LEVERAGING THERMODYNAMIC INTERACTIONS TO ENHANCE DRUG DELIVERY ALAN DOGAN - by - CASE WESTERN RESERVE UNIVERSITY - OhioLINK ETD ...
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
LEVERAGING THERMODYNAMIC INTERACTIONS TO ENHANCE DRUG DELIVERY by ALAN DOGAN Submitted in partial fulfillment of the requirements for the degree of Master of Science Department of Biomedical Engineering CASE WESTERN RESERVE UNIVERSITY May, 2021
We hereby approve the thesis/dissertation of Alan Dogan Candidate for the degree of Master of Science*. Committee Chair Horst A. von Recum, PhD Committee Member Samuel Senyo, PhD Committee Member Anirban Sen Gupta, PhD Date of Defense April 1, 2021 *We also certify that written approval has been obtained for any proprietary material contained therein. 2
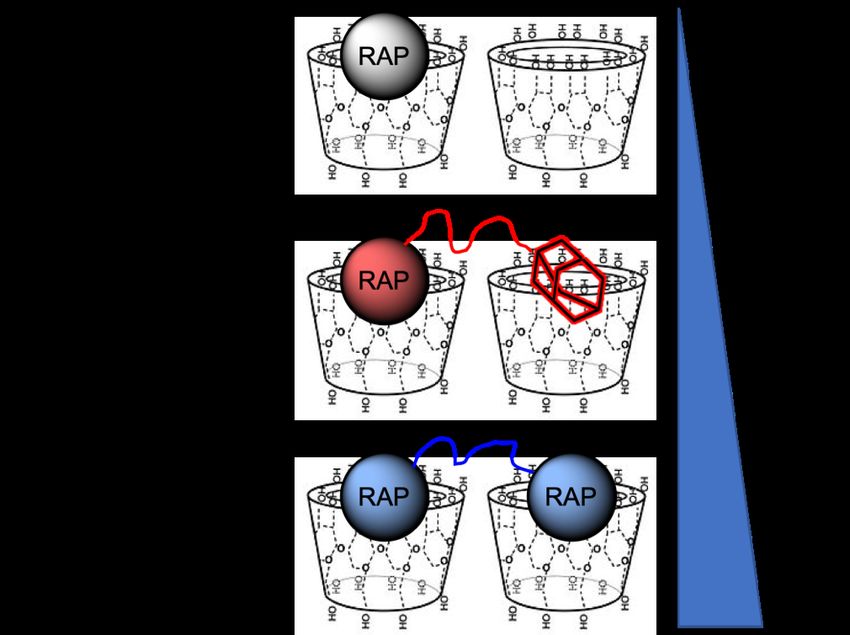
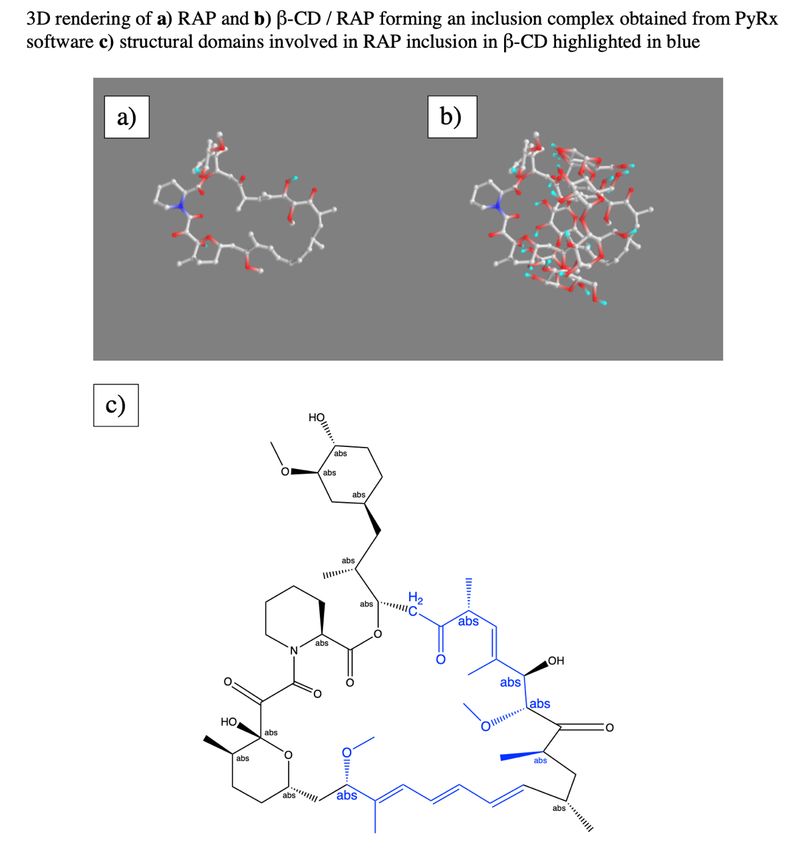
TABLE OF CONTENTS LIST OF TABLES ..................................................................................................................... 5 LIST OF FIGURES .................................................................................................................... 6 ACKNOWLEDGEMENTS ...................................................................................................... 10 ABSTRACT ............................................................................................................................. 11 1. INTRODUCTION ................................................................................................................ 12 1.1 Polymerized Cyclodextrin (pCD) as a Delivery Platform ............................................................. 12 1.2 Small-molecule Model Drug: Sirolimus (Rapamycin; RAP) ........................................................ 13 1.2.1 Previous attempts to deliver RAP ........................................................................................................ 14 1.2.2 RAP bioconjugation for enhanced delivery – molecular ‘anchors’........................................................ 14 1.3 Protein-based Model Drugs: Bovine Serum Albumin (BSA) and Monoclonal Antibodies (mAb) . 15 1.3.1 Issues with existing protein therapeutic delivery .................................................................................. 16 1.3.2 Altering “affinity” to enhance interactions between proteins and their delivery ‘host’ ........................... 16 2. MATERIALS AND METHODS .......................................................................................... 17 2.1 Materials ..................................................................................................................................... 17 2.2 Synthesis of Drug Conjugates ...................................................................................................... 18 2.2.1 Synthesis of rapamycin conjugates ...................................................................................................... 18 2.2.2 Synthesis of BSA and mAb conjugates ................................................................................................ 21 2.3 pCD Synthesis (MPs and SRPL).................................................................................................. 23 2.4 Chemical Structure Analysis........................................................................................................ 24 2.4.1 Nuclear Magnetic Resonance .............................................................................................................. 24 2.4.2 Fourier Transform Infrared Spectroscopy ............................................................................................ 24 2.4.3 (Ad-PEG5000-Mal) rate of hydrolysis.................................................................................................... 24 2.5 pCD “Affinity” Analysis ............................................................................................................. 25 2.5.1 PyRx docking simulation .................................................................................................................... 25 2.5.2 Surface plasmon resonance (SPR) ....................................................................................................... 25 2.6 Drug Loading and Release Kinetics ............................................................................................. 26 2.6.1 RAP, RAP-Ad, and “Dimeric” RAP loading/release kinetics ............................................................... 26 2.6.2 BSA and BSA-(PEG-Ad)x loading/release kinetics .............................................................................. 26 2.7 Drug conjugate structure/function analysis .................................................................................. 27 2.7.1 Rapamycin and conjugate mTOR activity ............................................................................................ 27 2.7.2 mAb specificity modified ELISA ........................................................................................................ 28 2.8 Statistical analysis ....................................................................................................................... 29 3. RESULTS: Small-molecule conjugation for enhancing pCD affinity .................................... 29 3.1 Synthesis of RAP-Ad and “Dimeric” RAP ................................................................................... 29 3.1.1 Succinylation of RAP.......................................................................................................................... 29 3.1.2 EDC/NHS coupling to PEG................................................................................................................. 31 3.1.3 CuAAC ‘clicked’ adamantane ............................................................................................................. 32 3.1.4 “Dimeric” RAP synthesis .................................................................................................................... 32 3.2 Molecular tethers increased RAP’s affinity for β-CD ................................................................... 33 3.3 Drug loading & release kinetics were enhanced in pCD MPs ....................................................... 34 3
