Structure-Based Stabilization of Insulin as a Therapeutic Protein Assembly via Enhanced Aromatic-Aromatic Interactions
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
JBC Papers in Press. Published on June 8, 2018 as Manuscript RA118.003650
The latest version is at http://www.jbc.org/cgi/doi/10.1074/jbc.RA118.003650
Structure-Based Stabilization of Insulin as a Therapeutic Protein Assembly via Enhanced
Aromatic-Aromatic Interactions
Nischay K. Rege1, Nalinda P. Wickramasinghe1, Alisar N. Tustan2, Nelson F.B. Phillips1, Vivien C. Yee1,
Faramarz Ismail-Beigi2, & Michael A. Weiss1,3,*
1
Department of Biochemistry, Case Western Reserve University, Cleveland, OH 44106
2
Department of Medicine, Case Western Reserve University, Cleveland, OH 44106
3
Department of Biochemistry, Indiana University School of Medicine, Indianapolis, IN, 46202
*Running title: Stabilization of Insulin Hexamer
*To whom correspondence should be addressed: Michael A. Weiss, Department of Biochemistry, Indiana
University School of Medicine, Indianapolis, IN, 46202
Tel.: +317 274 7151; E-mail: weissma@iu.edu
Keywords: protein design, molecular pharmacology, diabetes mellitus
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
Key contributions to protein structure and stability
are provided by weakly polar interactions, which
arise from asymmetric electronic distributions
within amino acids and peptide bonds. Of Introduction
particular interest are aromatic side chains whose Weakly polar interactions are ubiquitous among
directional π systems commonly stabilize protein protein structures (1). Among such interactions, the
interiors and interfaces. Here, we consider relative packing of aromatic rings is of particular
aromatic-aromatic interactions within a model interest in relation to the organization of protein cores
protein assembly: the dimer interface of insulin. and subunit interfaces (2). Aromatic-aromatic
Semi-classical simulations of aromatic-aromatic interactions are governed by quantum-chemical
interactions at this interface suggested that properties, which underlie dispersion forces and give
substitution of residue TyrB26 by Trp would rise to asymmetric distribution of partial charges.
preserve native structure while enhancing Whereas aromatic stacking is prominent in nucleic-
dimerization (and hence hexamer stability). The acid structures, pairs of aromatic side chains in
crystal structure of a TrpB26-insulin analog proteins more often exhibit edge-to-face (ETF1)
(determined as a T3Rf3 zinc hexamer at a resolution contacts (3). Can such contacts be exploited in
of 2.25 A) was observed to be essentially identical therapeutic protein engineering? Here, we have
to that of wild-type insulin. Remarkably and yet in analyzed aromatic-aromatic interactions in the insulin
general accordance with theoretical expectations, hexamer (4) as a basis for designing improved long-
spectroscopic studies demonstrated a 150-fold acting (basal) analogs. This class of analogs is central
increase in the in vitro lifetime of the variant to the treatment of Type 1 and Type 2 diabetes mellitus
hexamer, a key pharmacokinetic parameter (5).
influencing design of long-acting formulations.
Functional studies in diabetic rats indeed revealed Classical crystal structures of insulin hexamers
prolonged action following subcutaneous (6,7) immediately suggested a pathway of assembly
injection. The potency of the TrpB26-modified (4). Pertinent to the mechanism of storage in the
analog was equal to or greater than an unmodified secretory granules of pancreatic β-cells (8), such
control. Thus exploiting a general quantum- assembly is also of pharmacologic importance (9).
chemical feature of protein structure and stability, Insulin assembly both protects the hormone from
our results exemplify a mechanism-based approach degradation in pharmaceutical formulations and
to the optimization of a therapeutic protein modulates its pharmacokinetic properties (10).
assembly. Indeed, the first use of insulin analogs in diabetes
1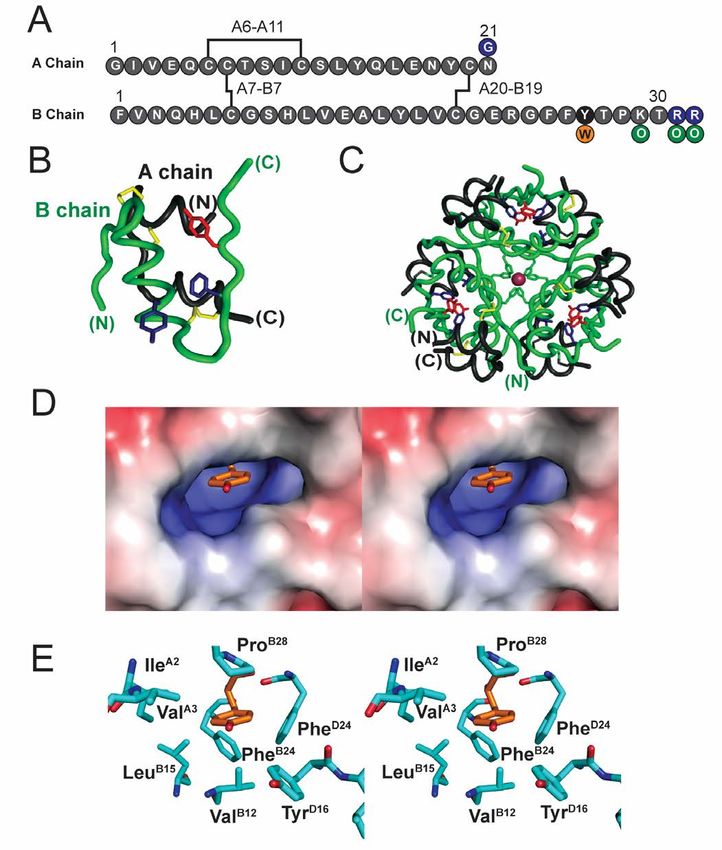
therapy reflected efforts to destabilize the insulin insulin hexamer and retard its disassembly while
hexamer and thereby accelerate absorption of preserving the biological activity of the monomeric
monomers and dimers from the subcutaneous (SQ) hormone. The crystal structure of a TrpB26-insulin
depot (summarized in Fig. S1; (11,12)). Because it is analog (as a zinc-insulin hexamer) was essentially
more straightforward to introduce unfavorable identical to that of WT insulin. Finally, we undertook
substitutions than favorable ones, such engineering studies in diabetic rats to obtain proof of principle that
(corresponding to rapid-acting analogs) was more this approach could extend the duration of insulin
successful than complementary efforts to enhance the action on SQ injection.
thermodynamic (and kinetic) stability of the insulin
To our knowledge, our results represent the first
hexamer (13). There are presently three rapid-acting
exploitation of aromatic-aromatic interactions to
insulin analogs in clinical use (14), but no basal
enhance the physical and biological properties of a
products designed on the basis of enhanced hexamer
therapeutic protein. Because standard MM
assembly despite extensive efforts (15). These
calculations employ a simplified model of aromatic
difficulties were circumvented by alternative
systems (i.e., approximating their quantum-
mechanisms of protracted action (acylation and pH-
mechanical (QM) properties via partial atomic charges
dependent SQ precipitation (14); Fig. S2).
