Listeria monocytogenes as a Vector for Cancer Immunotherapy: Current Understanding and Progress - MDPI
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
vaccines
Review
Listeria monocytogenes as a Vector for Cancer
Immunotherapy: Current Understanding
and Progress
John C. Flickinger Jr. 1 , Ulrich Rodeck 2 and Adam E. Snook 1, * ID
1 Department of Pharmacology and Experimental Therapeutics, Thomas Jefferson University,
1020 Locust Street, Philadelphia, PA 19107, USA; John.Flickinger@jefferson.edu
2 Department of Dermatology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA;
Ulrich.Rodeck@jefferson.edu
* Correspondence: Adam.Snook@jefferson.edu; Tel.: +1-215-503-7445
Received: 22 June 2018; Accepted: 23 July 2018; Published: 25 July 2018
Abstract: Listeria monocytogenes, a Gram-positive facultative anaerobic bacterium, is becoming a
popular vector for cancer immunotherapy. Indeed, multiple vaccines have been developed utilizing
modified Listeria as a tool for generating immune responses against a variety of cancers. Moreover,
over a dozen clinical trials testing Listeria cancer vaccines are currently underway, which will help to
understand the utility of Listeria vaccines in cancer immunotherapy. This review aims to summarize
current views on how Listeria-based vaccines induce potent antitumor immunity and the current
state of Listeria-based cancer vaccines in clinical trials.
Keywords: Listeria; cancer; vaccine; immunotherapy; bacteria
1. Introduction
The proficiency of the immune system to recognize and eliminate cancer cells, a process summarily
called immunosurveillance, is well documented [1]. Failure of immunosurveillance enables the clinical
development of cancer and has motivated the search for strategies to rearm and restore effective
immune responses to malignant cells. One such strategy includes vaccine development against
cancer. In general, cancer vaccines are composed of antigens found in tumor cells, often referred to
as tumor-associated antigens (TAA), paired with adjuvants designed to induce an immune response.
Antigen-specific T-cell responses induced by cancer vaccines have the potential to produce more
targeted elimination of cancer cells than conventional chemotherapy, as well as lead to durable
memory responses capable of challenging cancer recurrence. This review aims to summarize one
particular approach to cancer vaccination, employing Listeria monocytogenes vectors, and its current
progress in terms of preclinical development and clinical trials.
Listeria monocytogenes (Lm) is a Gram-positive bacteria most widely known for its ability to infect
humans and produce a variety of symptoms, including gastroenteritis, meningitis, and encephalitis [2].
In general, the human immune system mounts potent innate and adaptive immune responses capable
of controlling Lm infections. As a result, serious infection by Lm is rare and typically limited to elderly,
pregnant, or immunocompromised patients [2]. Decades worth of research on the properties that make
Lm immunogenic and how these properties can be exploited have led to its advancement as a vaccine
platform for cancer immunotherapy in clinical trials.
Lm has numerous features that make it an attractive vector for cancer immunotherapy. While other
vectors may be inhibited through neutralizing antibodies, Lm infection triggers only modest humoral
responses which fail to block reinfection [3,4]. This permits repeated administration of Lm-based
Vaccines 2018, 6, 48; doi:10.3390/vaccines6030048 www.mdpi.com/journal/vaccinesVaccines 2018, 6, 48 2 of 19
vectors to boost T-cell responses in patients as needed. Additionally, and in contrast to DNA or peptide
vaccines, Lm strongly induces both innate and adaptive immune responses. Indeed, the ability of
bacteria to activate innate immune mechanisms and elicit antitumor responses was first appreciated
over 100 years ago when William Coley observed instances of spontaneous cancer regression in sarcoma
patients who acquired bacterial skin infections [5]. Beyond innate immune mechanisms, and in contrast
to other bacterial vectors, such as Salmonella, Lm is a potent stimulator of cytotoxic lymphocytes and
cell-mediated immunity [4]. Over 50 years ago, George Mackaness further demonstrated that mice
exposed to sublethal doses of Lm developed long-lived, antibody-independent immune responses
which protected against a future Lm challenge administered at lethal doses [6]. These and other
observations eventually led to the exploration of Lm as a vaccine vector with the aim of inducing
similar cell-mediated immune responses towards foreign antigens. This concept was reduced to
practice by Paterson and colleagues using Lm-expressing β-galactosidase antigen to induce a cytotoxic
lymphocyte response that killed cancer cells expressing β-galactosidase [7]. Since the publication of
this seminal paper over 25 years ago, numerous Lm vaccines have been developed for the treatment
of a variety of cancers. Here, we will provide an overview of the mechanisms underlying Lm-based
cancer vaccines and summarize the current state of Lm vaccines in cancer clinical trials.
2. Pathogenesis of Listeria Infection
In nature, Lm typically enters through the gastrointestinal system after ingestion of contaminated food,
where it crosses the intestinal epithelium and spreads into the bloodstream [2]. However, in the context of
immunotherapeutic applications, Lm is largely administered intravenously, bypassing the intestinal
epithelium. Once in the bloodstream, Lm can enter a variety of organs, including the liver, brain,
and placenta [2,8]. While it can survive in the extracellular environment, its preferred niche is the
cell cytoplasm where it undergoes replication [9]. As Lm transitions into intracellular compartments,
numerous immune mechanisms are activated. These mechanisms are essential for understanding Lm
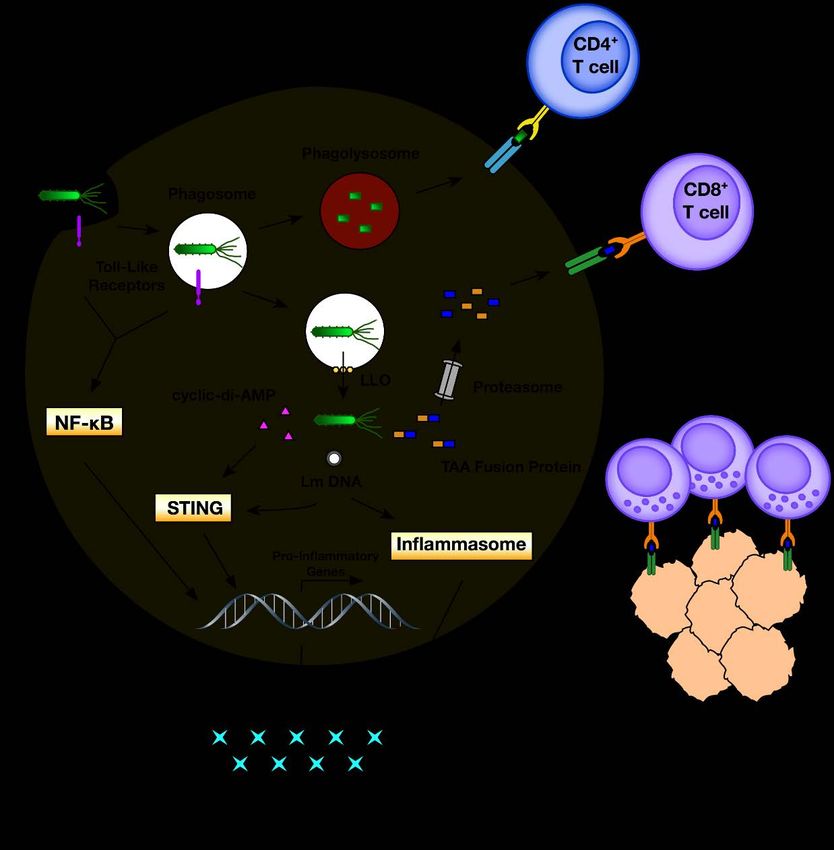
vaccine therapy and are illustrated in Figure 1.