3.4 RAP retains biofunctionality after selective modification ............................................................. 36 4. RESULTS: Protein conjugation for enhancing pCD affinity ................................................. 38 4.1 Synthesis of BSA-(PEG5000-Ad)x & mAb-(PEG5000-Ad)x .............................................................. 38 4.1.1 PEG-Adamantane (PEG-Ad) “tether” synthesis ................................................................................... 38 4.1.2 PEG-Ad “tethers” stable for up to 40 days ........................................................................................... 39 4.1.3 PEG-Ad ‘tethers’ successfully conjugated to reduced proteins ............................................................. 40 4.2 PEG-Ad ‘tethers’ greatly increased conjugate affinity for β-CD ................................................... 41 4.3 PEG-Ad conjugation to BSA greatly increases loading capacity into pCD, regardless of form ..... 42 4.4 Protein-polymer conjugates can be delivered for up to 65 days in pCD polymers ......................... 43 4.5 All mAb-polymer conjugates maintained at least 70% of antigen-recognition ability ................... 44 5. DISCUSSIONS .................................................................................................................... 46 5.1 RAP-Ad and “Dimeric” RAP - selective small-molecule drug bioconjugation.............................. 46 5.2 Protein-polymer conjugates can be designed to multiplex affinity interaction with pCD to promote controlled release .............................................................................................................................. 49 5.3 Regulatory considerations for drug-polymer combinations........................................................... 52 6. CONCLUSIONS .................................................................................................................. 55 7. FUTURE WORKS ............................................................................................................... 55 7.1 Improving bioconjugation for scalability ..................................................................................... 55 7.2 Investigating impact of PEG length on multiplexing with host ..................................................... 55 7.3 Impact of locally immobilized drugs and distribution of synthesized chemical species ................. 56 7.4 Clinical applications: closing the gap between drug half-lives and treatment durations ................. 57 8. PUBLICATION USAGE...................................................................................................... 58 Appendix .................................................................................................................................. 59 Bibliography............................................................................................................................. 62 4

LIST OF TABLES Table 1. SPR kinetics results for unmodified and modified RAP against β-CD immobilized CM5 chip, 1% DMSO:diH2O running buffer. Reported KD values were within the model confidence interval with Chi2 values below 10% of the maximum SPR response. PyRx docking simulation was used to predict β-CD / RAP interactions………………………..34 Table 2. SPR kinetics results for unmodified and modified protein-polymer conjugates against β- CD immobilized CM5 chip, diH2O running buffer. Indicated (*) KD values were within the model confidence interval with Chi2 values below 10% of the maximum SPR response…………………………………………………………………………………..42 Table 3. Top selling pharmaceuticals in 2020 (worldwide) along with Therapeutic Class (SM = small-molecule, P = protein/peptide), and common Administration Method (IV = intravenous infusion/injection, Tab = oral tablet, SC = subcutaneous injection). Targets, half-life, and treatment duration were obtained from Prescribers’ Digital Reference (PDR)1……………………………………………………………………………………58 5
LIST OF FIGURES Figure 1. RAP traditionally complexes with cyclodextrin 1:1 (ligand:host). The conjugation of additional “affinity” groups to RAP is expected to improve affinity by leveraging multiple complexations in pCD hydrogels, as both 1:1 and 1:2 interactions will occur...15 Figure 2. Illustration comparing the loading of small-molecule drugs, protein therapeutics, and Protein-PEG-Ad conjugates. Increased conjugation of ‘PEG-Ad’ groups should decrease KD, increase thermodynamic interactions between payload and pCD matrix, and ultimately increase loading and prolong release…………………………………………17 Figure 3. a) Molecular structure of Rapamycin with cartoon representation isolating hydroxyl group on C40. b) Synthesis outline for RAP-Ad and “Dimeric” RAP. X functional group represents an alkyne for Cu(II) ‘click’ chemistry and an amine for EDC/NHS coupling.21 Figure 4. Synthesis overview for Protein-PEG-Ad conjugates a) synthesis of Ad-PEG5000-Mal, a nucleophilic addition/elimination reaction b) synthesis of Protein-(PEG5000-Ad)x where X represents the number of PEG5000-Ad ‘tethers’ conjugated to each molecule of protein. BSA (PDB entry 4F5S) is used as a model protein, mAb (PDB entry 1IGT) is used as a ‘functional example’ of an antibody therapeutic………………………………………...22 Figure 5. a) FT-IR of RAP and succinylated RAP. v = 1740 (C=O), 3100 (broadened O-H) cm-1 suggests terminal carboxylic acid formation. b) 1H-NMR (DMSO-D6). [*] δ = 12.2 ppm (- COOH) corresponding to addition of acidic hydrogen after succinic anhydride opening...23 Figure 6. 1H-NMR (DMSO-D6) EDC/NHS RAP intermediate: [▴] δ = 10.7 ppm (-OH) peak of NHS and [◆] δ = 7.8 ppm (amine group of EDC). PEG1000 [*] δ = 3.6 ppm (CH2-CH2-O) repeat units of PEG………………………………………………………………………31 6

Figure 7. 1H-NMR (DMSO-D6) RAP-PEG1000-Ad: [*] adamantane peaks δ = 1.7 & 1.6 ppm, [◆] tetrazole ring peaks δ = 7.0 ppm…………………………………………………….32 Figure 8. a) 1H-NMR of “Dimeric” RAP: PEG3000 [◆] δ = 3.6 ppm peak and main RAP peak [*] δ = 2.6 ppm b) EDC/NHS reaction yield products determined from HPLC peak integral analysis c) Nonpolar-phase HPLC UV-Vis spectrum for “Dimeric” RAP synthesis………………………………………………………………………………….33 Figure 9. Total drug released after loading in pCD microparticles with concentrations of a) 3.7 mM (“low concentration”) [n=3] and b) 21 mM (“high concentration”) [n=3]. Error bars represent the standard error of the mean…………………………………………………35 Figure 10. a) “Low concentration” (3.7 mM ) cumulative release profile from pCD microparticles for RAP, RAP-Ad, and “Dimeric” RAP. * All groups were statistically significant in first 40 days of release (p
compared between groups [n=3] and control after 28 hours of Alamar blue incubation. * indicates p