(19,20)), ab initio QM simulations of the aromatic
The dimer interface of insulin (repeated three times cluster and their incorporation in QM/MM simulations
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
in the hexamer) contains a cluster of eight conserved (21) promise to establish a rigorous foundation for
aromatic rings (TyrB16, PheB24, PheB25, TyrB26, and their therapeutic protein design, including further
dimer-related mates in subunit D; Fig. 1A,B,C). Of optimization through incorporation of modified or
these, successive ETF contacts are formed by B16- non-standard amino acids (22-24). The present results
D26, B24-B26, B24-D24, and B26-D16; the B25 side suggest that insulin’s conserved aromatic cluster can
chain is peripheral to this network. Whereas variation provide a natural laboratory for such foundational
at these sites is in general constrained by the structure analysis and its therapeutic translation.
of the hormone-receptor interface (16,17), our
attention focused on the B26 side chain because of its Results
functional tolerance to diverse substitutions (18) and Molecular mechanics calculations suggested that
because of its partial exposure in the monomer, dimer, augmented aromatic-aromatic interactions were
and hexamer (4) (Fig. 1 C, D). We sought to possible at the TrpB26 dimer interface.
investigate whether variant B16-D26 and B26-D16
ETF contacts across the dimer interface might in MM simulations were employed to estimate the
strength of aromatic-aromatic interactions of TrpB26 at
principle modulate—in either direction—the strength
the insulin dimer interface in relation to those of the
of these interactions. We hypothesized that enhanced
native Tyr. These calculations employed the
ETF contacts at this interface might provide the long- CHARMM empirical energy function in which
sought approach to stabilize the insulin hexamer and aromatic rings contain partial atomic charges,
so improve the pharmacokinetc (PK) properties of parametrized to mimic the electrostatic properties of
basal formulations. the π system (19,20). Working models of the variant
dimer were obtained by local energy minimization (see
The present study had three parts. The first
Experimental Procedures).
employed local modeling, using the standard
CHARMM empirical energy function, to probe Energies of interaction between the eight aromatic
possible effects of a TyrB26→Trp substitution on residues at the insulin dimer interface (TyrB16, PheB24,
aromatic-aromatic interactions within the wild-type PheB25, TyrB26, and their symmetry-related mates)
(WT) aromatic cluster. These molecular mechanics were calculated using local models in which TrpB26
(MM) calculations suggested that substitution of was substituted within WT T2, R2, and TRf reference
TyrB26 by Trp could enhance dimer-related ETF dimers ( extracted from PDB structures 4INS, 1ZNJ,
and 1TRZ) (25). The total interaction energy between
contacts and yet otherwise preserve a native-like
B26 and the other aromatic residues at the
interface. We next prepared this analog to examine TrpB26 interface, which was calculated using the full
whether this substitution might indeed stabilize the CHARMM potential energy function, was augmented
2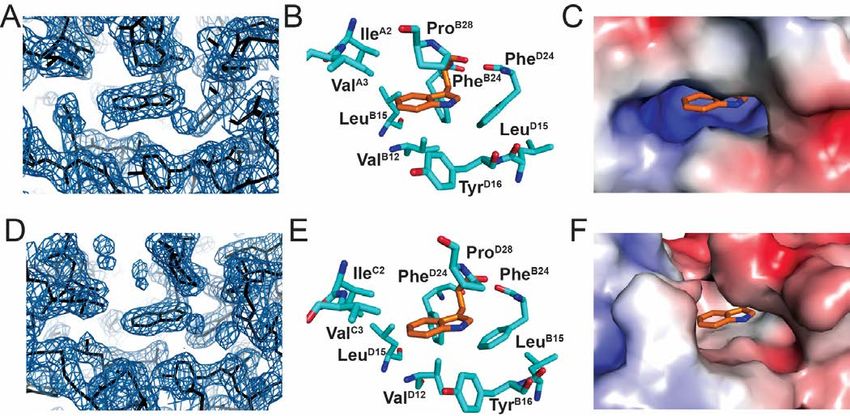
by 2.0 and 0.8 kcal/mol relative to the minimized WT and phenol-free mobile phase. Subsequent
interface in the context of R2 and TRf structures, dissociation of the R6 hexamers was monitored in the
respectively, and diminished by 1.5 kcal/mol in the chromatograms (Fig. 4A, Table 1). Absence of a void-
context of the T2 structure. Results are summarized in volume signal (V0) indicated that none of the proteins
Table S1. formed large non-specific aggregates. Whereas WT
and OrnB29-insulin eluted as a broad peak representing
A minimal model was utilized to further evaluate an association state intermediate between monomer
the potential impact of a TrpB26 substitution on and dimer (9.7 and 8.2 kDa respectively) and whereas
aromatic-aromatic interactions at the dimer interface. insulin lispro eluted essentially as a monomer (5.1
To this end, a structural model of the dimer interface kDa), TrpB26, OrnB29-insulin eluted in two distinct
(extracted from a T6 insulin hexamer; PDB ID: 4INS) peaks. The larger peak corresponded to a trimeric or
was first built containing residue B26 (TyrB26 or tetrameric association state (MW 28 kDa), and the
TrpB26) and its nearest aromatic neighbors (PheB24, smaller corresponded to a monomeric state (4 kDa;
PheD24, and TyrD16). Simulations predicted the Fig. 4A,B; see Fig. S4 for reference chromatograms of
orientation of the B26 ring corresponding to free- monomer and hexamer). These findings suggest that
energy minima of electrostatic interactions between oligomers intermediate between hexamer and dimer
the aromatic residues (the partial-charge may delay dissociation of the TrpB26 analog; in
parametrization of aromatic residues in CHARMM is particular, the absence of broad elution tails implies
shown in Fig. S3) (23). When substituted at position that TrpB26 imposes significant barriers to rapid
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
B26, Trp displayed improved electrostatic interactions dissociation.
with its three aromatic neighbors (relative to the WT
Tyr) over a broad range of conformations (Fig. 2). TrpB26 analog retained native biological activity.
This trend extended to conformations that are
sterically permitted in the context of the WT insulin The in vitro affinity of the TrpB26 analog for the
hexamer. lectin-purified insulin receptor (IR; isoform B
holoreceptor) was determined to be 0.14(±0.02) nM,
TrpB26 analog exhibited markedly decreased approximately 50% that of OrnB29-insulin (0.07(±0.02)
hexamer dissociation rate. nM) and WT insulin (0.08(±0.03) nM) (Fig. 5A). The
potencies of these analogs, evaluated by intravenous
The effect of the TrpB26 substitution on the lifetime (IV) injection in diabetic rats, were nonetheless
of insulin hexamers under formulation conditions was indistinguishable from WT insulin (Fig. 5B).
assessed in the context of OrnB29-insulin, a structural
equivalent of WT insulin that is amenable to TrpB26 analog displayed a zinc-dependent delay in
production by trypsin-catalyzed semisynthesis (26). onset of biological activity.
The lifetime of Co2+-substituted, phenol-stabilized
(R6) hexamers of TrpB26, OrnB29-insulin was assessed Hexamer assembly delays absorption of insulin
at equilibrium in relation to native TyrB26, OrnB29- from its SQ injection site (11,12). To assess the onset
insulin. Optical absorbance spectra of these analogs and duration of TrpB26, OrnB29-insulin relative to
(characteristic of Co2+ with tetrahedral coordination) OrnB29-insulin, the pharmacodynamics (PD) profile of
were similar to WT (Fig. 3A,B). Assessment of R6 these proteins (made 0.15 mg/ml, corresponding to a
dissociation rates (summarized in Table 1) revealed a monomer concentration of 27 µM and a putative
150-fold increase in hexamer half-life of the TrpB26 hexamer concentration of 4.5 µM) were evaluated as
analog relative to its parent OrnB29-insulin (Fig. 3C,D). zinc-free solutions or as pre-assembled phenol-
This increase is remarkable given that the difference stabilized R6 hexamers in the presence of excess zinc
between the half-lives of a rapidly dissociating analog ions (0.30 mM ZnCl2; 70 zinc ions per hexamer). A
in clinical use (lispro2 (27-29)) and WT is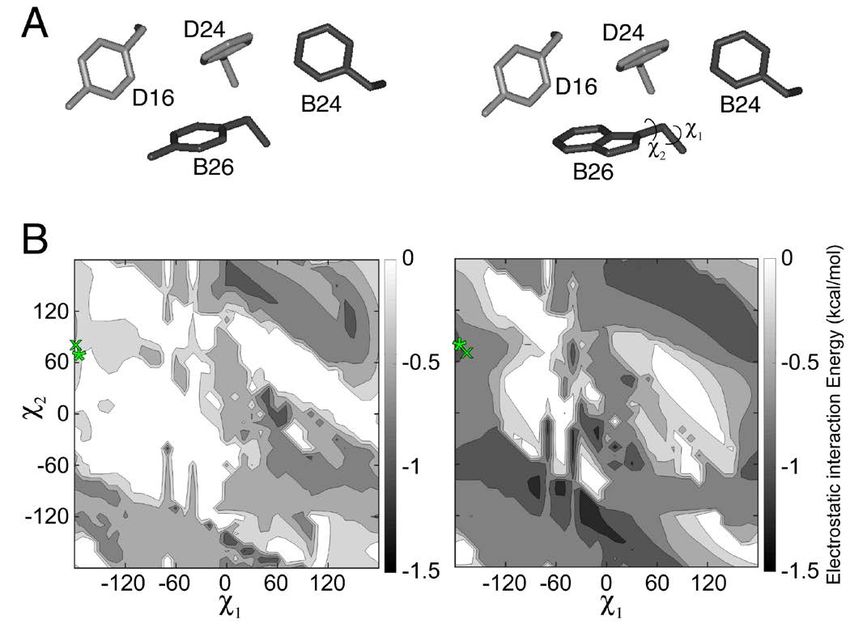
results, these findings suggest that the prolonged range of the native Tyr in WT crystal structures
lifetime of the TrpB26 R6 hexamer (as inferred from the (Tables S5 and S6, respectively) with a slight deviation
above kinetic studies of the Co2+ -substituted hexamer) in the positioning of the peptide backbone to
are responsible for the inferred zinc-dependent delay accommodate the larger indole side chain.
in SQ absorption.