Lm entry into mammalian cells is primarily mediated through a family of proteins called
internalins. Of particular interest are the internalin A (InlA) and internalin B (InlB) proteins,
which guide Lm invasion through binding to receptors on host cells and triggering receptor-mediated
endocytosis [8]. Importantly, InlA and InlB bind to different receptors and promote infection of
different cell types. InlA binds E-cadherin which is found on epithelial cells, while InlB interacts with
the hepatocyte growth factor, Met [10,11]. In addition to entry via cell surface receptors, Lm is actively
taken up via phagocytosis by antigen-presenting cells (APCs) [2]. Importantly, as Lm interacts with
the outside surfaces of mammalian cells, innate immune responses against Lm begin to mount. On the
surface of the cell, as well as inside phagosomes, pathogen-associated molecular patterns (PAMPs)
expressed by Lm are recognized by mammalian Toll-like receptors (TLRs), leading to the activation of
NF-κB signaling and the expression of pro-inflammatory genes [2,12].
Upon internalization into phagosomes, it appears that Lm can undergo one of two fates. It is
estimated that the majority of bacteria are killed upon phagosome–lysosome fusion [13], providing a
source of antigens for MHC class II-dependent presentation and priming of Lm-specific CD4+ T-cell
responses [4]. However, Lm has evolved mechanisms to escape lysosomal degradation and enter
the cytosols of infected cells by a well-defined mechanism. As Lm transitions from the extracellular
environment into the host cell, it begins to express the transcription factor PrfA [9]. PrfA activates
numerous virulence genes, including those responsible for phagosome escape, such as the pore-forming
toxin listeriolysin O (LLO) and two different phospholipases, PlcA and PlcB. Secreted LLO molecules
oligomerize at the phagosomal membrane to form a β-barrel pore-like structure through which Lm
can pass [14], while phospholipases mediate exit through direct hydrolysis of membrane lipids [15].
Once outside the phagosome, Lm-secreted peptides enter the host cell cytosol where they can be
degraded by proteasomes and loaded onto MHC class I molecules for presentation to cytotoxic TVaccines 2018, 6, 48 3 of 19
cells [4]. The direct secretion of antigens into the cytosol, as well as prior degradation in phagosomes,
Vaccines 2018, 6, x FOR PEER REVIEW 3 of 18
results in the induction of potent CD4+ and CD8+ T-cell responses to Lm antigens [4,16,17].
Figure
Figure 1. 1. Innate
Innate andand adaptive
adaptive immune
immune responses
responses to recombinant Listeria.
to recombinant Listeria. Listeria
Listeria monocytogenes
monocytogenes (Lm)(Lm)
is internalized by
is internalized by antigen-presenting
antigen-presenting cells cells into
into phagosomes.
phagosomes. During
During entry,
entry, Lm
Lm is sensed by
is sensed toll-like
by toll-like
receptors
receptors leading
leading to the activation
to the activation of of NFκ-B
NFκ-B andand synthesis
synthesis of of pro-inflammatory
pro-inflammatory genes.genes. Phagosomes
Phagosomes
may then fuse with lysosomes to form phagolysosomes where Lm can be killed, leading to to
may then fuse with lysosomes to form phagolysosomes where Lm can be killed, leading loading
loadingof
+
its antigens
of its antigens onto MHC
onto MHC class II for
class activation
II for of CD4
activation +
of CD4 helper T cells.
helper Alternatively,
T cells. Alternatively,Lm Lmcan can
express the
express
pore-forming
the pore-forming toxin listeriolysin
toxin listeriolysin O (LLO)
O (LLO) to to
perforate
perforate phagosomes
phagosomes and
andgain
gainentry
entryinto
intothe
the cytosol.
cytosol.
Once in the cytosol, recombinant Lm secretes tumor-associated tumor-associated antigens
antigens (TAA) as fusion proteins
with Lm
with Lmantigens
antigensinto intothe
thecytosol
cytosol where
wherethey cancan
they be degraded
be degradedby proteasomes
by proteasomesand loaded onto MHC
and loaded onto
+ cytotoxic T lymphocytes. Additionally, cytosolic Lm
class I for activation of TAA-specific CD8 cytotoxic T lymphocytes. Additionally, cytosolic Lm
MHC class I for activation of TAA-specific CD8
+
triggers
triggers further
further induction
induction of of pro-inflammatory
pro-inflammatory pathways
pathways through
through secretion
secretion of
of the
the cyclic
cyclic dinucleotide
dinucleotide
cyclic-di-AMP
cyclic-di-AMP and and detection
detection of of Lm DNA. Secreted
Lm DNA. Secreted cyclic-di-AMP
cyclic-di-AMP directly
directly stimulates
stimulates the STING
the STING
pathway and negatively regulates the NF-κB inhibitor RECON (not shown) [18], while the presence
pathway and negatively regulates the NF-κB inhibitor RECON (not shown) [18], while the presence of
of genomic Lm DNA can lead to the activation of STING and inflammasome pathways, both of which
genomic Lm DNA can lead to the activation of STING and inflammasome pathways, both of which
contribute
contribute to to the
the transcription
transcription of of pro-inflammatory
pro-inflammatory genes genes and
and cytokines.
cytokines.
Having escapedthe
Having escaped the phagosome,
phagosome, LmLm is to
is free free to throughout
move move throughout
the host the host cell
cell cytosol viacytosol via
expression
expression of the factor
of the virulence virulence factor
actin actin assembly-inducing
assembly-inducing protein Anchored
protein (ActA). (ActA). Anchored to the surface
to the surface of Lm,
of Lm, ActA protein interacts with the Arp2/3 complex to promote nucleation and polymerization of
actin monomers into filaments [19]. Forces generated via actin polymerization are capable of
projecting Lm throughout the cytosol as well as into the plasma membrane of the infected cell,Vaccines 2018, 6, 48 4 of 19
ActA protein interacts with the Arp2/3 complex to promote nucleation and polymerization of actin
monomers into filaments [19]. Forces generated via actin polymerization are capable of projecting Lm
throughout the cytosol as well as into the plasma membrane of the infected cell, forming a protrusion
that can be internalized by a neighboring cell and leading to dissemination of the infection. [19].
Similar to the sensing of Lm on the cell surface and inside phagosomes by toll-like receptors,
additional pattern recognition receptors recognize Lm in the cytosol and further stimulate
pro-inflammatory pathways. One route is through the activation of retinoic acid-inducible gene
I (RIG-I)-like receptors by Lm RNA, leading to the activation of the mitochondrial antiviral signaling
protein (MAVS) and transcription of interferon (IFN) genes [20,21]. Further transcription of IFN genes
is mediated through activation of the STING pathway through at least two mechanisms. First, Lm can
secrete cyclic diadenosine monophosphate (cyclic-di-AMP), directly activating STING [22]. Second,
Lm lysis can expose genomic DNA leading to activation of the cyclic GMP-AMP synthase, production
of cGAMP, and activation of STING [23]. Regardless of the route, activation of the STING pathway
promotes the transcription of type I IFN genes, including IFN-β [22,24]. Importantly, IFN-β and
activation of the STING pathway improve the priming of CD8+ T cells in the tumor microenvironment,
leading to potent antitumor responses [25,26]. Paradoxically, Lm mutants, secreting higher levels of
cyclic-di-AMP, elicit reduced T-cell responses, suggesting that the role of STING activation in Lm
infection is not fully understood [24]. Finally, Lm infection can activate NLRP3 and AIM2 receptors,
leading to activation of the inflammasome and further increasing pro-inflammatory cytokines [2,27].