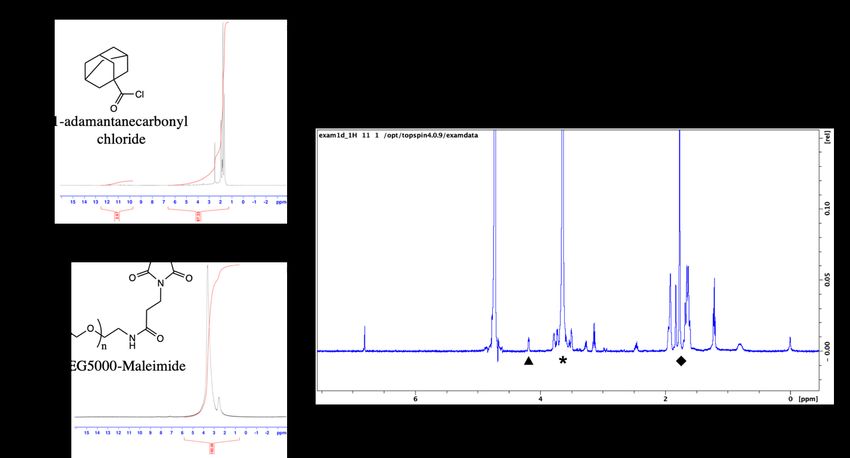
Figure 18. Quantitative estimation of mAb (anti-IL10) structure/function after PEG5000-Ad conjugation [n=3]. Positive controls (according to MabTag protocol) were assumed to have 100% antigen recognition…………………………………………………………..45 9

ACKNOWLEDGEMENTS I want to thank my family for their unconditional support throughout my undergraduate and graduate studies. I would also like to thank Dr. Horst von Recum for providing me guidance during my work and giving me the freedom to pursue what I found interesting. I would also like to thank Dr. Sam Senyo and Dr. Anirban Sen Gupta for serving on my committee, Dr. Valentin Rodionov and Dr. James Faulk for their guidance in chemistry, Dr. Nathan Rohner and Kathleen Young for their mentorship, and all of my friends who supported me during my time at Case Western Reserve University. 10
Leveraging Thermodynamic Interactions to Enhance Drug Delivery By ALAN DOGAN ABSTRACT Polymerized cyclodextrin (pCD) has been used as a drug delivery system for small-molecule (often hydrophobic) drugs, resulting in 2-6 weeks of controlled delivery. However, pCD is largely limited to drugs that match the chemical and structural characteristics that are compatible with pCD and cannot be used to deliver many modern therapeutics. Herein, we investigate the impact of conjugating “high-affinity” groups to drug payloads with low- affinity to pCD, to ultimately increase the drug-conjugate’s affinity for pCD promoting both enhanced loading and prolonged release kinetics. Utilizing the small-molecule drug Rapamycin (RAP), bovine serum albumin (BSA), and monoclonal antibodies (mAb), it was shown that relatively low-affinity drugs and protein therapeutics can be altered into conjugates with an enhanced affinity for pCD, resulting in controlled delivery up to 65 days. This work greatly increases pCD’s utility for delivering protein therapeutics and can be used to purposely extend the delivery of drugs historically difficult to administer. 11
1. INTRODUCTION Both small-molecule and protein therapeutics have made significant advances in the past decade. Although protein and peptide therapeutics have become increasingly popular for historically challenging diseases, small-molecule drugs remain a major player in today’s clinically-relevant targeted therapeutic treatments. Despite an unprecedented rate of new drug development, many novel therapeutics suffer from unfavorable delivery kinetics. Especially in chronic treatment situations – such as long-term immunosuppression, treatment of diabetes, and chronic pain management – many breakthrough drugs are not effectively utilized as they are restricted to intravenous (IV) or oral administration, which frequently results in high systemic and/or low local concentrations. One method to alleviate these delivery shortcomings is by utilizing polymer-based biomaterials to extend the window of delivery during administration, however many rely upon diffusion-based thermodynamics2. While diffusion-based delivery systems can circumvent certain metabolic and clearance issues associated with IV injection, the rate of delivery is difficult to control and rarely constant3–5. 1.1 Polymerized Cyclodextrin (pCD) as a Delivery Platform Polymerized cyclodextrin (pCD) is an alternative affinity-based drug delivery system that utilizes hydrophobic interactions between cyclodextrin’s hydrophobic “interior” pocket and hydrophobic payloads. Typically, this interaction has been used to enhance loading and extend the release of small molecule, hydrophobic drugs, as the inclusion pocket of cyclodextrin only ranges from 0.5- 0.8 nm in diameter and is greatly dependent on drug structure6. pCD has shown to have drug loading and drug release kinetics directly associated with the KD (dissociation constant) of its 12
included payload, and has the ability to refill in vivo, making it especially useful for long-term therapeutic timelines7–11. While pCD provides a platform for long-term drug delivery, the subtype of drugs that are structurally compatible with pCD are limited to a select few small-molecule, hydrophobic drugs, typically with aromatic or hydrocarbon moieties12. Small-molecule drugs can exhibit a wide range of thermodynamic interactions with pCD and innately have either a ‘low’ or ‘high’ affinity for the polymer, corresponding with a short, burst release or a long, controlled release, respectively. However, for many small-molecule drugs, structure and function are directly correlated, and non- specific chemical alteration can attenuate or eliminate cellular action. Therefore, to make low- affinity drugs compatible with pCD this work investigates bioconjugation methodology to increase alter affinity for pCD without sacrificing structure/function relationships. Herein, small-molecule (i.e. Sirolimus) and protein-based (i.e. bovine serum albumin & anti-IL10 mAb) species were selectively chemically modified with high-affinity groups to increase their affinity for pCD to, in turn, extend their time of delivery by leveraging their interactions. 1.2 Small-molecule Model Drug: Sirolimus (Rapamycin; RAP) Sirolimus (Rapamycin; RAP), a commonly-used small-molecule mTOR inhibitor, is notoriously “tricky” to conjugate and modify, as much of its chemical structure is involved in binding to its target, FK506 binding protein, which impacts downstream processes involving cell survival, migration, and cell signaling13. This leaves the sterically-hindered hydroxyl group on C40 as the optimal location for modification, and many have taken advantage of this by synthesizing “rapalogs” (Rapamycin analogs) including Temsirolimus and Everolimus14. Mainly, these 13