Spectroscopic probes revealed native-like
TrpB26 protracted the PD profile of a model pI- structure and thermodynamic stability of TrpB26
shifted analog. analogs in solution.
Insulin analogs with isoelectric points (pI) shifted The native-like crystal structure of TrpB26, OrnB29-
to neutral pH generally exhibit prolonged activity due insulin is in accordance with its unperturbed circular
to precipitation in the SQ depot (30). To determine dichroism (CD) spectrum and thermodynamic stability
whether TrpB26 might further prolong the activity of under monomeric conditions (Fig. 7A). Free energies
such analogs, this substitution was introduced into a of unfolding (ΔGu 3.3 ± 0.1) kcal/mole at 25 °C as
GlyA21, OrnB29, OrnB31, OrnB32-insulin. This “glargine- inferred from two-state modeling of chemical
like” framework was designed to recapitulate the pI denaturation (33)) were indistinguishable due to small
shift of glargine with greater ease of semisynthesis3. and compensating changes in transition midpoint and
The proteins (formulated at 0.6 mM with 0.3 mM slope (m value) (33,34) (Fig. 7B, Table 2). Further
ZnCl2, corresponding to 3 zinc ions per hexamer) were evidence that the crystal structure extends to the
each injected SQ in diabetic rats. monomer in solution was provided by 2D 1H-
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
NMR studies of TrpB26 substituted within an
The pI-shifted parent analog displayed peak engineered insulin monomer (lispro (35)). Whereas
activity at ca. 120 min with blood-glucose levels the spectrum of lispro (at pD 7.6 and 37 oC) exhibits
returning to baseline after about 360 min. By contrast, sharp resonances for each aromatic spin system
its TrpB26 derivative displayed a prolonged PD profile: (Fig. 8A), as expected for a monomeric analog (35),
peak activity occurred 180 min with slow return to the spectrum of its TrpB26 derivative exhibits
baseline >800 min (Fig. 5E; See Fig. S6 for IV broadening of resonances at the dimer interface (B16,
potency). Such a marked delay in peak activity was B24-B26). The latter spin systems can be observed on
not observed in the parent glargine-like analog or the TOCSY spectrum (Fig. 8C) but not in the
TrpB26 derivative when administered in the absence of corresponding DQF-COSY spectrum due
zinc (Fig. S7). These results suggest that TrpB26 may to antiphase cancellation. Like the aromatic
favorably be incorporated into current basal analogs as ring TyrB26 in spectra of insulin lispro (Fig. 8A,B), the
a complementary mechanism of prolonged SQ indole ring exhibited regiospecific nonlocal nuclear
absorption. Overhauser enhancements (NOEs) from its six-
Crystal structure of TrpB26 analog demonstrated member moiety to the methyl resonances of ValB12 and
native-like dimer interface. IleA2 (Fig. 8B,C,D).
The crystal structure of TrpB26, OrnB29-insulin was The pattern of secondary shifts in the variant is
determined as a zinc-coordinated hexamer in the similar to that in the parent monomer. In particular,
presence of phenol to a resolution of 2.25 Å. the aromatic 1H-NMR resonances of TrpB26 (red cross
Diffraction and refinement statistics are provided in peaks in Fig. 8C) exhibit upfield features (relative to
Table S2. The asymmetric unit constituted a “TRf” Trp in the isolated B23-B30 octapeptide; dashed lines)
dimer4 (31,32). The overall structures of the T- and Rf similar to those of TyrB26 in the parent
protomers were essentially identical to those of WT spectrum (purple cross peaks in Fig. 8A versus dotted
insulin (Fig. S8) with respective RMSDs 1.11 ± 0.30 lines) (36). Dilution of the TrpB26 sample partially
and 1.36 ± 0.30. Additional RMSDs are given in mitigated resonance broadening but preserved these
Tables S3 and S4. Side-chain packing near the B26 trends in dispersion. Indole-specific NOEs indicated
position was largely unperturbed. In both protomers that the side chain assumes one predominant and
the TrpB26 indole group was oriented with its six- asymmetric conformation within a native-like crevice
member ring packing against conserved core residues between A- and B-chain α-helices (Table S7).
IleA2, ValA3, and ValB12 (Fig. 6); the indole NH group Because TyrB26 undergoes rapid ring rotation about the
is exposed to solvent in the TR dimer and T3Rf3 Cβ-Cγ bond axis ("ring flips"), analogous side-chain
hexamer. The TrpB26 side chain in both R- and T specific NOEs (inferred in prior studies from
protomers also displayed dihedral angles within the molecular modeling) cannot be observed directly
(Table S8).
4
MM calculations suggested improved aromatic- six of which engaged in a successive set of aromatic-
aromatic interactions within the variant crystal aromatic interactions. Quantum-chemical simulations
structure. of model systems have suggested that nearest-
The contribution of aromatic-aromatic interactions neighbor interactions predominate even in the
involving TrpB26 to the stability of the variant dimer presence of multiple rings (46). Pairwise dissection of
interface of the T3Rf hexamer was evaluated through insulin’s dimer interface has highlighted the potential
calculation of non-bonded interaction energies among opportunity to enhance its stability through
aromatic residues B16, B24, B25, and B26 in the TRf substitution of TyrB26 by Trp. Whereas our
dimer. These calculations, which employed the crystallographic analysis verified that this substitution
variant crystal structure, were in overall accordance preserves native architecture, a TrpB26 insulin analog
with expectations based on our initial local MM-based exhibited a dramatic increase in hexamer lifetime in
modeling (above). In particular, based on aromatic- vitro. Results of animal testing demonstrated native
aromatic interactions alone, the TrpB26, OrnB29 dimer intrinsic potency (i.e., on IV bolus injection) but with
displayed an increase in interaction energy of 1.4 prolonged activity on SQ injection, presumably due to
kcal/mol relative to WT TRf reference structure 1TRZ;
delayed dissociation of the variant zinc hexamers in
the results of these calculations are given in Table
the SQ depot.
S9a,b. Although the standard CHARMM empirical
energy function, when applied to analyze either Protein engineering of insulin analogs is
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
crystallographic and MM-minimized models of TrpB26 constrained by the complexity of insulin’s
insulin, suggested that the electrostatic properties of “conformational lifecycle”: from oxidative folding
the Trp side chain were the primary contributors to the
intermediates and self-assembly in the pancreatic β-
increased stability of the dimer, this physical
interpretation may reflect the limitations of the partial- cell (8) to adoption of an active, “open” conformation
charge representation (23,37). Indeed, preliminary ab on receptor binding (Fig. 9A,B) (16). Specific residues
initio QM simulations of a minimal model (consisting may play distinct roles at each stage. In particular,
of two aromatic rings in vacuo) predict that enhanced because interfaces within the insulin hexamer overlap
Van der Waals interactions may also make a the hormone’s receptor-binding surface—essentially
significant contribution (Fig. S9) (see Discussion). invariant among vertebrates (47)—modifications often
impair activity (13). A given WT residue may
Discussion represent a compromise among competing structural
The physical origins of protein stability and tasks. A recent survey of 18 substitutions at position
recognition define a foundational problem in B26 demonstrated that Tyr is suboptimal with respect
biochemistry (38) with central application to to IR-binding affinity but enhances self-assembly
molecular pharmacology (39). The zinc-insulin relative to more active alternatives (Ala, Ser or Glu)
hexamer provides a favorable system for structure- (18). The latter side chains destabilize the “closed”
based design due to its long history of crystallographic dimer interface but are favorable at the solvated B26-
investigation (40). Indeed, the hexamer’s rigidity, as related edge of the open hormone-receptor interface
interrogated by NMR spectroscopy (41), renders the (Fig. 9C).
overall structure robust to diverse amino-acid Protective self-assembly is of key pharmacologic
substitutions (42,43), even those that destabilize the importance.
dimer interface5 (32,44). This rigid framework has
often enabled analysis of discrete interactions without Insulin self-assembly protects the hormone from
complications due to the long-range transmission of degradation and toxic misfolding in pancreatic β-cells6
conformational change (31,32). (8,48) and in pharmaceutical formulations (49).