3. Development of Attenuated Listeria Strains for Vaccination
Because wild-type Listeria is pathogenic and not suitable for clinical use, considerable effort
has been expended to improve Lm safety. The ideal Lm strain would minimize pathogenicity while
maximizing immunogenicity towards the target antigen. Multiple strategies to attenuate Lm have
been developed towards that end.
3.1. Deletion of Virulence Genes
A widely employed strategy to attenuate Lm involves deleting genes responsible for Lm tropism
and cell-to-cell transmission. As mentioned previously, Lm tropism into non-phagocytic cells, such as
hepatocytes, is mediated by the virulence factor internalin B. Therefore, deletion of inlB is expected to
limit liver toxicity and render phagocytic antigen-presenting cells the primary target of Lm infection.
Indeed, ∆inlB mutants display reduced hepatocyte entry while infecting monocytes as effectively as
wild-type strains [28]. Further attenuation can be achieved by deleting the virulence factor actA [28].
In wild-type Lm, ActA expression allows Lm to polymerize host actin filaments to maneuver through
the host cytosol and spread from one cell into another [2,9]. Deletion of actA is thus expected to confine
Lm to the cytoplasm of the cell it initially infects, limiting the spreading of infection and reducing
toxicity. Evidence for this is supported by experiments demonstrating that ∆actA strains have a much
higher LD50 (>1000-fold) than wild-type strains [29]. By deleting both inlB and actA, it is possible to
selectively infect antigen-presenting cells and reduce off-target toxicity. Vaccines using ∆actA/∆inlB
strains also exhibit rapid clearance of infection from the liver and spleen compared to single ∆actA or
∆inlB mutants [28]. Importantly, despite a rapid clearance of Lm, ∆actA/∆inlB strains demonstrate
comparable antitumor responses [28]. The Lm ∆actA/∆inlB strain, also known as Live Attenuated
Double-Deleted (LADD) (Aduro BioTech Inc., Berkeley, CA, USA), forms the basis for many vaccines
in clinical trials.
3.2. Episomal Replacement of Virulence or Metabolic Genes
While attenuation can be achieved through gene deletion, the deletion of a gene followed by
reintroduction of that gene via plasmid can also result in attenuation. It is likely that episomal gene
expression leads to different quantities or spatial patterns of gene product than expression from the host
genome [17]. As described above, PrfA is a master transcription factor that regulates the transcriptionVaccines 2018, 6, 48 5 of 19
of multiple Lm virulence factors, including LLO, ActA, and phospholipases [9]. Indeed, Lm strains
deficient in prfA, such as XFL-7, are severely attenuated [30,31]. Moreover, due to the lack of LLO and
phospholipase expression, prfA mutants are incapable of phagolysosome escape and invasion into
the host cytosol [31]. Partial restoration of virulence is achieved by the addition of plasmid-encoded
PrfA into which the tumor-associated antigens (TAA) are cloned as well [30,32,33]. The result is an
immunogenic strain that is significantly attenuated compared to wild-type Lm [30,32,33].
A similar attenuation strategy has been employed utilizing the ∆dal/∆dat strain (Lmdd).
Lm requires the dal and dat genes to synthesize D-alanine to build peptidoglycan and lipoteichoic
acid [34]. Strains that are deficient in both of these genes fail to replicate in vitro unless supplemented
with exogenous alanine [35,36]. Furthermore, immunogenicity of the Lmdd strain in vivo depends on
co-administration of alanine [37]. However, by transforming the Lmdd strain with a plasmid encoding
the dal gene under a constitutively active promoter, it is possible to partially restore virulence and
achieve immunogenicity without alanine supplementation [38]. Often, the Lmdd strain is further
attenuated by the deletion of actA creating what is often called the LmddA strain [38,39]. Both the
LmddA and the XFL-7 strain are currently being evaluated in clinical trials by Advaxis Inc., Princeton,
NJ, USA.
3.3. Killed but Metabolically Active
Heat-killed Lm and strains deficient in phagolysosomal escape have long been noted to be poor
inducers of CD8+ T-cell responses [40,41], suggesting that Lm entry into the host cytosol is required
for the effective activation of CD8+ T cells. As a result, Lm vaccine development has relied on
attenuated, rather than killed, Lm formulations. However, live attenuated Lm strains are capable of
inducing listeriosis in some cases and may not be safe to use in immunocompromised patients [4,42].
In an effort to further improve the safety profile of Lm vaccines, killed but metabolically active
(KBMA), Lm vaccines have been developed. Lm strains deficient in nucleotide excision repair can be
photochemically inactivated by the DNA crosslinking agent psoralen coupled with UV irradiation [43].
This process induces irreversible DNA damage and renders Lm incapable of replication. However,
for a period of hours after photochemical treatment, Lm is capable of invading host cells, escaping
into the cytosol, and secreting TAA products to elicit antitumor CD8+ T-cell responses [43,44]. Initially,
KMBA vaccines were found to invoke inferior protective responses compared to live, attenuated
vaccines [45]. However, enhanced CD8+ T-cell responses have been achieved using KBMA vaccines
with a constitutively activating prfA mutation [44]. While promising, KBMA Lm vaccines have yet to
enter clinical trials.
4. Fusion with Listeria Antigens Enhances Antitumor Responses
While it is possible to generate therapeutic immune responses by engineering Lm to express a
TAA alone, nearly all Lm vaccines express TAAs as chimeric proteins fused to a native Lm antigen.
Initial studies piloting Lm vaccines fused TAAs to highly-secreted Lm proteins as a method to increase
the delivery of TAAs into the cytosols of host cells [46]. However, it is now well appreciated that certain
Lm proteins, when fused to TAA, can also act as adjuvants, boosting therapeutic efficacy. The two
most common fusion partners with TAAs include modified versions of LLO and ActA.
4.1. Listeriolysin O
Listeriolysin O (LLO) is a virulence factor in the Lm life cycle and perforates the phagosome
to aid Lm entry into the cytosol. Because LLO could also perforate the plasma membrane, vaccines
incorporating LLO fusion proteins are typically engineered to express it as a truncated version (tLLO)
which is deficient in the hemolytic domains necessary for pore formation [30,33]. Effective antitumor
immunity using tLLO-TAA constructs was first demonstrated by Gunn et al. [30]. In these studies,
mice were injected with a cancer cell line expressing the human papillomavirus (HPV) antigen E7 and
then vaccinated with Lm expressing E7 alone or E7 as a fusion protein with tLLO (tLLO-E7). While E7Vaccines 2018, 6, 48 6 of 19
expressing Lm had almost no effect on tumor growth, immunization with tLLO-E7-expressing Lm
induced complete regression of 75% of the tumors [30]. Since these initial observations, numerous
studies examining the role of tLLO in antitumor immunity have been conducted, and it is believed
that tLLO enhances vaccine responses through a combination of different mechanisms.