modifications induce limited changes in solubility and in vivo half-life and rapalogs still require daily oral or IV injection for sufficient systemic treatment14. Even worse, for immunosuppression and cancer treatments, bolus, systemic administration of RAP and rapalogs can result in adverse events including lowered platelet counts, pulmonary complications, and cardiac irregularities15–17. 1.2.1 Previous attempts to deliver RAP Efforts have been made to increasingly localize RAP therapies by utilizing polymer-mediated delivery systems, however these attempts are largely limited by the maximum loading of drug into the polymer matrices and release profiles of usually much less than 20 days18. As many patients require treatment on the scale of weeks to months, there is a need to increase the time duration of polymer-controlled release of RAP. Our group has previously used pCD to deliver RAP to promote antifibrotic activity after esophageal cancer resection resulted in a 28-day window of release, which satisfies the time frame for wound healing and cancer therapeutic delivery but not for increasingly chronic applications like immunosuppression19,20. 1.2.2 RAP bioconjugation for enhanced delivery – molecular ‘anchors’ To improve RAP’s suboptimal affinity towards pCD complexation we selectively tethered the drug to a high-affinity adamantane (Ad) functional group using a poly(ethylene) glycol (PEG) spacer. Traditionally, small-molecule drugs interact with CD pockets in a 1:1 fashion – one ligand is included in one CD “host”. Increasingly complicated interactions, such as a 1:2 ligand:host interactions and polymer “threading” through CD pockets, can be leveraged to increase these “affinity” interactions21–23. Explicitly, RAP was region-specifically tethered to adamantane (RAP- Ad) or an additional RAP molecule (“Dimeric” RAP) utilizing a biocatalyst, and the conjugates 14
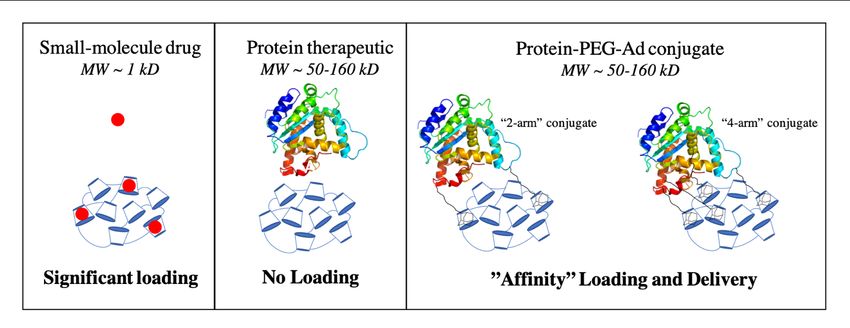
exhibited enhanced affinity towards pCD hydrogels, ultimately leading to enhanced loading and extended release, all without loss of biofunctionality (Figure 1). “Dimeric” RAP is distinct from a dimeric analogue version of FK506, known as FK1012, which lacks a flexible PEG hinge region, reducing the capacity to interact with two CD groups24. Figure 1. RAP traditionally complexes with cyclodextrin 1:1 (ligand:host). The conjugation of additional “affinity” groups to RAP is expected to improve affinity by leveraging multiple complexations in pCD hydrogels, as both 1:1 and 1:2 interactions will occur. 1.3 Protein-based Model Drugs: Bovine Serum Albumin (BSA) and Monoclonal Antibodies (mAb) As protein and peptide therapeutics now constitute the current, and possibly future, direction of pharmaceuticals, we proposed to apply the idea of modulating affinity to hydrophilic payloads, like proteins, so that they can take advantage of the “affinity-driven” loading and release kinetics common to pCD hydrogels. 15
1.3.1 Issues with existing protein therapeutic delivery Many protein therapeutics suffer from short in vivo half-lives and high accumulation in the liver and spleen25,26. This translates to clinical protocols that typically involve parenteral drug administration; however, frequent injections decrease patient compliance, increases risk of infection, and promotes nonspecific drug targeting -- all of which greatly decreases therapeutic efficacy. Therapeutic efficacy of protein-based drugs with bolus administration is also highly dose- dependent, which can also decrease positive patient outcomes. Therefore, there is a need for extending and controlling the delivery of protein and peptide therapeutics. While many groups have investigated pCD to deliver small molecules like Rifampicin and other smaller therapeutics like RNA, the delivery proteins, to our knowledge, has never been achieved due to their large molecular weights and hydrophilic nature27,28 1.3.2 Altering “affinity” to enhance interactions between proteins and their delivery ‘host’ Herein, we used bovine serum albumin (BSA) as a model protein, and monoclonal antibodies (mAb) as a functional example to show that proteins can be delivered from pCD hydrogels by increasing the protein’s affinity for the system through the conjugation of polymer-Ad groups (Figure 2). Additionally, we explore the impact that conjugating polymer-Ad groups have on the functionality of mAbs to help assess if pCD can be a viable platform for the clinical delivery of therapeutic proteins and peptides. 16
Figure 2. Illustration comparing the loading of small-molecule drugs, protein therapeutics, and Protein-PEG-Ad conjugates. Increased conjugation of ‘PEG-Ad’ groups should decrease KD, increase thermodynamic interactions between payload and pCD matrix, and ultimately increase loading and prolong release. 2. MATERIALS AND METHODS 2.1 Materials β-cyclodextrin (β-CD) prepolymer, lightly crosslinked with epichlorohydrin, was purchased from Cyclolab (Budapest, Hungary). HyperSep C18 SPE Cartridges was purchased from Sigma-Aldrich (St. Louis, MO). Sirolimus (RAP) was purchased from Biotang (Lexington, MA). Propargyl- PEG1000-amine and PEG3000-diamine was purchased from BroadPharm (San Diego, CA). Cuprisorb was purchased from Seachem Laboratories (Madison, GA). 7” NMR tubes were purchased from Bruker (Billerica, MA). Phenomenex Luna 5u C18 250x4.6 mm column was purchased from Phenomenex (Torrance, CA). Bovine Serum Albumin (BSA) heat shock fraction, protease free, fatty acid free, essentially globulin free, pH 7, >98% and QuantiPro BCA Assay Kit were purchased from Sigma-Aldrich 17
(St. Louis, MO). IL-10 ELISA Kit (Interleukin 10) was purchased from Antibodies-online.com (Limerick, PA; MabTag). β-cyclodextrin (β-CD) prepolymer, lightly crosslinked with epichlorohydrin, was purchased from Cyclolab (Budapest, Hungary). 4-20% Mini-PROTEAN TGX Precast Protein Gels and Precision Plus Protein Dual Color Standards were purchased from Bio-Rad (Hercules, CA). Maleimide PEG Hydroxyl (MW=5,000 g/mol) was ordered from JenKem Technology USA (Plano, TX). All other reagents, solvents, and chemicals, including dialysis bags were purchased from Fisher Scientific (Hampton, NH) in the highest grade available. 2.2 Synthesis of Drug Conjugates 2.2.1 Synthesis of rapamycin conjugates Due to RAP’s functionality as a mTOR (mammalian target of rapamycin) protein inhibitor, it has been found that only specific modification of C40 yields RAP analogs with retained biofunctionality (Figure 3). Also, targeting the C40 carbon ensures that RAP’s ability to bind with CD will be retained after modification (Figure S1). Therefore, immobilized Candida antarctica lipase B (NOVOZYM 435 Immobilized) is used to hold RAP in a conformation in which only the C40 hydroxide group is exposed29. Briefly, 250 mg of NOVOZYM 435 Immobilized was added to a 1.85:1 mass ratio of RAP to succinic anhydride in 4:1 toluene:dichloromethane and allowed to react for 48 hours at 37 ℃ in an inert, N2 atmosphere at 200 rpm. The solvent was degassed for 5 minutes before the reaction took place and subsequently sonicated for 5 minutes at 40 ℃ to ensure solubilization of reactants. 18