Because the zinc insulin hexamer exhibits delayed SQ
Our studies, stimulated by the seminal recognition absorption relative to monomers and dimers,
of aromatic-aromatic interactions by Burley and mutational destabilization of the hexamer (10,12) led
Petsko more than 30 years ago (2), focused on the to development of rapid-acting insulin analogs (14).
classical dimer interface, a basic building block of the Efforts to stabilize the insulin hexamer—as a converse
hexamer (32). Long appreciated as "a thing of beauty" strategy to obtain protracted action—were less
(4,45), this interface contains eight aromatic residues, successful (13). Current long-acting insulin analogs
5
rely instead on higher-order self-association of (60). Photo-activatable aromatic probes (para-azido-
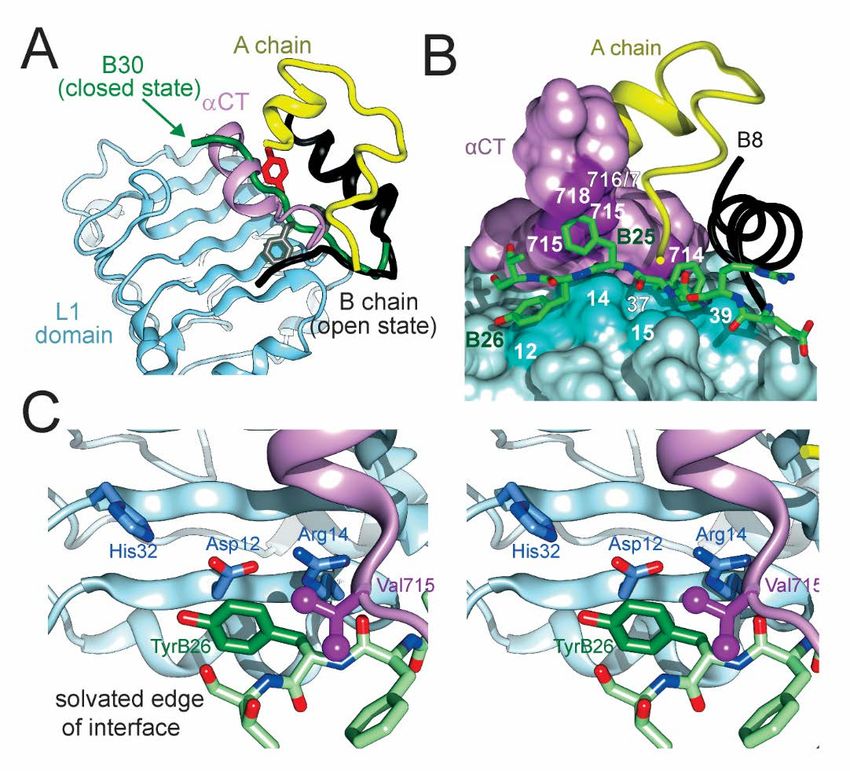
hexamers within the SQ depot (30) and binding Phe) at any of these sites exhibited efficient cross-
acylated monomer to albumin as a circulating depot linking to the IR (61,62). The co-crystal structure of
(highlighted in Fig. S2) (50,51). an insulin monomer bound to a fragment of the IR
ectodomain (the μIR model) revealed distinct binding
The problem of how to improve a protein interface
sites at the surface of the L1 domain of the IR β-
is in general more subtle than its opposite, for
subunit (B16, B24 and B26) or its αCT element (B24
destabilizing substitutions abound at conserved
and B25). PheB24 packs within a nonpolar pocket near
interfaces whereas stabilizing substitutions can be rare
aromatic residues in L1 (residues Leu37, Phe39) and
(13). Structure-based candidate substitutions may
αCT (residue Phe714); one wall of this pocket is defined
encounter entropy-enthalpy (EEC) compensation
by the aliphatic side chains of LeuB15, CysA20, and
(52,53) or cause unintended biological perturbations
CysB19 as in free insulin (16). This environment differs
(54). The challenge posed by insulin is magnified by
in detail from those in the insulin dimer but exhibits
the structural elegance of its self-assembly (as
analogous general features. Although aromaticity is
emphasized by Hodgkin and colleagues in a classic
not required to fill the B24-binding pocket (33), its
review) (55). Diverse structure-based strategies were
specific size and shape constrain potential
previously undertaken with only limited success.
substitutions. TyrB16 lies at the periphery of L1 (near
Alternate strategies previously used to create basal
Phe39 and Lys40). Its substitution by Ala or other
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
insulin analogs are depicted in Fig. S2 and summarized
aromatic residues preserves activity (63).
in Table S10. One approach focused on the general
PheB25 occupies a cleft between αCT residues (Val715,
nonpolar character of dimer interface: additional
Pro716, Arg717, and Pro718) that can only
hydrophobic substitutions were introduced in an effort
accommodate trigonal γ-carbons (22). B25-
to enhance this feature (56). Such designs were not
related aromatic-aromatic interactions are limited due
successful and also impaired biological activity,
to the peripheral location of this residue (Fig. S11).
although analogs were identified whose sparing
solubility slowed SQ absorption (56). A second We chose to focus on position B26 because of its
approach exploited the classical TR transition among broad functional tolerance of diverse substitutions
insulin hexamers (57). At the pivot point of this (18). As illustrated above (Fig. 8C), TyrB26 binds at
allosteric transition (Fig. S10), an invariant glycine the solvated periphery of the μIR interface. Indeed,
(GlyB8) was substituted by Ser in an effort to stabilize substitution of small, polar, or charged amino acids,
the more stable R-state hexamer (58). This analog was (such as Ala, Ser, or Glu) enhances receptor affinity —
unstable as a monomer (54) and exhibited reduced but at the price of impaired self-assembly and
activity (58). Yet another approach sought to stabilize decreased thermodynamic stability with heightened
the hexamer by relieving electrostatic repulsion susceptibility to physical degradation (16). These
created by the internal clustering of six acidic side findings highlighted the evolutionary importance of
chains (GluB13): their isosteric substitution by Gln the native dimer interface and dual role of TyrB26. We
indeed promoted assembly of zinc-free hexamers but thus hypothesized that an aromatic substitution at the
impaired biological activity (59). Overlap between the B26 position might enhance self-assembly without
self-assembly surfaces of insulin and its receptor- loss of biological activity.
binding surfaces thus compounded the optimization
The classical structure of the insulin dimer
problem.