It has been suggested that tLLO enhances the delivery of TAA to the proteasome, facilitating
antigen processing and MHC class I presentation [47]. Indeed, LLO contains PEST-like sequences
and destabilizing N-terminal residues that enhance protein degradation and affect cytosolic LLO
levels [48–50]. This hypothesis has been supported by direct comparison of Lm vaccines expressing
either E7 alone, E7 fused to the isolated PEST sequence (PEST-E7), E7 fused to a version of tLLO lacking
the PEST sequence (∆PEST-E7), or the full tLLO sequence fused to E7 (tLLO-E7). Immunization with
PEST-E7 elicited increased CD8+ T-cell responses and antitumor immunity compared to vaccination
with E7 alone [47], suggesting that PEST sequences can improve vaccine responses towards TAAs.
Consistent with this view, vaccination with ∆PEST-E7 generated reduced CD8+ T-cell responses
and antitumor immunity compared to tLLO-E7 [47]. However, immunization with ∆PEST-E7 also
elicited superior antitumor responses compared to the E7 vaccine, suggesting that tLLO may enhance
responses through additional mechanisms independent of PEST sequences [32,47]. One possibility
is that tLLO may signal through pattern recognition receptors. Indeed, LLO and other pore-forming
toxins can activate TLR4 [51]. Initially, these immunogenic properties were thought to depend on
LLO cholesterol-binding domains which are involved in LLO-induced pore formation [51]. However,
later experiments indicated that even non-hemolytic forms of LLO are capable of inducing dendritic cell
maturation and the synthesis of proinflammatory cytokines, suggesting that LLO might be signaling
through pattern recognition receptors independent of its cytotoxic attributes [52]. However, this study
was unable to identify a specific pattern recognition receptor responsible for these effects.
Since many TAAs targeted by vaccines are also expressed by normal tissues, effective vaccines
must overcome tolerance [4]. Importantly, multiple lines of evidence suggest that tLLO-TAA fusion
constructs can remodel the immunosuppressive tumor microenvironment and overcome tolerance
mechanisms. While Lm expressing E7 alone elicits poor antitumor responses, its effectiveness
is substantially increased by regulatory T-cell depletion [30]. Moreover, experiments examining
tumor-infiltrating CD4+ CD25+ regulatory T cells (Tregs) revealed that vaccination with Lm expressing
E7 alone increased the abundance of Tregs, while the tLLO-E7 vaccination decreased Tregs [53,54],
supporting the hypothesis that tLLO may enhance antitumor responses through changes in Tregs.
These results may be partly explained by a preferential expansion of CD4+ Foxp3− cells upon
vaccination with tLLO-E7 [54]. Interestingly, vaccination with Lm strains expressing tLLO alone
appears to affect the CD4+ effector and regulatory T-cell numbers similarly to those obtained with the
tLLO-E7 fusion protein [54]. In addition, tLLO-TAA constructs reportedly reduce the number and
functional activity of myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment,
potentially contributing to the ability of Lm to break peripheral tolerance against TAAs [55].
Interestingly, immunity induced by tLLO–TAA fusion constructs does not appear to be limited
by central tolerance mechanisms. Experiments using transgenic mice expressing the E7 antigen
in the thymus, resulting in central tolerance to E7, demonstrated that tLLO-E7 vaccines could expand
low-avidity E7-specific T cells and produce antitumor responses [56]. Additionally, tLLO fusions widen
the breadth of T-cell receptors (TCRs) recognizing self-antigen. In support of this notion, DNA vaccines
containing a fusion of the self/tumor antigen HER2 with tLLO induced T-cell responses towards
subdominant epitopes [32]. In this study, DNA vaccines encoding HER2 elicited responses to only
two HER2 epitopes, while a tLLO-HER2 vaccine elicited responses against four HER2 epitopes [32].
However, Lm-based, rather than DNA-based, vaccines expressing HER2 fragments fused with tLLO
induced response against nine HER2 epitopes, suggesting that even broader responses are generated
in the context of Lm [32].Vaccines 2018, 6, 48 7 of 19
4.2. ActA
A different widely used strategy for Lm vaccination involves fusing TAAs to ActA. Similar to
fusion with tLLO, these approaches tend to rely on truncated ActA sequences of varying lengths.
Some investigators have used the first 100 amino acids of ActA [44,57], while others used the
first 420 amino acids of the protein [58,59]. Similar to tLLO, fusion of E7 with ActA produces
superior antitumor immunity compared to E7 alone [60]. However, explanations for superior
therapeutic responses are incomplete. Like tLLO, ActA contains N-terminal PEST domains which may
facilitate proteasomal degradation and enhance TAA processing [4,61]. However, experiments have
also suggested that ActA may exert immunostimulatory effects independent of ActA–TAA fusion.
Using protein vaccines containing either ActA, E7, or ActA covalently conjugated to E7 (ActA-E7),
immunization with a mixture of ActA and E7 produced similar CD8+ T-cell responses and antitumor
immunity compared to vaccination with ActA-E7 fusion protein [60]. It has been speculated that
ActA may possess PAMP-like attributes, leading to the production of pro-inflammatory cytokines
and maturation of antigen-presenting cells [60]. Much like tLLO, the precise mechanisms for its
adjuvant-like properties are not entirely understood.
At least two studies have directly compared Lm vaccines expressing ActA-E7 to those expressing
tLLO-E7 [56,59]. Vaccination with either ActA-E7 or tLLO-E7 resulted in similar levels of circulating
E7-specific CD8+ T cells and yielded similar antitumor effects [59]. Interestingly, vaccination
with tLLO-E7 produced higher percentages of tumor-infiltrating E7-specific CD8+ T cells [58,59].
Both vaccines generated similar levels of E7-specific responses in transgenic mice expressing E7,
indicative of a comparable potency in breaking tolerance [56]. While it is unclear which strategy,
if any, is superior, it is well-established that fusion of TAA to either ActA or tLLO can boost immune
responses towards TAAs and enhance antitumor immunity [4,30].
5. Listeria Vaccines in Clinical Trials
To date, over 30 clinical trials testing 10 different Lm cancer vaccines have been initiated (Table 1).
Two companies have been at the forefront of advancing Lm-based vaccines through clinical trials,
with each using unique approaches. In general, Lm-based vaccines developed by Aduro BioTech Inc.
are derived from the LADD® strain. TAA genes are cloned into integration vectors allowing for stable
incorporation into the Lm genome [62]. Expression of TAAs is regulated by the ActA promoter and
occurs as a fusion protein with a modified form of ActA [17]. In contrast, Lm vaccines manufactured
by Advaxis Inc. are based on either the XFL-7 or the LmddA strains described above. Expression of
TAAs occurs episomally as a fusion protein with tLLO under control of the hly (LLO) promoter [38,39].
5.1. ADXS11-001 (Axalimogene Filolisbac [AXAL])
ADXS11-001, also known as Axalimogene Filolisbac or AXAL, is a vaccine against cancers caused
by the human papillomavirus (HPV). HPV expresses the E6 and E7 oncoproteins which directly
promote cell division, genomic instability, and tumorigenesis by interfering with the functions of
the tumor suppressor proteins p53 and RB [63]. Approximately 5% of all cancers world-wide are
caused by HPV infection, with most cases being attributed to HPV subtypes 16 and 18 [64]. AXAL is
based on the XFL-7 strain, engineered to secrete the E7 protein from HPV 16 fused to tLLO [30,58,59].
Numerous preclinical studies have demonstrated its ability to induce regression of HPV-transformed
tumors, leading to its advancement to multiple clinical trials [30,58,59].Vaccines 2018, 6, 48 8 of 19
Table 1. Listeria cancer vaccines in clinical trials.