After completion, the enzyme was filtered off with dichloromethane. The solution was then filtered through a silicon column with dichloromethane:methanol (13:1) as eluent to remove residual succinic anhydride. Following the succinylation of RAP, the free carboxylic acid group was activated through EDC/NHS chemistry. Briefly, 10 molar excess EDC (1-ethyl-3-(3-dimethylaminopropyl) - carbodiimide) and 4 molar excess NHS (N-hydroxysuccinimide) was reacted with succinylated RAP in DMSO:NaHCO3 solution (0.1 M, pH 8.3, 1:1.4 v/v) with excess EDC/NHS for 5 hrs at room temperature while stirred. Once reacted, excess EDC/NHS was removed by running the solution through a HyperSep C18 Column. First the cartridge was washed with 10 mL methanol/water (90:10) and then 10 mL water as preconditioning, and 20 mg sample in 2 mL acetonitrile/water was loaded on column. Extraction was carried out by acetonitrile/water (70:30) as eluent. Several fractions (4 fractions, each 3 mL) were collected, and UV absorbance was checked at 280 nm. The fractions containing “activated” RAP was dried by lyophilization and used for further steps. For RAP-Ad synthesis, EDC/NHS activated RAP was then reacted with 2 molar equivalents of propargyl-PEG1000-amine at room temperature overnight in DMSO:NaHCO3 solution (0.1 M, pH 8.3, 1:1.4 v/v). For “Dimeric” RAP synthesis, 0.5 molar equivalent of PEG3000-diamine was reacted under similar conditions. To functionalize the opposite end the PEG during RAP-Ad synthesis, an adamantane group was “clicked” to PEG’s free alkyne end using Copper-Catalyzed Azide-Alkyne Cycloaddition 19
(CuAAC) chemistry. RAP-PEG1000-propargyl was reacted with 5 molar equivalents of azido- adamantane in the presence of 5 molar excess copper sulfate and 10 molar excess NaAsc ((+)- sodium L-ascorbate) for 12 hours in DMSO:H2O (2:1). A green color was observed if sufficient Cu(II) ions was present during reaction. To remove excess copper ions and other unreacted reagents after reaction completion, the sample dialyzed by a 1 kD cutoff molecular weight dialysis membrane for 48 hours in 1 liter of deionized H2O (diH2O) solution with a 5 g bed of Cuprisorb to act as a sink for excess copper ions To functionalize the second amine group of the RAP-PEG3000-amine conjugate, RAP-PEG3000- amine was reacted with two molar equivalents of EDC/NHS “activated” RAP overnight in in DMSO:NaHCO3 (pH 8.3, 1:1.4 v/v) with excess EDC/NHS. Reaction was then dialyzed by a 3.5 kD cutoff molecular weight dialysis membrane for 48 hours in 1 L of diH2O. 20
Figure 3. a) Molecular structure of Rapamycin with cartoon representation isolating hydroxyl group on C40. b) Synthesis outline for RAP-Ad and “Dimeric” RAP. X functional group represents an alkyne for Cu(II) ‘click’ chemistry and an amine for EDC/NHS coupling. 2.2.2 Synthesis of BSA and mAb conjugates To synthesize adamantane-capped PEG (Ad-PEG5000-Mal), a 1:1 (w/w) ratio of adamantane carbonyl chloride and maleimide-PEG5000-hydroxyl was dissolved in anhydrous chloroform (5 mL chloroform per 1 g mixture), according to a previous protocol30. Equal molar amounts of triethylamine to PEG was then added dropwise and the solution was allowed to react for 24 hrs. at room temperature (RT) under agitation (Figure 4a). Once reacted, the product was precipitated with diethyl ether three times (using half of the total volume of chloroform) and solvent was removed in vacuum at 25 oC overnight (ON). As pCD is a covalently bonded polymer network with closely-packed monomer units, PEG5000 was selected for adamantane conjugation as its estimated hydrodynamic radius (~2 nm) was predicted to sufficiently “space out” the adamantane 21
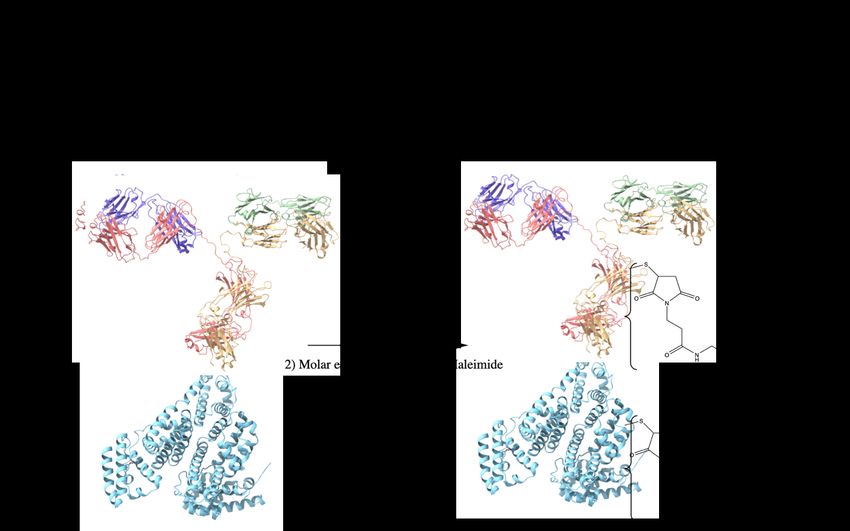
groups from the hydrophobic, interior of folded proteins (3-14 nm). This was done to help ensure adamantane groups were available for complexation within cyclodextrin’s interior. Protein- PEG5000-Ad conjugates were synthesized with sulfhydryl-reactive crosslinker chemistry. Protein samples (Bovine Serum Albumin; BSA, or anti-interleukin 10 monoclonal antibody; mAb) were incubated with 10x molar excess TCEP (tris(2-carboxyethyl)phosphine) slurry for 1 hr at RT on an end-over-end mixer. Once disulfide bonds were reduced, a variable molar excess (2x, 4x, 8x) of Ad-PEG5000-Mal was added to the protein samples, and were allowed to react 4 hrs at RT (or ON at 4 oC) (Figure 4b). The protein-polymer conjugates were then dialyzed for 48 hrs. (MWCO = 14 kD). In this study, BSA-PEG5000-Ad and mAb-PEG5000-Ad were investigated. Figure 4. Synthesis overview for Protein-PEG-Ad conjugates a) synthesis of Ad-PEG5000-Mal, a nucleophilic addition/elimination reaction b) synthesis of Protein-(PEG5000-Ad)x where X 22
represents the number of PEG5000-Ad ‘tethers’ conjugated to each molecule of protein. BSA (PDB entry 4F5S) is used as a model protein, mAb (PDB entry 1IGT) is used as a ‘functional example’ of an antibody therapeutic. 2.3 pCD Synthesis (MPs and SRPL) pCD polymer microparticles (MPs) and Sustained Release Polymeric Liquid (SRPL), a fluidic pCD formulation, were synthesized to serve as insoluble drug delivery vehicles31. As we aimed to investigate the impact of polymer topology on drug loading capabilities for protein payloads, SRPL was used as an example of crosslinked, linear branch topology while MPs were used to represent crosslinked, spherical topology. Briefly, microparticles were synthesized from epichlorohydrin-crosslinked β-cyclodextrin prepolymer solubilized in 0.2 M potassium hydroxide (25% w/v) pre heated in a 60 ℃ oil bath for 10 minutes. Light mineral oil was then added to a beaker with Tween 85/Span 85 (24%/76%) and stirred at 500 rpm. Ethylene glycol diglycidyl ether (0.01 M) was added drop-wise to the prepolymer solution dropwise and then mixed. After vortexing, the prepolymer solution was added to the oil/Span/Tween 85 solution and heated in a 70℃-oil bath. The stir speed was increased and then the mixture was stirred for 3 hours. After incubating the particles were taken out and centrifuged at 200 x g to be separated from the oil mixture and then washed with excess hexanes twice, excess acetone twice, and finally diH2O twice. The microparticles were then frozen and lyophilized before further use. Based on previous studies, we can estimate that this protocol generates pCD microparticles with a size of 81.88 ± 36.86 µm with a polydispersity index of 0.2 32 . 23