motivated study of successive aromatic-aromatic
The present study revisited the architecture of the interactions as a physical mechanism of stability
insulin hexamer in light of recent insights into the (6,64). The increased stability of ETF aromatic-
hormone’s receptor-binding surface (18). Prominent aromatic interactions involving Trp over those
roles are played by a quartet of aromatic residues in the involving Tyr contributes to the increased stability of
C-terminal B-chain β-strand: TyrB16, PheB24, PheB25 the TrpB26 hexamer. The larger size of the delocalized
and TyrB26 (33). These four residues—and the π orbital of Trp in relation to that of Tyr causes a
clustering of eight dimer-related side chains—have stronger negative charge to accumulate on the “face”
long been the focus of structure-activity relationships of the indole ring. For this reason, hydrogen atoms
6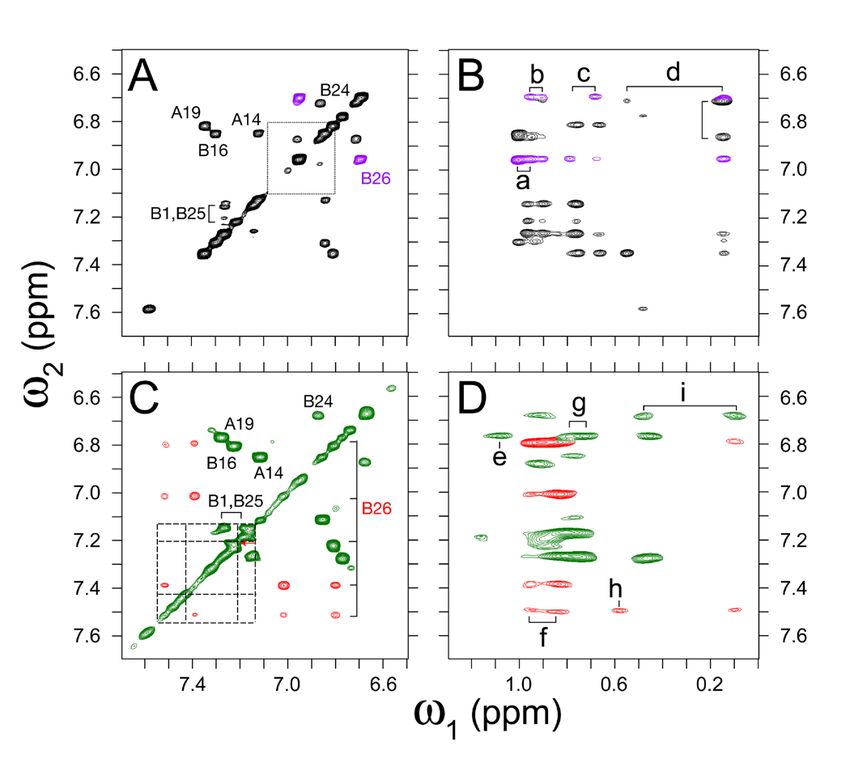
surrounding the aromatic rings of local residues form between TrpD26 and TyrB16 was 62° whereas that
stronger electrostatic interactions with the face of the between TrpD26 and PheB24 was 48°. Previous studies
indole ring of Trp than with the phenol ring of Tyr have suggested that Trp residues may form aromatic-
(65,66). Aromatic pairs involving Trp residues are less aromatic interactions over longer distances than those
common than those involving Tyr or Phe (1). formed by Tyr-Phe pairs (68). Thus, the two intra-
However, the strength of aromatic-aromatic chain aromatic pairs, TrpB26/PheB24 and TrpD26/PheD24
interactions involving Trp is evidenced by functional (respectively separated by 7.8 Å and 7.1 Å) may
importance of Trp-based interactions; an example is interact more efficiently than the corresponding
provided by a Trp-Tyr aromatic “lock” that stabilizes Tyr/Phe pairs in WT insulin (ring geometries are
the active conformation of the ghrelin receptor (67). summarized in Table S11a,b).
Indeed, comparison of electrostatic interactions of
CHARMM calculations of aromatic-aromatic
TrpB26 and TyrB26 within the minimized model of the
interactions across the dimer interface of TrpB26,
B16/B24/B26 aromatic network of insulin revealed
OrnB29-insulin revealed 1.4 kcal/mol increased
that interactions involving Trp were favored over those
interaction energy. Residue-by-residue analysis of
involving Tyr across a broad variety of orientations of
each component of the aromatic network indicated that
the respective aromatic rings. Extension of the
the increased strength of interaction across the dimer
aromatic "lock" metaphor (introduced by Holst, B. et
interface was the result of the interactions involving
al. to describe conformational “trapping” in GPCR
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
TrpD26 (see Table S9b). This result suggests that
structure (67)) to the insulin hexamer highlights the
TyrB26→Trp may only display stabilizing properties
kinetic effect of the B26 substitution on the rate of
when in an R-state protomer. If so, the T6 hexamer
hexamer disassembly (as probed by the Co2+-
formed by TrpB26 insulin would be expected to have
EDTA sequestration assay), which was more dramatic
dissociation kinetics similar to WT insulin whereas the
than effects on equilibrium association (as probed by
corresponding R6 hexamer may be expected to have
SEC). It would be of future interest to measure
markedly increased stability. The R→T transition,
activation energies for disassembly. Insight into the
which is rapid in WT insulin on release of phenol, may
structural origins of the prominent TrpB26-associated
represent the kinetic barrier responsible for the meta-
kinetic lock may be provided by activated molecular-
stable association state observed in SEC experiments.
dynamics simulations of hexamer disassembly.
The packing of TrpB26/D26 within respective cores of
TrpB26 side chain exhibited an orientation similar
T- and Rf crystallographic protomers is similar in each
to native Tyr.
case to the WT TyrB26/D26 and oriented such that the
The TrpB26 side chain in the crystal structure of indole's nonpolar six-membered portion projects more
Trp , OrnB29-insulin displayed an orientation similar
B26
deeply into a crevice between A- and B-chains than
to that of the native Tyr. A slight main-chain shift in does its proximal heterocycle. Our 1H-NMR studies
the B24-B28 β-strand (0.2 [±0.03] Å) was sufficient to of TrpB26 within an engineered monomer (35)
enable native-like packing of the larger indole ring provided evidence that this overall conformation does
against the core of a protomer (Fig. S12). In both T not require self-assembly. Analogous partial burial
and Rf subunits, the six-membered component of the of TyrB26 and TrpB26 in respective protein structures
indole side chain was oriented towards IleA2, ValA3, would in itself be expected to augment
and ValB12. Based on the classical 3.5-6.5 inter- the variant's stability due to enhanced solvation free
centroid distance, residue TrpB26 (of the T protomer) energy7 (69) (i.e., as predicted by water-
showed potential interactions with residue TyrD16 and octanol transfer studies of free Tyr and Trp (69); Table
PheD24. An interplanar angle of 78° between TrpB26 and S12). Guanidine-denaturation nonetheless indicated
TyrD16 was indicative of classical aromatic-aromatic that their stabilities are indistinguishable8.
packing within proteins (2), which generally range
We speculate that the predicted residue-specific
from 50-90°. TrpB26 and PheD24 displayed an
differences in solvation free energy are attenuated by
interplanar angle of 36°, however; this orientation is
differences in protein dynamics leading to EEC (70).
less common. Similarly, in the Rf protomer TrpD26
Although the two B26 side chains each exhibit upfield
packed near TyrB16 and PheB24. The interplanar angle
secondary 1H-NMR shifts and analogous inter-residue
7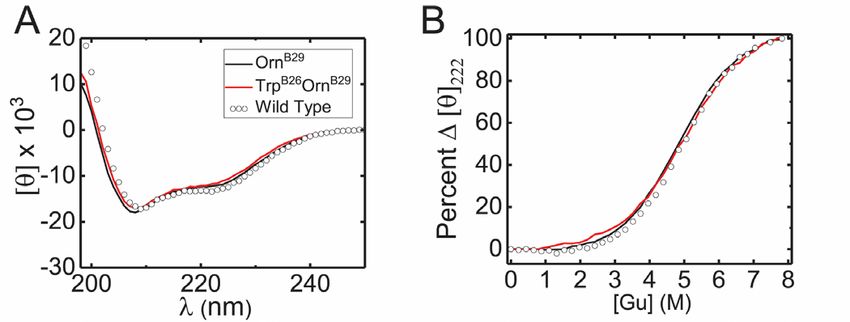
NOEs, it is possible that the substitution is associated to affect only the local structure of the insulin hexamer.
with local or non-local differences in protein Thus, the effects of the mutation were amenable to
dynamics. It would be of future interest to investigate initial analysis in simplified (eight-ring) models of the
dynamic features by amide proton 1H-2H exchange dimer interface of insulin.
and heteronuclear NMR relaxation methods (70).