Vaccine Antigen Cancer Indication Drug Combination Phase Completion Studies Identifier Company
I 2009 [65] N/A
I/II 12/2018 1 NCT02164461
Vaccine alone II 04/2016 NCT01116245
Cervical
II 10/2018 1 NCT01266460
III 06/2021 NCT02853604
ADXS11-001 HPV 16 E7 Vaccine + chemotherapy II 2017 [66] CTRI/2010/091/001232
Cervical and Oropharyngeal Vaccine + αPD-1 I/II 12/2019 NCT02291055
I 11/2014 NCT01598792
Oropharyngeal Vaccine alone Advaxis
II 08/2019 NCT02002182
Vaccine + chemoradiation I/II 02/2018 [67] NCT01671488
Anal
Vaccine alone II 03/2022 NCT02399813
Lung Vaccine + chemotherapy II 03/2019 1 NCT02531854
ADXS31-142 PSA Prostate Vaccine + αPD-1 I/II 12/2019 [68] NCT02325557
ADXS31-164 HER2 HER2 + Solid Tumors Vaccine alone I/II 12/2018 NCT02386501
ADXS-NEO Personal Neo-antigens Colon, Lung, Head and Neck Vaccine alone I 09/2020 NCT03265080
CRS-100 (ANZ-100) None Hepatic metastases Vaccine alone I 02/2008 [69] NCT00327652
Pancreatic, Lung,
Vaccine alone I 02/2009 [69] NCT00585845
Ovarian and Mesothelioma
Vaccine + Cy/GVAX II 08/2016 [70] NCT02004262
Vaccine + Cy/GVAX II 02/2017 [71] NCT01417000
Pancreatic Vaccine + αPD-1 + Cy/GVAX II 01/2019 NCT02243371
CRS-207 Mesothelin Vaccine + αPD-1 + αCTLA-4 + Cy/GVAX II 10/2019 NCT03190265
Vaccine + αPD-1 + IDO1 inhibitor + Cy/GVAX II 06/2023 NCT03006302 Aduro
Ovarian, Fallopian and Peritoneal Vaccine + αPD-1 + IDO1 inhibitor I/II 12/2018 NCT02575807
Gastroesophageal Vaccine + αPD-1 II 05/2019 NCT03122548
Vaccine + chemotherapy I 12/2018 [72] NCT01675765
Mesothelioma
Vaccine + αPD-1 II 03/2019 1 NCT03175172
ADU-623 EGFRvIII and NY-ESO-1 Brain Vaccine alone I 12/2018 [73] NCT01967758
pLADD Personal Neo-antigens Colorectal Vaccine alone I 12/2020 NCT03189030
Vaccine alone I 03/2020 [74] NCT02592967
JNJ-64041757(ADU-214) EGFRvIII and mesothelin Lung
Vaccine + αPD-1 I/II 03/2022 NCT03371381 Janssen 2
Vaccine alone I 06/2018 NCT02625857
JNJ-64041809(ADU-741) Multiple prostate antigens Prostate
Vaccine + anti-androgen II 09/2018 NCT02906605
1 Primary completion; 2 Licensed from Aduro.Vaccines 2018, 6, 48 9 of 19
Most clinical trials studying AXAL have been in patients with cervical cancer. After a Phase I
clinical trial demonstrated safety [65], multiple Phase II trials were initiated. Results from a Phase II trial
in India comparing AXAL and AXAL plus cisplatin to a historical control were recently published [66].
In this trial, patients with refractory cervical cancer received either three doses of AXAL or four
doses of AXAL with five infusions of cisplatin. While median overall survival (OS) was equivalent,
patients receiving AXAL or AXAL plus cisplatin experienced 1.5 to 2-fold increases in overall survival
at 12 months (34.9%) and 18 months (24.8%) [66]. In addition to combination with chemotherapy,
AXAL is also being tested in combination with immune checkpoint inhibitors. Preclinical models
using AXAL with αPD-1 inhibitors have demonstrated a reduction in Treg and MDSC abundance
and an increased antitumor response compared to AXAL alone [75]. A Phase I/II trial combining
AXAL with the αPD-1 inhibitor durvalumab was initiated but briefly put on hold after a patient died
of respiratory failure (ClinicalTrials.gov identifier NCT02291055). After an investigation, the FDA
reinstated this trial and it is currently ongoing. Of great interest, AXAL was recently advanced into
the double-blind, randomized Phase III AIM2CERV clinical trial, comparing AXAL to a placebo in
patients with high-risk, locally-advanced cervical cancer (ClinicalTrias.gov identifier NCT02853604).
This is currently the only Phase III trial testing an Lm-based vaccine. Patients in the AIM2CERV
trial will receive chemoradiation with curative intent followed by multiple infusions of AXAL or
placebo. Dosing of AXAL or the placebo is to occur every 3 weeks for the first 3 months, followed by
an infusion every 8 weeks for a total of five doses or until disease recurrence. The primary endpoint
will compare disease-free survival (DFS) in patients receiving AXAL or placebo and is expected to be
met in June 2020.
While most trials with AXAL have been in cervical cancer, AXAL is also being pursued in patients
with HPV-associated oropharyngeal, anal, and lung cancers. In oropharyngeal cancer, a Phase I trial
was terminated early after a patient suffered a dose-limiting toxicity (ClinicalTrials.gov identifier
NCT01598792). Despite this setback, a Phase II trial testing AXAL as a monotherapy in anal cancer
continues to recruit patients (ClinicalTrials.gov identifier NCT02002182). In anal cancer, a Phase I
trial studying AXAL in combination with radiation and the chemotherapies mitomycin and 5-FU was
recently completed (ClinicalTrials.gov identifier NCT01671488). Of the nine patients who completed
treatments, eight were progression-free at a median follow-up time of 42 months [67]. Based on those
results, a subsequent Phase II trial assessing AXAL as a monotherapy in anal cancer was initiated
(ClinicalTrials.gov identifier NCT2399813). This trial is expected to be completed in March 2022.
In addition to AXAL, Advaxis is also pursuing a second-generation version called ADXS-DUAL
in collaboration with Bristol-Myers Squibb. ADXS-DUAL possess E7 from both HPV 16 and HPV 18
serotypes. Although the majority of clinical trials by Advaxis have focused on AXAL, Advaxis is also
advancing Lm vaccines against the tumor antigens PSA (prostate-specific antigen) and HER2 (human
epidermal growth factor 2).
5.2. ADXS31-142
PSA is a serine protease whose expression is normally confined to the prostate. While its utility
as a serum diagnostic marker in prostate cancer remains controversial, it is well-accepted that PSA
can act as an immunotherapeutic target in prostate cancer [76]. Early studies piloting tLLO-PSA
vaccines were based on the XFL-7 strain of Lm; however, clinical studies use ADXS31-142 based
on the LmddA strain. Both versions have been shown to modestly reduce tumor burden and the
number of tumor-infiltrating Tregs in mice inoculated with the prostate cancer cell line TPSA23 [33,38].
Based on the hypothesis that radiation therapy may induce immunogenic tumor cell death and promote
favorable inflammatory environments for T-cell activation, follow-up studies assessed ADXS31-142 in
combination with radiation therapy [77,78]. While ADXS31-142 or radiation alone slowed TPSA23
tumor growth, combination therapy led to multiple instances of complete tumor regression [77].