SRPL was synthesized with β -Cyclodextrin crosslinked with epichlorohydrin, dried for 4 hours at 70℃, and then stirred (150 rpm) in a 55℃-oil bath for 10 min. DMSO (4 mL per gram dried CD) was then added to the CD and incubate for 10 min. Hexamethylene diisocyanate (HDI) crosslinker (45 uL HDI per gram dried CD) was added into the CD mixture and capped with N2 gas. The speed of mixing was then increased and after two 15-minutes intervals the vial was checked to observe increases in viscosity until the solution appears viscous and glassy (~30 min). Then diH2O was added to quench the remaining crosslinker and synthesized polymer was lyophilized ON. 2.4 Chemical Structure Analysis 2.4.1 Nuclear Magnetic Resonance Nuclear magnetic resonance (NMR) was used to verify successful conjugation during synthesis steps. All spectra of presented chemical species were recorded by Bruker 300 MHz NMR system (Bruker, Germany) in DMSO-D6 or Chloroform-D solvent, as indicated. 2.4.2 Fourier Transform Infrared Spectroscopy Fourier Transform Infrared Spectroscopy (FT-IR) was used to verify bond formation during RAP succinylation. FT-IR spectrums were obtained with a Bio Rad Digilab FTS 3000-MX Excalibur Series FT-IR. Briefly, samples were mixed with potassium bromide (KBr) in a 30:70 w/w ratio. 13 mm pellets were formed by compacting the powder under 6-7 metric tons for thirty seconds. 2.4.3 (Ad-PEG5000-Mal) rate of hydrolysis For protein conjugates, the rate of hydrolysis was investigated for the ester bond present between PEG and its hydrophobic adamantane group. 10 mg of Adamantane-PEG5000-Mal was dissolved in 10 mL of DMSO-D6 and incubated to 37oC, agitated at 100 rmp. 600 µL aliquots were sampled at designated intervals and 1H-NMR spectra were obtained. Percent hydrolysis was 24
determined by comparing the integral of the “hydrolysis alcohol peak” at δ = 6.1 ppm and a fixed peak at δ = 4.2 ppm. Samples were capped with N2 after obtaining each sample. 2.5 pCD “Affinity” Analysis 2.5.1 PyRx docking simulation To test “affinity” we utilized a computer “docking” software to predict thermodynamic interactions between host and ligand based on spatial data. Molecular structure data files for RAP and β-CD were downloaded from the PubChem database. Structures were converted to PDBQT format and energy was minimized before loading RAP as a ligand and cyclodextrins as a host in PyRx (Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA). The Autodock Vina algorithm was used to predict the strength of the ligand:host interaction. 2.5.2 Surface plasmon resonance (SPR) Experimental “affinity” between β-CD monomers and protein-conjugates were measured experimentally through surface plasmon resonance (SPR) with a Biacore X100 system (GE 19,33 Healthcare Bio-Sciences, Pittsburgh, PA) according to previous protocols . The surface of a sensor chip CM-5 was conjugated with EDC (0.4 M) and NHS (0.1 M) followed by 10 mM 6- amino-6-deoxy- β-cyclodextrin (CycloLab) suspended in HBS-N buffer (a HEPES balanced salt solution with pH 7.4). The other channel was conjugated similarly with amino-dextran (Thermo Fisher Scientific) to determine specific versus nonspecific interactions with a chemically similar but non-affinity substrate. The remaining functional groups were capped with ethanolamine. A multi-cycle kinetic experiment was performed with drug dissolved in a MilliQ water solution (with 1% dimethyl sulfoxide for RAP samples) and was regenerated with 100 mM sodium hydroxide between samples. The differential responses between the channels were fit to steady state affinity 25
using Biacore evaluation software. Indicated KD values (*) were within model confidence interval (Chi2 values below 10% of the maximum SPR response)11. A concentration range of 0.125-10 nM was used. 2.6 Drug Loading and Release Kinetics 2.6.1 RAP, RAP-Ad, and “Dimeric” RAP loading/release kinetics Briefly, 100 ug of pCD was incubated in concentrated drug solutions (either ‘low’ [3.7 mM] or ‘high’ [21 mM] conditions) in DMSO for 72 hours. Particles were spun down (10,000 rpm), washed once with 1 mL of diH2O, spun down once again, and incubated in 500 uL of “physiological release buffer” (phosphate buffered saline [PBS], 0.1% Tween80) at 37 ℃. At recorded time points, the release buffer was removed and replaced with fresh buffer to better mimic in vivo conditions: each sample’s absorbance was measured at 278 nm to determine drug concentration based on standard curves. For RAP-Ad and “Dimeric” RAP species, calculations were based on predicted structural molecular weights of 2215 g/mol and 5026 g/mol. Drug loading efficiency was determined from the summation of drug present in release aliquots throughout the duration of release. 2.6.2 BSA and BSA-(PEG-Ad)x loading/release kinetics Affinity and drug loading and release kinetics of protein-(PEG5000-Ad)x was tested in vitro in both pCD MPs and SRPL. BSA was used as a model protein, as it is relatively inexpensive and easy to detect with BCA assay kits. 10 mg of dried pCD MPs and SRPL, respectively, were soaked for 48 hrs in drug solutions (100 ug/ml) of each protein-conjugate species. The loaded- pCDs were then washed 3 times with 1 mL of 0.1 sodium phosphate buffer and then incubated in 1 mL of a “physiological release buffer” (phosphate buffered saline [PBS] and 0.1% Tween80) at 26
37 ℃ on a rotary shaker. To simulate “infinite sink” in vivo conditions, aliquots were sampled frequently at recorded time intervals. BSA-conjugate concentrations were measured using a Micro BCA kit that gave a colorimetric readout that were compared to a standard curve for each BSA-conjugate species, respectively. Affinity interactions were ‘maximized’ by keeping the number of potential ‘affinity-groups’ under the theoretical maximum ‘host-groups’ (e.g. BSA- (PEG5000-Ad)2 has two potential binding sites). Calculations were based on the following assumptions: β-CD MW = 1,135 g/mol, Epichlorohydrin MW= 92 g/mol, BSA MW= 65,000 g/mol, and Mal-PEG5000-Ad at 5,330 g/mol. 2.7 Drug conjugate structure/function analysis 2.7.1 Rapamycin and conjugate mTOR activity As the mTOR pathway is responsible for regulating cellular migration, a fibroblast cellular migration assay was used as an indirect indicator for mTOR inhibition. PT-K75 porcine mucosal fibroblasts (ATCC, Manassas, VA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 15% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) at 37°C, 5% CO2. Cells were plated on a 96 well plate at ~19,200 cells per well and allowed to adhere for 2 hours in standard media, after which standard media was replaced with “starvation” media (DMEM, 0.5% FBS, 1% P/S) and incubated for 24 hrs. Plates were then “scratched” with a sterile 200 uL pipette tip, washed with “starvation” media, imaged, and treated with drug aliquots for 25 hours. At 25 hours, scratches were reimaged, compared to t=0 timepoint, and normalized to buffer-only controls. Images were analyzed in ImageJ with a variance-based macro to measure total area of “wound”. 27