Energy minimization of a local model of TrpB26-
Because TrpB26 promotes partial dimerization of
insulin (i.e., as substituted into a WT insulin dimer
insulin lispro under NMR conditions (as indicated by
1 extracted from a representative crystal structure of a
concentration-dependent H-NMR resonance
T3Rf3 zinc hexamer) yielded a native-like framework
broadening), such studies may require use of an
with enhanced nearest-neighbor B26-related aromatic-
alternative monomeric template.
aromatic interactions. The partial-charge (monopole)
Given the ubiquity of EEC as a confounding model of the aromatic rings in the CHARMM
general aspect of protein design (71), the profound empirical energy function—parametrized in
effects of TrpB26 on the properties of the insulin accordance with ab initio simulations (77)—predicted
hexamer seem all the more remarkable. We envision an increase of 0.8 kcal/mol. Although this calculation
that EEC is circumvented in this case by the rigidity of could in principle have been confounded by
the insulin hexamer (including the interlocked transmitted conformational perturbations and did not
aromatic residues at its dimer interfaces) with efficient consider potential changes in conformational entropy
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
burial of the WT and variant B26 side chains (72). or solvation, its conservative features were verified by
With similar internal structures and external solvation x-ray crystallography. That the structure of TrpB26,
properties, the variant hexamer would gain (relative to OrnB29-insulin is essentially identical to WT suggests
WT) two advantages from the Tyr → Trp substitution: that local properties of TrpB26 directly underlie the
(i) greater B26-related solvation transfer free energy observed increase in hexamer stability and lifetime.
(73) (Table S12) augmented by (ii) an uncompensated
The general asymmetry of the variant TRf dimer in
enthalpic advantage arising from more favorable ETF
the crystallographic hexamer was associated with
aromatic interactions as next discussed.
differences in the details of corresponding aromatic-
Molecular mechanics calculations rationalized aromatic interactions across the dimer interface.
physical and pharmacologic properties. Although CHARMM calculations predicted an
increase of a 1.4 kcal/mol in interaction energies (0.7
The structural rigidity of the R6 insulin hexamer
kcal/mole per protomer) in accordance with our initial
(42,74) motivated local MM-based modeling to assess
modeling, residue-by-residue decomposition ascribed
the interactions contributing to the stability of the
this increase primarily to TrpD26 (in the Rf protomer)
TrpB26-insulin hexamers. Analysis insulin oligomers
and not to TrpB26 (in the T protomer). It is formally
by Raman spectroscopy revealed dampened
possible that TrpB26 is only stabilizing in an R state, but
conformational fluctuations of the R6 hexamer in
additional studies would be required to resolve this
relation to lower-order oligomers and T6 hexamers
issue. Our cobalt-EDTA sequestration studies focused
(72). Moreover, the thermodynamic stability of the
on the R6 state as the preferred storage vehicle in a
assembly was evidenced by the lack of conformational
pharmaceutical formulation. On SQ injection, rapid
changes visualized by NMR spectroscopy over a
diffusion of phenolic ligands from the depot leads to a
temperature range of 10-80 °C (42). The resistance of
TR transition. That TrpB26 was found to delay
the R6 hexamer to structural perturbation has also been
subsequent absorption into the blood stream suggests
shown in the context of mutant insulin analogs:
that this substitution also enhances the kinetic stability
native-like x-ray crystal structures have been reported
and lifetime of the T6 hexamer.
of R6 containing a broad range of substitutions (Table
S13) (17,75,76). Even substitutions that were shown A seeeming paradox is posed by the evolutionary
to destabilize the dimer interface of insulin, such as the exclusion of Trp at position B28 of vertebrate insulins
substitution of PheB24 by the non-aromatic despite the evident compatibility of this aromatic side
cyclohexylalanine (Cha), were shown to have little chain with native structure and function. We speculate
impact on the global structure of the R6 hexamer. For that this exclusion reflects the biological importance of
this reason, the TyrB26→Trp substitution was expected the rapid disassembly of zinc insulin hexamers on their
8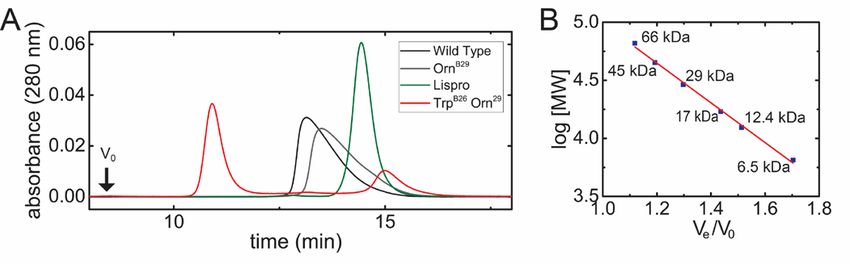
secretion by pancreatic β-cells. Whereas the enhanced analog exhibited a decreased dissociation rate (17).
thermodynamic and kinetic stabilities of TrpB26-insulin Because modified amino acids raise the cost and
hexamers would seem favorable for storage within complexity of protein manufacture, we wondered
secretory granules (as within pharmaceutical whether a natural amino acid might mimic, at least in
formulations) (8), delayed disassembly of the variant part, the structure of 3I-Y-insulin and so confer
hexamers in the portal circulation would be predicted favorable pharmacologic properties with conventional
to reduce the hormone's bioavailability on first pass manufacturing.
through the liver, as insulin dimers and hexamers
This translational goal motivated our initial MM-
cannot bind to the IR (78). Such delayed disassembly
based local modeling of TrpB26 insulin. Analysis of the
might also decrease the delivery of free zinc ions to the
crystal structure of 3I-Y insulin, determined as an R6
liver, recently predicted to constitute a regulatory
zinc hexamer (23), revealed that the large, nonpolar
signal in its own right (for review, see (79)).
iodo-substituent packed within the core of the insulin
The resulting impairment in hormonal regulation of protomer with preservation of a native-like dimer
hepatic metabolism (and accompanying systemic interface. Molecular-dynamics simulations,
hyperinsulinemia (80)) could in principle have undertaken with a multipolar electrostatic model of the
imposed a selective disadvantage in the course of modified aromatic ring (23), rationalized this
vertebrate evolution. Although to our knowledge, conformation: the iodine atom efficiently filled a
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
such a kinetics-based mechanism has not been cryptic packing defect in WT insulin, lined by the
observed in vivo, the converse—abnormally rapid conserved side chains of IleA2, ValA3, and TyrA19. The
clearance of insulin—has been found on release from enhanced packing efficiency of the modified insulin
zinc-deficient secretory granules; presumably zinc- and novel network of halogen-specific electrostatic
free insulin oligomers more rapidly dissociate on interactions (“weakly polar” interactions (1)) appear to
dilution in the portal circulation and so are more underlie the analog’s increased thermodynamic
efficiently cleared than WT zinc insulin hexamers stability and resistance to fibrillation. Whereas the
(81). Similarly, the exclusion of Trp at position B26 standard partial-charge model of aromatic systems
of vertebrate IGFs may reflect a disadvantageous failed to account for the conformation of iodo-Tyr
competition between self-assembly and binding to observed in the crystal structure, a multipolar
IGF-binding proteins, which are critical to the electrostatic model rationalized thermodynamic
integrated physiology of IGF function (for review, see stabilization of the dimer interface by this halogen
(82)). Such functional complexity may impose hidden “anchor” (23). Subtle changes in the geometry of
constraints on the evolution (and so divergence) of aromatic-aromatic interactions were observed both in
protein sequences. The conserved “aromatic triplet” the simulations and in the crystal structure. Although
of vertebrate insulins and IGFs provides a natural activated MD simulations were not undertaken to
laboratory to uncover such evolutionary constraints probe the process of dimer dissociation, we presume
(45). These considerations further suggest that that the above mechanisms of ground-state
structural features of a protein pertinent to its stabilization—enhanced core packing efficiency and a
endogenous function may in general be distinct from halogen-specific weakly polar network—also underlie
those biophysical properties that may re-engineered to the increased barrier to dissociation as indicated by the
optimize molecular pharmacology. prolonged lifetime of the variant R6 hexamer (23).