Synergistic antitumor responses were associated with significant increases in PSA-specific T cells in
the spleen and tumor [77] as well as enhanced intratumoral Th1 responses [78]. Recent reports haveVaccines 2018, 6, 48 10 of 19
demonstrated an additional therapeutic benefit by combining ADXS31-142 and radiation therapy with
αPD-1 or αPD-L1 inhibitors [78].
Currently, ADXS31-142 is being tested in a Phase I/II trial in patients with metastatic
castration-resistant prostate cancer. The trial (ClinicalTrials.gov identifier NCT02325557), also known
as the KEYNOTE-046 trial, is assessing progression-free survival following ADXS31-142 alone or in
combination with the αPD-1 antibody pembrolizumab. While the trial is not yet completed, an early
report found that patients vaccinated with ADXS31-142 generated T-cell responses against multiple
prostate antigens beyond PSA through a process known as epitope spreading [68]. In this context,
it is believed that PSA-specific T cells inducing prostate cancer cell death and the production of
inflammatory cytokines result in the release of additional tumor antigens and induction of immune
responses against additional epitopes, ultimately broadening the therapeutic scope of the vaccine.
5.3. ADXS31-164
HER2 is a receptor tyrosine kinase that is frequently overexpressed by cancers, including breast,
salivary gland, and bone [79,80]. HER2 expression is often associated with poor prognosis, as HER2
signaling has been shown to directly activate pathways responsible for tumor growth and survival [79].
While antibody-based therapies targeting HER2 have been developed, many patients still experience
recurrence after treatment. In addition to antigen escape, antibody-based therapies against HER2
primarily induce antibody-dependent cytotoxicity in the absence of adaptive immune responses.
Indeed, poor Th1 responses after anti-HER2 antibody therapy strongly correlated with recurrence,
leading to speculation that vaccines boosting Th1 responses could improve the standard of care [81,82].
Due to its large molecular mass, early Lm-based HER2 vaccine designs consisted of five different
Lm vaccines, with each expressing tLLO fused to a unique region of the rat HER2 extracellular or
intracellular domain [32]. Individually, each Lm strain antagonized the growth of the HER2 expressing
NT-2 tumor cell line to a similar extent. Based on this proof of concept, a single Lm vaccine against
human HER2, known as ADXS31-164, was subsequently developed [39,83]. ADXS31-164 is derived
from the attenuated LmddA strain and is engineered to secrete a chimeric tLLO fusion protein
containing immunogenic regions from two extracellular domain sequences and one intracellular
domain sequence of HER2 [83]. The chimeric HER2 protein is designed to encompass most of the
known human MHC class I epitopes of the HER2 protein. Preclinical studies with this vaccine
demonstrated the ability of ADXS31-164 to reduce the number of regulatory T cells in tumors and
increase the CD8+ T-cell/Treg ratio [39]. Impressive results with ADXS31-164 in preclinical studies has
led to its advancement to clinical trials in canines and humans.
Recently, results from a Phase I trial of ADXS31-164 in canine osteosarcoma were published [84].
In this trial, canines received three doses of ADXS31-64 following amputation and chemotherapy.
Out of 18 immunized dogs, 15 developed T-cell responses against HER2. Impressively, this led
to a significant reduction in metastatic disease and increase in survival. Canines vaccinated with
ADXS31-164 had a median survival time of 956 days, and 56% were still alive three years after
chemotherapy, compared to a historical control of 423 days and 22%, respectively [84]. Canines with
immune responses to the intracellular HER2 domain had the most robust effects on survival,
potentially related to the fact that the intracellular HER2 domain is indispensable for oncogenic
cell signaling [85]. In contrast to HER2/neu antibodies, which are encumbered by adverse effects,
including cardiotoxicity [86], ADXS31-164 was not associated with cardiotoxicity in canines [84].
In 2014, Advaxis licensed the use of ADXS31-164 in canines to Aratana Therapeutics Inc. where it
was renamed AT-014. The USDA, in early 2018, granted conditional clinical approval for the use
of AT-014 in canine osteocarcoma. Currently, AT-014 is the only Lm vaccine to receive any clinical
approval. While data from mice and dogs indicate promising potential, results from human trials have
yet to be released. Currently, an ongoing Phase I/II trial is examining the use of ADXS31-164 in HER2
expressing solid tumors. This trial is expected to be completed in December of 2018.Vaccines 2018, 6, 48 11 of 19
5.4. CRS-100 (ANZ-100)
While most antitumor effects of Lm vaccines are attributed to Lm secretion of TAAs leading to
TAA-specific T-cell responses, studies utilizing CRS-100 highlight the powerful effects that Lm vaccines
can have on the innate immune system. CRS-100 consists of the live, attenuated ∆actA/∆inlB strain of
Lm and does not express exogenous antigens. Despite this, the CRS-100 vaccination has been shown
to mediate antitumor effects, primarily through inducing antigen-independent NK cell activity. In
preclinical modes of mice with hepatic colorectal cancer metastases, the CRS-100 vaccination induced
recruitment of NK cells to the liver, leading to antitumor responses [87]. The therapeutic effects of the
vaccine were eliminated by depletion of NK cells. Interestingly, while initial antitumor immunity was
mediated by NK cells, challenge experiments in surviving vaccinated mice found that tumors were
rejected through memory CD8+ T-cell responses, suggesting that primary destruction of the tumor
by NK cells can trigger adaptive immune responses [87]. A Phase I dose escalation study of CRS-100
in patients with hepatic metastases showed similar effects on NK cells. A single injection of CRS-100
upregulated expression of the NK cell maturation marker CD38 and stimulated the production of
Th1 cytokines in a dose-dependent manner [69]. Moreover, this trial was the first to demonstrate the
safety and tolerability of the LADD® strain in a clinical setting. It appears that the development of
CRS-100 has been halted in favor of constructs that incorporate TAAs which may directly stimulate
antigen-specific CD8+ T-cell responses [17].
5.5. CRS-207
CRS-207 is an Lm vaccine expressing the tumor-associated antigen mesothelin as a fusion protein
with a modified form of ActA [69]. Mesothelin is a glycoprotein that is normally confined to serosal
cells lining the pleura, peritoneum, and pericardium [88]. Overexpression of mesothelin has been
observed in pancreatic, ovarian, lung, and multiple other cancers, making it an attractive target for
immunotherapy [88]. Clinical trials testing CRS-207 have primarily been performed in patients with
metastatic pancreatic ductal adenocarcinoma (PDAC). After Phase I testing demonstrated the safety
of CRS-207 in patients with pancreatic, mesothelioma, lung, and ovarian cancers (ClinicalTrials.gov
identifier NCT00585845), multiple Phase II trials in PDAC were initiated using a prime-boost strategy.
In these trials, CRS-207 is given as a boost after initial priming with GVAX and cyclophosphamide
(Cy/GVAX) therapy. GVAX is a cancer vaccine composed of irradiated pancreatic cancer cells
engineered to secrete GM-CSF [89]. Cyclophosphamide is typically given prior to GVAX based
on studies demonstrating inhibitory effects on regulatory T cells and an increased survival benefit
compared to GVAX alone [89]. The use of CRS-207 in combination with Cy/GVAX has been supported
by at least two observations. First, early Phase I testing of CRS-207 found an increased survival
benefit in a small cohort of PDAC patients who received prior GVAX therapy compared to those
who did not [69]. Moreover, preclinical studies in mouse PDAC models utilizing GVAX prime with
CRS-207 boost demonstrated synergistic induction of mesothelin-specific T cells and inhibition of tumor
growth [71]. Early Phase II testing in patients with metastatic PDAC demonstrated increased median
OS in patients receiving GVAX with CRS-207 compared to Cy/GVAX alone (6.1 vs. 3.9 months) [71].