mTOR signaling pathway also plays a vital role in cell metabolism and proliferation. Therefore, cellular proliferation is closely linked to mTOR activation. PT-K75 porcine mucosal fibroblasts were cultured according to previously stated methods. Cells were plated on a 96 well plate at around 7,000 cells per well and allowed to adhere in growth media for 2 hours. Aliquoted concentrations of each drug were added to wells (n=3) and incubated for 24 hours. According to manufacturer protocols, 10 µl of 0.15 mg/ml resazurin, which is used to quantify fibroblast metabolic activity, viability and proliferation19,34, was added per treatment well. After incubation for an additional 21 hours, fluorescence (530/590 excitation/emission) was measured with a Synergy H1 Microplate Reader (BioTek Instruments, Inc., Winooski, VT) and normalized to buffer controls. 2.7.2 mAb specificity modified ELISA To quantify antibody functionality after conjugation of affinity groups, we utilized a modified ELISA protocol to investigate mAb antigen recognition after PEGylation. Briefly, coating-capture antibody, and blocking steps were performed according to the MabTag ELISA protocol and 500 pg/ml of rhIL-10 standard was added to each well. Detection-antibody was treated for 1 hour with 10x molar excess TCEP and incubated for 4 hrs. at RT with varying molar excess of Mal-PEG- Ad. Upon reaction completion, 100 uL of modified detection-antibody was added to each well, and each group (n=3) was normalized to unmodified detection-antibody. ‘Antibody functionality’ was reported as a percent of positive controls, which were assumed to have 100% antigen recognition. 28
2.8 Statistical analysis All statistics were calculated in Origin (OriginLab, Northampton, MA). Statistically significance was defined as p < 0.05 with further specifications stated in figure captions. Drug release curves were analyzed by one-way ANOVA and Tukey post hoc test (p
Figure 5. a) FT-IR of RAP and Succinylated RAP. v = 1740 (C=O), 3100 (broadened O-H) cm-1 suggests terminal carboxylic acid formation. b) 1H-NMR (DMSO-D6). [*] δ = 12.2 ppm (-COOH) corresponding to addition of acidic hydrogen after succinic anhydride opening. Succinylated RAP was further observed 1H-NMR: the introduction of δ =12.2 ppm corresponds to the acidic hydrogen found after the opening of succinic anhydride during the reaction, suggesting succinylation was successful. Comparing the integrated peak values of featured groups in both succinic acid and RAP - δ =2.5 ppm corresponding to succinic acid’s -CH2 protons and δ =3.3 ppm corresponding to RAP’s -OCH3 protons - we reported that 85.6% of RAP molecules were successfully succinylated (Figure 5b). Succinylation efficacy was confirmed to range from 73-85% using Equation 1, which agreed with previously reported values (molecular weight for RAP and succinylated RAP assumed to be 914.172 g/mol and 1000 g/mol, respectively)29 H 123456 78 9:329 ;?@A629 BCD ÷FGGG = H IJK ∗ 100 (1) 123456 78 BCD ÷LFM.FOP IJK 30
3.1.2 EDC/NHS coupling to PEG After the introduction of a carboxylic group to RAP, we aimed to conjugate succinylated RAP to the free amine group on bifunctional PEGs. The intermediate EDC/NHS coupling of PEG to RAP was also confirmed with 1H-NMR. EDC/NHS intermediate formation success can be seen in the disappearance of δ =12.2 ppm peak (-COOH) in RAP and the introduction of peaks at δ =7.8 ppm, corresponding to the amine peak (-NH(CH2)2) of protonated EDC and δ =10.7 ppm corresponding to excess NHS’s hydroxyl groups (-OH) (Figure 6). RAP-NHS formation is evident by the relatively low peak intensity of NHS’s hydroxyl group (δ =10.7 ppm) despite introducing excess NHS to the reaction mixture. Successful conjugation of PEG1000 to RAP can be seen from the introduction of a large peak at δ =3.6 ppm corresponding to the (CH2-CH2-O) repeat units of PEG. The disappearance of the δ =7.8 ppm and δ =10.7 ppm peaks after dialysis suggests RAP-PEG1000 products were formed and excess EDC and NHS, respectively, were successfully filtered. Figure 6. 1H-NMR (DMSO-D6) EDC/NHS RAP intermediate: [▴] δ = 10.7 ppm (-OH) peak of NHS and [◆] δ = 7.8 ppm (amine group of EDC). PEG1000 [*] δ = 3.6 ppm (CH2-CH2-O) repeat units of PEG. 31
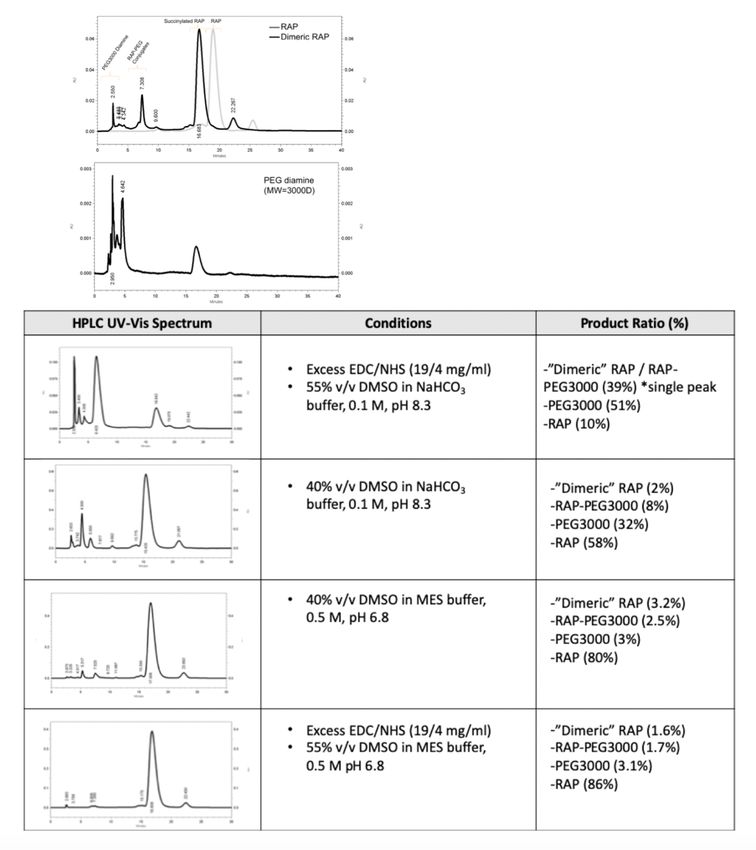
3.1.3 CuAAC ‘clicked’ adamantane For RAP-Ad synthesis, we aimed to “click” an adamantane functional group to the free alkyne end of RAP-PEG1000-alkyne. Adamantane conjugation to the PEG1000 chain yielded two distinct peaks at δ = 1.6 & δ =1.7 ppm, corresponding to adamantane’s -CH2 and -CH groups, respectively. In addition, the formation of the triazole ring resulted in a characteristic peak at δ = 7 ppm (Figure 7). Comparing the area of the PEG1000 and adamantane peaks, there appears to be approximately one adamantane for each PEG molecule. Figure 7. 1H-NMR (DMSO-D6) RAP-PEG1000-Ad: [*] adamantane peaks δ = 1.7 & 1.6 ppm, [◆] tetrazole ring peaks δ = 7.0 ppm. 3.1.4 “Dimeric” RAP synthesis To complete the synthesis of “Dimeric” RAP, we sought to conjugate an additional “activated” RAP molecule to the free amine end of RAP-PEG3000-amine. Conjugation of an additional RAP molecule to RAP-PEG3000-amine via EDC/NHS coupling was confirmed via 1H-NMR and HPLC UV-Vis (Beckman HPLC, Phenomenex Luna 5u C18 250x4.6 mm column, λ= 280nm). EDC/NHS reaction efficiency heavily effected the conjugation yield and purification of the “Dimeric” RAP. 32
As EDC/NHS coupling reactions have previously been shown to be improved by altering buffer type and pH, several alternative solvents were tested to see if the reaction could be pushed to favor increased conjugation35. The corresponding 1H-NMR and HPLC UV-Vis of formulation that yielded the greatest conjugation of “dimer” (29.4% “Dimeric” RAP, 20.7% RAP-PEG3000) (Figure 8). Other attempts at increasing conjugation yield are show in Figure S2. Figure 8. a) 1H-NMR of “Dimeric” RAP: PEG3000 [◆] δ = 3.6 ppm peak and main RAP peak [*] δ = 2.6 ppm b) EDC/NHS reaction yield products determined from HPLC peak integral analysis c) Nonpolar-phase HPLC UV-Vis spectrum for “Dimeric” RAP synthesis. 3.2 Molecular tethers increased RAP’s affinity for β-CD Historically, cyclodextrin drug delivery leverages the “affinity” a drug (ligand) has for pCD’s inclusion complexes. Dissociation constants (KD) between small molecule drugs and cyclodextrins have previously been used to predict both loading efficiency and release profiles, as it is a representation of the thermodynamics of complexing drugs with cyclodextrin33,36,37. RAP was 33