Comparison of TrpB26-insulin and a Although the profound QM effects of halo-
corresponding iodo-Tyr analog. aromatic substitutions, including weakly polar
interactions and “halogen bonding” (84), cannot be
Design of TrpB26-insulin was in part motivated by recapitulated by natural amino acids, it seemed
prior studies of an analog containing 3-iodo-Tyr at the possible that enhanced core packing efficiency might
B26 (17,23,83). The latter analog (“3I-Y-insulin”) be achieved by analogy to 3-I-Y-insulin. Indeed, the
exhibited a variety of favorable properties: increased steric profile of the asymmetric indole side chain of
affinity for the IR (83), increased thermodynamic Trp is similar in size to 3-iodo-Tyr. Accordingly, we
stability and augmented resistance to fibrillation (17). imagined that the offset six-membered portion of the
Moreover, when formulated as an R6 hexamer, this
9bicyclic indole ring might pack within the core of sufficient for characterizing aromatic-aromatic
insulin in a manner similar to the iodine atom. In this interactions (88), more recent work has highlighted its
intuitive picture the extended π-system of TrpB26 was limitations with respect to delineating underlying
envisioned to interact with neighboring residues to physical mechanisms (85,89). The limits of
recapitulate, at least in part, the favorable electrostatic parametrized classical force fields are of particular
properties of iodo-TyrB26 (23,37). importance when applications are sought in
nonstandard systems (such as unnatural protein
The above line of reasoning led to the present set of
mutagenesis (90)) for which the parameters were not
studies. In accordance with our original intuition and
intended. Examples are provided by halogen-modified
local MM-based modeling, the crystal structures of 3I-
aromatic systems (widely employed in medicinal
Y-insulin and TrpB26, OrnB29-insulin exhibited similar
chemistry) (91), which are associated with marked
features. Nevertheless, salient differences between the
changes in quantum-chemical properties (92). Formal
quantum-chemical properties of iodo-Tyr and Trp
QM/MM simulations of proteins may nonetheless be
might make the similar structures of these B26 analogs
circumvented through force fields incorporating
a fortuitous outcome of distinct mechanisms. Whereas
multipolar electrostatic models of aromatic-aromatic
effects of 3-iodo-Tyr on aromatic-aromatic
networks (85).
interactions at the insulin dimer interface are subtle,
presumably reflecting indirect inductive effects of The correspondence between our initial local MM-
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
iodo-substitutent (23), substitution of Tyr by Trp based modeling and our experimental findings,
introduces a larger aromatic surface at this interface. It however striking, may be coincidental (93): rigorous
is possible that this feature underlies the more marked elucidation of the physics of the variant aromatic
impact of TrpB26 on insulin oligomerization relative to cluster at insulin’s dimer interface may require
3-iodo-Tyr. application of free-energy MD-based simulations with
explicit inclusion of water molecules (“molecular
Aromatic-aromatic interactions exemplify alchemy” (94)). This approach may also provide
limitations of classical models. insights into whether or how EEC may be
The quantum origins of aromatic-aromatic circumvented. Nevertheless, standard MM
interactions are complex, and so classical electrostatic calculations can guide initial biochemical analysis of
models (such as partial-charge model of Tyr in the protein structure as a guide for protein engineering. In
standard CHARMM empirical energy function) can be the present study such initial modeling reinforced our
incomplete (85). Although QM calculations more structural intuition by highlighting the plausibility that
fully capture this complexity, a trade-off is substitution of TyrB26 by Trp might preserve a native-
encountered between rigor and computational like interface and in fact enhance its weakly polar
feasibility, especially in complex systems such as properties. Because the rigid hexameric framework
proteins (86), but even in ab-initio simulations of provided a favorable context for local modeling of
benzene-benzene interactions (46). Standard MM and amino-acid substitutions at subunit interfaces and yet
MD methods thus employ parametrized force fields to it is the monomer that functions as a hormone in the
approximate quantum-chemical interactions (21). bloodstream, it would be of future interest to apply
Although parameters (e.g., partial charges assigned to more sophisticated computational techniques to
an aromatic ring) have been chosen to provide simulate the structure and dynamics of TrpB26 insulin
reasonable protein models, such use of “monopolar” as a monomer in solution. Such predictions may in
electrostatics neglects the polarizability of aromatic principle be tested through biophysical studies of an
systems and so omits dispersion forces (87). engineered insulin monomer containing TrpB26.
Parametrized classical model may thus Although this substitution is favorable in the context
mischaracterize the strength or directionality of of the hexamer (and so of potential pharmacologic
intermolecular interactions, particularly those benefit), design of a monomeric NMR model will need
involving more than one aromatic group or an aromatic to overcome the confounding effects of TrpB26, as
group and a charged moiety. Although the pioneering defined in this experimental context, to promote self-
studies of Burley and Petsko suggested that the partial- association.
charge model of aromatic rings in proteins was
10Concluding Remarks (Sanofi-Aventis, Paris, FR). Reagents for peptide
synthesis were as described (100).
To our knowledge, this study represents the first
exploitation of aromatic-aromatic interactions to Preparation of Insulin Analogs
enhance the biophysical properties of a therapeutic Variant insulins were prepared by semisynthesis
protein (95). Our approach may be broadly applicable (101). In brief, synthetic peptides were coupled to a
in protein engineering (as ETF interactions are tryptic fragment of insulin (des-octapeptide [B23-B30]
ubiquitous) and generalizable to non-standard insulin) in aqueous/organic solvent using trypsin as a
aromatic moieties. The latter would be likely to catalyst. Following rp-HPLC purification, predicted
require QM/MM methods rather than classical force molecular masses were confirmed by mass
fields parametrized with partial charges (90). Overall spectrometry (33).
effects of such substitutions on protein stability and
self-assembly will require an integrated analysis Hexamer Disassembly Assays
of solvation free energies (86), changes in protein Disassembly of phenol-stabilized (R6) Co2+-
dynamics (96) and potential EEC (97). Three- and substituted insulin hexamers was monitored as
four-dimensional heteronucelar NMR experiments described (102). In brief, WT insulin or variants were
would be expected to provide higher-resolution made 0.6 mM in buffer containing 50 mM Tris-HCl
information regarding the local and non-local (pH 7.4), 50 mM phenol, and 0.2 mM CoCl2 (18) and
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
interactions of the optimized aromatic system. In the incubated overnight at room temperature to attain
present application, analyses of 15N relaxation and 1H- conformational equilibrium. Spectra (400-750 nm)
2
H amide-proton exchange are expected to improve were obtained to monitor tetrahedral Co2+ coordination
understanding of the molecular dynamics of the (27) through its signature absorption band at 574 nm
TrpB26-modified aromatic cluster and so provide a (27). Co2+ sequestration initiated by addition of EDTA
more rigorous biophysical context for its enhanced to a concentration of 2 mM. Dissociation was probed
self-association properties (70,96). via attenuation of 574 nm band (27); data were fit to a
monoexponential decay equation (29).
The present application to insulin demonstrates a
direct relationship between stabilization of the insulin Protein Crystallography
hexamer and prolonged activity of a basal
Crystals were obtained by hanging-drop vapor
analog. Continuous and flat 24-hour insulin activity
diffusion at room temperature in the presence of a
("peakless" basal formulations) is of clinical interest in
1:1.7 ratio of Zn2+ to protein monomer and a 3.5:1 ratio
the treatment of Type 1 and Type 2 diabetes mellitus
of phenol to protein monomer in Tris-HCl. Diffraction
to reduce the risk of hypoglycemia (especially at night)
was observed using synchrotron radiation at a
at a given level of glycemic control (98). Because this
wavelength of 0.9795 Å at the Stanford Synchrotron
mechanism is unrelated to present strategies to achieve
Radiation Light Source (Beamline BL7-1; Stanford,
protracted action, we envision that TrpB26-related
CA); crystals were flash frozen to 100 K. Structure
enhancement of dimer-specific aromatic-aromatic
determination was carried out using molecular
interactions could favorably be introduced into current
replacement using CCP4 (103) and Phenix structure-
basal insulin formulations. In the future a combination
determination suites (104). The resulting structure
of orthogonal molecular strategies might enable
was validated using PDB Redo server (105). The
development of a once-a-week basal insulin therapy
lattice contained one TRf dimer per asymmetric unit.
analogous to that of GLP-1 agonists
The main-chain conformations of the 97 residues in the
(99). Optimization of weakly polar interactions may
refined model of the TRf dimer in the asymmetric unit
thus assume a central place in the toolkit of molecular
(excluding 2 Thr, 1 Orn, and 1 Phe residues) each
pharmacology.
resided in a most favored Ramachandran region.