The induction of mesothelin-specific T cells was found to correlate with increased overall survival [71].
Interestingly, there was no observed difference in the number of circulating mesothelin-specific
CD8+ T cells between Cy/GVAX and Cy/GVAX + CRS-207. Based on the results from this study,
a Phase IIb study (ECLIPSE) was initiated which compared CRS-207 vs. Cy/GVAX with CRS-207
vs. conventional chemotherapy. Data presented at a conference hosted by the American Society
for Clinical Oncology indicated no survival benefit from either CRS-207 alone (5.4 months), or the
combination of CRS-207 with Cy/GVAX (3.8 months), compared to standard-of-care chemotherapy
(4.6 months) [70]. Additional trials exploring CRS-207 and Cy/GVAX in combination with αPD-1 and
αCTLA-4 immune checkpoint inhibitors as well as with indoleamine dioxygenase (IDO) inhibitors
are underway.Vaccines 2018, 6, 48 12 of 19
These are complemented by other trials using CRS-207 in patients with mesothelioma, gynecologic,
and gastroesophageal cancers, and data in mesothelioma patients have been published. These
data showed that, after two infusions of CRS-207 in combination with six cycles of chemotherapy,
59% of patients exhibited a partial response, and an additional 35% experienced stable disease [72].
Further testing demonstrated an increase in tumor-infiltrating lymphocytes post-vaccination [72].
While numerous trials with CRS-207 are currently ongoing, Aduro has discontinued further clinical
advancement of CRS-207.
5.6. ADU-623
ADU-623 is currently the only Lm vaccine being developed for the treatment of brain cancers.
ADU-623 is a bivalent vaccine that expresses EGFRvIII and NY-ESO-1 antigens [73]. EGFRvIII is the
most commonly occurring EGFR (epidermal growth factor receptor) mutation, producing a neoantigen
formed from the spontaneous deletion of exons 2–7 which encode sequences in the extracellular
domain [90]. Oncogenic EGFRvIII signaling may directly promote tumor growth and is frequently
present in glioblastoma, lung, and other cancers [90]. NY-ESO is a cancer-testis antigen, the expression
of which is restricted to germ cells in healthy adults. However, aberrant expression of NY-ESO-1
has been observed in neuroblastoma, esophageal, and other cancers [91]. Currently, a Phase I trial
assessing ADU-623 in patients with high-grade astrocytomas is ongoing (ClinicalTrials.gov identifier
NCT01967758). The trial is expected to be completed in December 2018.
5.7. JNJ-64041757 and JNJ-64041809
In 2014, Aduro licensed the Lm vaccines JNJ-64041757 and JNJ-64041809 to Janssen Biotech.
JNJ-64041757 (previously known as ADU-214) is a bivalent Lm vaccine expressing EGFRvIII
and mesothelin antigens [4]. Early reports from a Phase I trial in non-small cell lung cancer
patients suggest that JNJ-64041757 exhibits a safety profile similar to CRS-207 and is capable of
generating mesothelin-specific T-cell responses [74]. Based on these results, a subsequent Phase
I/II trial assessing JNJ-64041757 alone or in combination with the αPD-1 inhibitor nivolumab was
launched (ClinicalTrials.gov identifier NCT03371381). The trial is expected to be completed in
March 2020. JNJ-64041809 (previously known as ADU-741) contains Lm expressing multiple prostate
cancer-associated antigens [4]. Currently, it is undergoing Phase I clinical testing in patients with
metastatic castration-resistant prostate cancer. A Phase II trial testing JNJ-64041809 in combination with
the nonsteroidal antiandrogen apulatamide was initiated, but it was withdrawn prior to enrollment
(ClinicalTrials.gov identifier NCT02906605).
5.8. pLADD and ADXS-NEO
During tumorigenesis, cancer cells undergo genetic alterations which can lead to the cumulative
production of mutated protein sequences not found in healthy tissues [92]. These mutated proteins,
termed neoantigens, may undergo proteasomal degradation and MHC loading, forming unique epitopes
presented only by the tumor cells harboring the underlying mutations. In theory, vaccines targeting
neoantigens may generate stronger T-cell responses due to the absence of tolerance mechanisms as well as
fewer off-target effects [92]. Based on this rationale, two Lm vaccines targeting personalized neoantigens,
ADXS-NEO and pLADD, were recently approved for Phase I testing. Proof of concept for ADXS-NEO
was demonstrated using CT26 and MC38 colorectal cancer cell lines, and this was presented at the
American Association for Cancer Research annual meeting [93,94]. The development of personalized
vaccines involved whole-exome sequencing to identify tumor-specific mutations followed by analysis
with predictive algorithms to identify potential neoepitopes. After screening to verify immunogenicity,
the neoantigens were cloned into Lm and found to induce tumor regression upon immunization [93,94].
A Phase I trial testing ADXS-NEO in patients with metastatic head and neck, colon, and lung cancer
is underway (ClinicalTrials.gov identifier NCT03265080). The personalized Lm vaccine pLADD is
similarly undergoing Phase I testing in patients with microsatellite stable metastatic colorectal cancerVaccines 2018, 6, 48 13 of 19
(ClinicalTrials.gov identifier NCT03189030). In addition to personalized vaccines, Advaxis is currently
developing ADXS-HOT. Because sequencing for neoantigens is a time-consuming and expensive process,
ADXS-HOT is designed to be an “off-the-shelf” therapy containing a collection of frequently-mutated
somatic, cancer testis, and oncofetal antigens [95]. However, the clinical details concerning this vaccine
have not been released.
5.9. Adverse Effects of Listeria Vaccine Therapy
In general, the most common side effects of Lm vaccines are grade 1 or 2 adverse events, including
transient fever, chills, nausea, vomiting, and hypotension [66,67,69,71]. Increases in circulating liver
enzymes, as well as lymphopenia, have also been reported upon administration of vaccines [65,69,71].
In at least two cases, systemic listeriosis was reported after administration but was adequately treated
with antibiotics [42,96]. Overall, multiple clinical trials have demonstrated an excellent safety profile
with attenuated Lm vaccines in humans.
6. Conclusions and Future Directions
While preclinical models have shown impressive therapeutic benefits of Lm vaccines, currently,
no Lm vaccines are FDA-approved. It is important to note that Lm vaccines are still relatively nascent,
with most clinical trials only being initiated in recent years. As these trials advance, further improvements
to Lm vaccine technology will likely be made in the coming years. Indeed, after initial clinical trials
used Lm expressing a single tumor antigen, Lm vaccine technology has evolved to include vaccines
expressing multiple tumor antigens. By targeting multiple antigens, it may be possible to induce
therapeutic responses that are less susceptible to antigen escape. Yet, even vaccination against multiple
tumor antigens may still not be sufficient to induce optimal therapeutic responses. Tumors consist
of many cell types and the tumor microenvironment has been hypothesized to be a protective niche
that may limit antitumor responses. Thus, many strategies moving forward are likely to combine Lm
vaccines with approaches that remodel the tumor microenvironment. One such strategy has been
to directly target angiogenesis using Lm vaccines. Indeed, Lm vaccines targeting tumor-associated
angiogenic proteins, including CD105 and VEGFR2, have demonstrated inhibited tumor growth [97–100]
as well as the ability to induce secondary antitumor immune responses via epitope spreading [98,99].
