Reduction Reduction Reduction Reduction
≥ 33% ≥ 50% ≥ 33% ≥ 50%
• Significantly more patients treated with luspatercept vs placebo achieved reductions in RBC transfusion
burden of ≥ 33% and ≥ 50% during any 12-wk or 24-wk interval
Cappellini. ASH 2018. Abstr 163.BELIEVE: Safety Summary
Luspatercept Placebo
Treatment-Emergent AEs, n (%)
(n = 223) (n = 109)
≥ 1 TEAE of any grade 214 (96.0) 101 (92.7)
≥ 1 TEAE of grade ≥ 3 65 (29.1) 17 (15.6)
≥ 1 serious TEAE 34 (15.2) 6 (5.5)
TEAE-related death 0 1 (0.9)*
TEAE-related study drug discontinuation 12 (5.4) 1 (0.9)
*Due to acute cholecystitis.
• Among grade ≥ 3 TEAEs, no single organ system or class was predominant
• Only serious TEAE occurring in > 1% of patients in either arm was anemia:
luspatercept, n = 3 (1.4%); placebo, n = 0
Cappellini. ASH 2018. Abstr 163.N Engl J Med 2020;382:1219-31.
Luspatercept Registration Luspatercept has been approved by the US Food and Drug Administration (FDA) in 2019 and by the European Medicines Agency (EMA) in 2020 to treat anemia in adult patients with b-thalassemia who require regular red blood cell transfusions.
BEYOND trial: luspatercept vs placebo in non-
transfusion-dependent β-thalassaemia Week
48
• Randomized phase 2 trial
Adults with non-transfusion-dependent Luspatercept 1 mg/kga s.c. q21d
β-thalassaemia or HbE/β-thalassaemia received: + BSC
• ≤ 5 RBC units within the 24 weeks before randomization
• No RBC transfusion within 8 weeks before randomization Randomized 2:1
• Hb ≤ 10 g/dL (planned N = 150)
Placebo s.c. q21d
+ BSC
Secondary endpoint
Primary endpoint
Patient-reported β-thalassaemia symptoms (NTDT-PRO), functional and
Proportion of patients with increase in mean Hb concentration of health-related QoL (FACIT-F score, SF-36), physical activity (6MWT); iron
≥ 1 g/dL in absence of RBC transfusion from Week 13 to 24 vs baselineb chelation therapy daily dose, LIC, serum ferritin; PK; AEs
Luspatercept is FDA and EU approved for adult patients with transfusion dependent anaemia.
a May be dose escalated to 1.25 mg/kg.
b Baseline: average of 2 or more measurements ≥ 1 week apart within the 4 weeks prior to randomization.
6MWT, 6-minute walk test; AE, adverse events; FACIT-F, Functional Assessment of Chronic Illness Therapy – Fatigue; NTDT-PRO, non-transfusion-dependent thalassemia patient-reported outcomes; PK, pharmacokinetics; q21d, every 21 days;
QoL, quality of life; SF-36, 36-Item Short Form Survey.
NCT03342404. Available from: https://clinicaltrials.gov/ct2/show/NCT03342404. Last updated April 2020. Accessed October 2020.
62Pathophysioly of Thalassemia
Cappellini MD,Motta I.Hematology 2017
Taher A., Weatherall DJ,Cappellini MD Lancet 2017www:thalasasemia.org.cy NEJM in press REVIEW The β-Thalassemias Ali T. Taher, MD, PhD, FRCP1; Khaled M. Musallam, MD, PhD2; Maria Domenica Cappellini, MD, FRCP, FACP3
You can also read

















