Experimental Procedures Receptor-Binding Assays
Materials Analog affinities for detergent-solubilized IR-B
Insulin was purchased from BioDel® (Danbury, holoreceptor were measured by a competitive-
CT). Insulin glargine was obtained from Lantus® displacement assay (18). Successive dilutions of WT
11insulin or analogs were incubated overnight with phosphate (pH 7.4) and 50 mM KCl (33). Free
WGA-SPA beads (PerkinElmer Life Sciences®), energies of unfolding (ΔGu) were inferred at 25 °C
receptor, and radio-labeled tracer before counting (18). from two-state modeling of protein denaturation by
1
To obtain dissociation constants, competitive binding guanidine-HCl (33,34). H-NMR spectra were
data were analyzed by non-linear regression by the acquired at 700 MHz at pH 8.0 or pD 7.6 (direct meter
method of Wang (106). reading) at 37 °C (36). Chemical shifts of aromatic
protons in residues B24-B24 (Phe-Phe-Tyr or Phe-
Rat Studies
Phe-Trp) were evaluated in relation to corresponding
Male Lewis rats (mean mass ~300 g) were chemical shifts in respective octapeptides B23-B30,
rendered diabetic by streptozotocin. Effects of insulin presumed to represent random-coil shifts.
analogs formulated in Lilly® buffer (18) on blood-
Molecular Mechanics Calculations
glucose concentration following SQ injection were
assessed in relation to WT- or OrnB29-insulin (18). Calculations were performed using CHARMM
OrnB29 insulin and TrpB26, OrnB29 insulin were made in (kindly provided by Prof. M. Karplus). Its standard
the above buffer. GlyA21, OrnB29, OrnB31, OrnB32, empirical energy function was employed (in whose
TrpB26 and GlyA21, OrnB29, OrnB31, OrnB32 were development aromatic rings were parametrized by
dissolved in dilute HCl (pH 4) containing meta-cresol, partial atomic charges in accordance with ab initio QM
glycerol, and a 1:2 ratio of ZnCl2: insulin monomer. simulations (21)). Representative WT insulin dimers
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
Rats were injected SQ with 3.44 nmoles of insulin or were obtained from PDB entries 4INS, 1ZNJ and
insulin analogs (~12-13.7 nmoles). 1TRZ. These structures and corresponding TrpB26
homology models were subjected to local energy
Animals used in this study were housed in the
minimization (100 steps of Steepest Descent followed
Association for Assessment and Accreditation of
by Adopted Basis Newton-Raphson with gradient
Laboratory Animal Care (AAALAC)-accredited
tolerance tolg 0.0008/10000 steps). Minimizations
facilities of Case Western Reserve University
were halted either at 1000 steps or when the above
(CWRU) School of Medicine. All procedures were
tolerance was reached. Changes in conformation were
approved by the Institutional Animal Care and Use
allowed only to eight side chains (TyrB16, PheB24,
Committee (IACUC) Office at CWRU, which
PheB25, TyrB26 (or TrpB26), and their dimer-related
provided Standard Operating Procedures and reference
mates; the remaining atoms in the respective dimers
materials for animal use (in accordance with the NIH
were fixed. Total interaction energies and respective
Guide for the Care and Use of Laboratory Animals).
electrostatic components were obtained between the
The animal health program for all laboratory animals
side chain of residue B26 and the neighboring three
was directed by the CWRU Animal Resource Center.
aromatic side chains (PheB24 and dimer-related TyrD16
Animal care and use was further monitored for
and PheD24). Following this survey of crystallographic
Training and Compliance issues by Veterinary
dimers, such energies were further evaluated in a
Services.
simplified molecular model that contained only the
Size-Exclusion Chromatography side chains of residues TyrB26 (or TrpB26) and the same
Analogs were made 0.6 mM in 10 mM Tris-HCl neighboring three aromatic residues as extracted
(pH 7.4), 1.6% glycerol (v/v), 0.3 mM ZnCl2, and 7 from PDB entry 4INS; this yielded the electrostatic
mM phenol (33). Insulin samples (20 µl) were loaded interaction energy map shown in Figure 2, in which
on an Enrich® SEC70 column (10 mm x 300 mm with B26 χ1 and χ2 dihedral angles were systematically
fractionation range 3-70 kDa); the mobile phase varied without energy minimization.
consisted of 10 mM Tris-HCl (pH 7.4), 140 mM NaCl, Quantum Mechanics Calculations
and 0.02% sodium azide. Elution times were
monitored by absorbance at 280 nm. Molecular Electron density and Molecular Electrostatic
masses and void volume (V0) were inferred in Potential (MEP) of Tyr and Trp side chains were
reference to standard proteins (18). calculated using B3LYP and 6-31G(d) basis set using
Gaussian utility Cubegen in Gaussian09 (77).
Spectroscopy The isosurface map was generated using Jmol (107).
CD spectra were acquired in 10 mM potassium
12Ab initio energies of interaction between pairs of
isolated aromatic rings were determined by calculating
interaction energies using the MP2 method with aug-
cc-pVDZ basis set in Gaussian 09 (77).
Acknowledgements. We thank K. El-Hage, M. Lawrence, M. Meuwly and V. Pandyarajan for helpful
discussion; K. Carr, R. Grabowski, P. Macklis, and M. Swain for assistance with rat studies; V. Kumar
and F. van den Akker for advice regarding x-ray crystallography; J. Whittaker and L. Whittaker for advice
regarding IR–binding assays; P. De Meyts (Novo Nordisk) for gift of radiolabeled insulin; and M. C.
Lawrence for assistance preparing Fig. 9. N.K.R. is a Pre-doctoral Fellow of the National Institutes of
Health (Medical Scientist Training Program 5T32GM007250-38 and Fellowship 1F30DK112644).
N.F.B.P. was supported in part by the American Diabetes Association (grant no. 7-13-IN-31 and 1-08-
RA-149). This work was supported in part by a grant from the NIH to MAW (R01 DK040949). Use of
the Stanford Synchrotron Radiation Lightsource at the SLAC National Accelerator Laboratory is
supported by the United States Department of Energy (DOE), Office of Science and Office of Basic
Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology
Program is supported by the DOE Office of Biological and Environmental Research and by the National
Downloaded from http://www.jbc.org/ by guest on November 18, 2018
Institutes of Health NIGMS Grant P41GM103393. The authors dedicate this article to the memory of the
late Prof. Colin Ward (Eliza and Walter Hall Institute, Melbourne, AU).
Database Deposition of Structures. The atomic coordinates and structure factors of TrpB26, OrnB29-
insulin (code 2CK2) have been deposited in the Protein Data Bank (http://www.rcsb.org).
Disclosure of Competing Interests. M.A.W. has equity in Thermalin, Inc. (Cleveland, OH) where he
serves as Chief Innovation Officer; he has also been a consultant to Merck Research Laboratories and
DEKA Research & Development Corp. N.F.B.P. is a consultant to Thermalin, Inc. F. I. –B. serves has
equity in Thermalin, Inc. and is a consultant to Sanofi and Novo Nordisk.
Author Contributions. Receptor-binding assays were performed by N.K.R.; rat studies were performed
by N.K.R., A.N.T., N.F.B.P. and F.I.-B.; biophysical assays and crystallization trials were performed by
N.K.R.; structure determination was performed by N.K.R. and V.C.Y.; analysis of the crystal structure
was undertaken by N.K.R. and N.P.W. with advice from M.A.W. The manuscript was written by N.K.R.
and M.A.W. with input from F. I-B. and N.F.B.P. Biophysical experiments were supervised by N.F.B.P.
The overall program of research was directed by M.A.W.
13You can also read

















