While exciting, these vaccines have not yet been tested clinically. Other studies are likely to explore
combination approaches using Lm vaccines with radiation, chemotherapy, immune checkpoint inhibitors,
and other drugs. These studies are expected to firmly establish the utility of Lm vaccines in the emerging
immunotherapeutic landscape for the benefit of patients with a variety of malignancies.
Author Contributions: J.C.F.J. and A.E.S conceived the review, J.C.F.J. wrote the manuscript, and A.E.S. and U.R.
provided critical comments.
Funding: This research received no external funding. The APC was funded by the Department of Defense
Congressionally Directed Medical Research Programs (#W81XWH-17-1-0299 to A.S).
Acknowledgments: The authors are supported, in part, by the Department of Defense Congressionally Directed
Medical Research Programs (#W81XWH-17-1-0299 to A.S), the Alfred W. and Mignon Dubbs Fellowship Fund (to
JCF), and Advaxis Inc. (to U.R. and A.S.).
Conflicts of Interest: The authors are supported, in part, by sponsored research projects from Advaxis Inc.
Advaxis had no role in the preparation of this manuscript.
References
1. Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape.
Immunology 2007, 121, 1–14. [CrossRef] [PubMed]
2. Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and
pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [CrossRef] [PubMed]Vaccines 2018, 6, 48 14 of 19
3. Leong, M.L.; Hampl, J.; Liu, W.; Mathur, S.; Bahjat, K.S.; Luckett, W.; Dubensky, T.W.; Brockstedt, D.G.
Impact of preexisting vector-specific immunity on vaccine potency: Characterization of Listeria
monocytogenes-specific humoral and cellular immunity in humans and modeling studies using recombinant
vaccines in mice. Infect. Immun. 2009, 77, 3958–3968. [CrossRef] [PubMed]
4. Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of
tumor immunotherapy. Front. Cell. Infect. Microbiol. 2014, 4, 51. [CrossRef] [PubMed]
5. Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten
original cases. Am. J. Med. Sci. 1893, 105, 487–511. [CrossRef]
6. Mackaness, G.B. Cellular resistance to infection. J. Exp. Med. 1962, 116, 381–406. [CrossRef] [PubMed]
7. Schafer, R.; Portnoy, D.A.; Brassell, S.A.; Paterson, Y. Induction of a cellular immune response to a foreign
antigen by a recombinant Listeria monocytogenes vaccine. J. Immunol. 1992, 149, 53–59. [PubMed]
8. Pizarro-Cerdá, J.; Kühbacher, A.; Cossart, P. Entry of Listeria monocytogenes in mammalian epithelial cells: An
updated view. Cold Spring Harb. Perspect. Med. 2012, 2. [CrossRef] [PubMed]
9. De las Heras, A.; Cain, R.J.; Bielecka, M.K.; Vázquez-Boland, J.A. Regulation of Listeria virulence: PrfA
master and commander. Curr. Opin. Microbiol. 2011, 14, 118–127. [CrossRef] [PubMed]
10. Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met
receptor tyrosine kinase. Cell 2000, 103, 501–510. [CrossRef]
11. Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R.-M.; Cossart, P. E-cadherin is the receptor for internalin,
a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 1996, 84, 923–932. [CrossRef]
12. Aubry, C.; Corr, S.C.; Wienerroither, S.; Goulard, C.; Jones, R.; Jamieson, A.M.; Decker, T.; O’Neill, L.A.J.;
Dussurget, O.; Cossart, P. Both TLR2 and TRIF contribute to interferon-β production during Listeria infection.
PLoS ONE 2012, 7, e33299. [CrossRef] [PubMed]
13. Sun, R.; Liu, Y. Listeriolysin O as a strong immunogenic molecule for the development of new anti-tumor
vaccines. Hum. Vaccines Immunother. 2013, 9, 1058–1068. [CrossRef] [PubMed]
14. Köster, S.; Van Pee, K.; Hudel, M.; Leustik, M.; Rhinow, D.; Kühlbrandt, W.; Chakraborty, T.; Yildiz, Ö.
Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation.
Nat. Commun. 2014, 5, 3690. [CrossRef] [PubMed]
15. Tattoli, I.; Sorbara, M.T.; Yang, C.; Tooze, S.A.; Philpott, D.J.; Girardin, S.E. Listeria phospholipases subvert
host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013, 32, 3066–3078. [CrossRef]
[PubMed]
16. Miles, B.A.; Monk, B.J.; Safran, H.P. Mechanistic insights into ADXS11-001 human papillomavirus-associated
cancer immunotherapy. Gynecol. Oncol. Res. Pract. 2017, 4, 9. [CrossRef] [PubMed]
17. Le, D.T.; Dubenksy, T.W.; Brockstedt, D.G. Clinical development of Listeria monocytogenes-based
immunotherapies. Semin. Oncol. 2012, 39, 311–322. [CrossRef] [PubMed]
18. McFarland, A.P.; Luo, S.; Ahmed-Qadri, F.; Zuck, M.; Thayer, E.F.; Goo, Y.A.; Hybiske, K.; Tong, L.;
Woodward, J.J. Sensing of bacterial cyclic dinucleotides by the oxidoreductase RECON promotes NF-κB
activation and shapes a proinflammatory antibacterial state. Immunity 2017, 46, 433–445. [CrossRef]
[PubMed]
19. Lambrechts, A.; Gevaert, K.; Cossart, P.; Vandekerckhove, J.; Van Troys, M. Listeria comet tails: The
actin-based motility machinery at work. Trends Cell Biol. 2008, 18, 220–227. [CrossRef] [PubMed]
20. Hagmann, C.A.; Herzner, A.M.; Abdullah, Z.; Zillinger, T.; Jakobs, C.; Schuberth, C.; Coch, C.; Higgins, P.G.;
Wisplinghoff, H.; Barchet, W.; et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during
infection in non-immune cells. PLoS ONE 2013, 8, e62872. [CrossRef] [PubMed]
21. Abdullah, Z.; Schlee, M.; Roth, S.; Mraheil, M.A.; Barchet, W.; Böttcher, J.; Hain, T.; Geiger, S.; Hayakawa, Y.;
Fritz, J.H.; et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J.
2012, 31, 4153–4164. [CrossRef] [PubMed]
22. Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP secreted by intracellular Listeria monocytogenes
activates a host type I interferon response. Science 2010, 328, 1703–1705. [CrossRef] [PubMed]
23. Hansen, K.; Prabakaran, T.; Laustsen, A.; Jørgensen, S.E.; Rahbæk, S.H.; Jensen, S.B.; Nielsen, R.; Leber, J.H.;
Decker, T.; Horan, K.A.; et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and
STING-dependent pathway. EMBO J. 2014, 33, 1654–1666. [CrossRef] [PubMed]
24. Archer, K.A.; Durack, J.; Portnoy, D.A. STING-dependent type I IFN production inhibits cell-mediated
immunity to Listeria monocytogenes. PLoS Pathog. 2014, 10, e1003861. [CrossRef] [PubMed]You can also read

















