previously reported to have an affinity of around 34 µM, which corresponded to a 28 day window of release19. A decrease in KD suggests a higher affinity and subsequent increased loading and prolonged release. Both RAP-Ad (KD=12.41 µM) and “Dimeric” RAP (KD=11.82 µM) exhibited a significantly decreased KD against β-CD monomers, determined from steady state affinity equation curve fitting according to standards set in Biacore’s evaluation software (Table 1). Table 1. SPR kinetics results for unmodified and modified RAP against β-CD immobilized CM5 chip, 1% DMSO:diH2O running buffer. Reported KD values were within the model confidence interval with Chi2 values below 10% of the maximum SPR response. PyRx docking simulation was used to predict β-CD / RAP interactions. 3.3 Drug loading & release kinetics were enhanced in pCD MPs In vitro drug loading and drug release studies have been used to predict future in vivo delivery efficacy. Two drug loading conditions were chosen: one “low concentration” in which the number of pCD complexation sites outnumbered the amount of drug, and one “high concentration” in which drug was added in large excess to pCD complexation sites. We observed a significant increase in high concentration loading groups, and low concentration loading, with both RAP-Ad and “Dimeric” RAP loading nearly 17 and 6 times more efficiently than RAP, respectively (Figure 9). 34
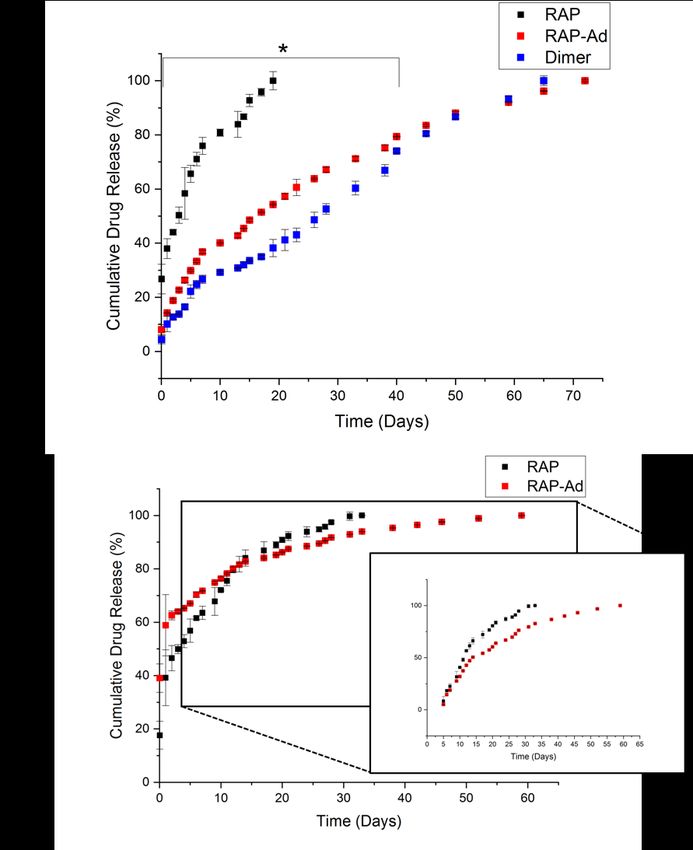
Figure 9. Total drug released after loading in pCD microparticles with concentrations of a) 3.7 mM (“low concentration”) [n=3] and b) 21 mM (“high concentration”) [n=3]. Error bars represent the standard error of the mean. All “low concentration” drug release profiles exhibited a limited “burst” release and released relatively consistently over time. RAP behaved similarly as previously reported, releasing over a timespan of around 20 days19. RAP-Ad and “Dimeric” RAP, however, both released to up to 65 days and had a reduced slope, correlating to the increase in affinity (Figure 10a). Additionally, the daily release of RAP-Ad was, on average, 6 times more concentrated than RAP control and “Dimeric” RAP was on average 2 times more concentrated than RAP control. This is a consequence of both increased affinity and drug solubility as a result of PEGylation (Figure S3). “High concentration” releases for both RAP and RAP-Ad had a two-phase release: a solvent- exchange “burst” release within days 1-4 followed by an “affinity-based” release thereafter. Due to RAP-Ad’s higher affinity, drug delivery took place over a timespan of over an extended 60 days, similar to the release at low concentration while delivering comparable amounts of drug daily after day 20 (Figure 10b). 35
Figure 10. a) “Low concentration” (3.7 mM ) cumulative release profile from pCD microparticles for RAP, RAP-Ad, and “Dimeric” RAP. * All groups were statistically significant in first 40 days of release (p
known not to interrupt protein binding. We utilized porcine fibroblasts as a model-mTOR pathway system, as fibroblasts have been shown to highly depend on mTOR for migration, proliferation, and survival39,40. Using a wound-healing “scratch” assay as a measure of mTOR activity (Figure 11) we found no significant differences in drug activity between RAP and RAP-Ad, while “Dimeric” RAP seemed to have diminished activity at lower concentrations (< 2 µM). We also found that RAP functioned comparably to both RAP-Ad and “Dimeric” RAP when inhibiting proliferation, with both groups showing improved proliferation prevention at concentrations lower than 1 µM (Figure 12). Figure 11. Scratch assay wound closure of PT-K75 porcine mucosal fibroblasts after 24-hours RAP, RAP-Ad, or “Dimeric” RAP treatment relative to PBS controls in serum starvation media. Area of each “scratch” was analyzed at time 0- and 25-hours utilizing ImageJ. Areas were compared to manual ROI selection for confirmation [n=3]. * indicates p
Figure 12. Proliferation of PT-K75 porcine mucosal fibroblasts after incubation with RAP, RAP-Ad or “Dimeric” RAP for 24 porcine mucosal fibroblasts hours. Fluorescence was compared between groups [n=3] and control after 28 hours of Alamar blue incubation. * indicates p
hydrogens) and the last carbon of PEG (δ = 4.2 ppm; 2 hydrogens), and a range of 73-96% conversion (n=2 batches) was observed (Figure 13). Figure 13. a) 1H-NMR (DMSO-D6) of 1-adamantanecarbonyl chloride which confirmed a 97.33% purity b) 1H-NMR (DMSO-D6) of maleimide-PEG5000-hydroxyl c) Ad-PEG5000-Mal 1H- NMR (DMSO-D6), with unique peaks at δ =1.7-1.9 ppm (Ad hydrocarbons, ◆), δ = 4.2 ppm (terminal -CH2- of PEG, ▴), and δ = 3.6 ppm (-CH2- PEG repeat units, *). 4.1.2 PEG-Ad “tethers” stable for up to 40 days As Ad-PEG5000-Mal contains an ester group prone to hydrolysis, the stability of the chemical species was observed over time via 1H-NMR. The integral of the terminal hydroxyl peak (-OH) at δ = 6.1 ppm was compared to a constant PEG (-CH2-) peak at δ = 4.2 ppm, and a percent species of hydrolysis was obtained. Over time, the integral of the terminal hydroxyl peak increased until it equaled half of the integral of the δ = 4.2 ppm, indicating 100% species hydrolysis. We found that at around 40 days, the adamantane is completely dissociated from the PEG linker, which is 39
most likely attributed to the partially-solubility of Ad-PEG5000-Mal (Figure 14). Hydrolysis was confirmed to have zero-order kinetics. Figure 14. a) hydrolysis mechanism for PEG-Ad b) 1H NMR (DMSO-d6) with Bruker 300 MHz, noting a hydrolysis peak at δ = 6.1 ppm and a reference peak at δ = 4.2 ppm (-CH2-) c) Ad species percentage was quantitively obtained from hydrolysis peak δ = 6.1 ppm [n=1]. Error bars are representative of the standard error of peak integrals. 4.1.3 PEG-Ad ‘tethers’ successfully conjugated to reduced proteins To ensure maleimide-sulfhydryl chemistry was successfully taking place, nonreducing SDS- PAGE gels were used to quantify the molecular weight of our BSA and mAb protein-polymer conjugates (Figure 15). An increase in molecular weight was observed for all protein-polymer species, and estimated conjugation ratio (ECR) of each species was obtained by the following equation: WXYJZ[\H]^_ `WXabJ^_cZ = WXaJKdI_b e^_^f_be (2) 40
You can also read