SHIMMER: PRIVACY-AWARE ALIGNMENT OF GENOMIC SEQUENCES WITH SECURE AND ECIENT HIDDEN MARKOV MODEL EVALUATION
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
SHiMMer: Privacy-Aware Alignment of Genomic Sequences with Secure and E cient Hidden Markov Model Evaluation Miran Kim ( mirankim@unist.ac.kr ) Ulsan National Institute of Science and Technology Yongsoo Song Seoul National University Xiaoqian Jiang University of Texas Health Science Center at Houston https://orcid.org/0000-0001-9933-2205 Arif Harmanci University of Texas Health Science Center Article Keywords: Posted Date: October 4th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-954109/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License
SHiMMer: Privacy-Aware Alignment of Genomic
Sequences with Secure and Efficient Hidden Markov
Model Evaluation
Miran Kim1,2, * , Yongsoo Song3 , Xiaoqian Jiang4 , and Arif Harmanci5, *
1 Department of Computer Science and Engineering, Ulsan National Institute of Science and Technology, Ulsan,
44919, Republic of Korea.
2 Graduate School of Artificial Intelligence, Ulsan National Institute of Science and Technology, Ulsan, 44919,
Republic of Korea.
3 Department of Computer Science and Engineering, Seoul National University, Seoul, 08826, Republic of Korea.
4 Center for Secure Artificial intelligence For hEalthcare (SAFE), School of Biomedical Informatics, University of
Texas Health Science Center, Houston, TX, 77030, USA.
5 Center for Precision Health, School of Biomedical Informatics, University of Texas Health Science Center, Houston,
TX, 77030, USA.
* Corresponding authors: mirankim@unist.ac.kr, Arif.Harmanci@uth.tmc.edu
ABSTRACT
As the cost of DNA sequencing is decreasing, personal genomic data is becoming more abundant. Genomic data is known to
be very identifying; even a few genetic mutations can identify an individual. Therefore, leakage of genetic information and the
associated metadata create privacy risks. While these risks are well-known, most of the basic methods are not privacy-aware.
One of these fundamental methods is the Hidden Markov Models (HMMs), which are especially important for comparative
genomics because genetic data are sequential in nature, e.g., DNA/RNA nucleotides sequences and protein residues. HMMs
are used mainly for comparing and aligning DNA/RNA and protein sequences, such as viral genomes or gene sequences,
whereby similar portions of sequences are identified and they are defined to be conserved throughout evolution, whereas
non-matching portions of the sequences indicate a divergence. HMM-based inference of sequence alignment is therefore
a vital component of sequence analysis. Here, we describe SHiMMer, Secure HMM evaluation method that can guarantee
cryptographic security while HMMs are used for sequence comparison. We used simulated data for alignment of genomic
sequences to demonstrate that SHiMMer can perform sequence alignment efficiently. We present the scaling of time/memory
requirements with increasing numbers of alignment states and lengths of sequences.
Introduction
DNA sequencing (WGS)1, 2 is the standard technique in research and clinical settings for building the complete personal
genomic sequence of individuals. These data are invaluable in research and recreational genomics. Population-scale genomic
sequence databases are established by concerted efforts of researchers such as The 1000 Genomes Project, HAPMAP, and
TOPMeD consortium 3–5 , which can enable studying ancestry, and complex genotypes6, 7 , as well as rare8–10 and chronic
diseases11 . There has been a multitude of efforts from smaller communities of researchers to build genomic databases of
human and non-model organisms12 . DNA sequencing also revolutionized the field of microbiology , virology, and disease
research13 . Billions of DNA sequences can be publicly accessed from GenBank14 , which hosts one of the largest genomic
sequence databases. These data are deposited in databases such as GISAID15 and NCBI so that they can be publicly accessed by
researchers worldwide. Most notably, the COVID-19 pandemic is making the most use of these databases whereby researchers
share viral strains through GISAID.
As DNA sequencing technologies are becoming cheaper16, 17 , the data is also becoming more prevalent for recreational
purposes such as genetic genealogy, e.g., connecting with unknown relatives. Companies such as 23andMe, AncestryDNA,
provide genetic information and genealogy tracing from users. It is anticipated that millions of genomes have been accumulated
in the corporate databases. While increasing data sharing is vital for new scientific discoveries, there are numerous challenges
around the uncontrolled sharing of genetic data. Unlike clinical tests that are heavily regulated by the FDA and other institutions,
a lot of the genealogy services work as black-boxes and lack reproducibility of results as there are no standards or oversight
for these services. Also, it is not clear how the data is shared with 3rd parties by genealogy companies: Users generally do
not have an adequate understanding of how and when their data can be shared and how it can be used. While companies suchas Nebula Genomics aim at using blockchain for this purpose18 , it is not clear how effective these approaches are and how
well they will be adopted. On another front, law enforcement has recently started using genetic data to identify the identities
of cold-cases19 . GEDMatch maintains a database that is dedicated to law enforcement. Although this approach helped save
some high-profile cases, there is no clear ethical framework about how the data is being used by agencies. After users sign the
consent forms20 , they lose control of their data. These privacy issues also apply to researchers: Legislation on personal data
sharing varies substantially around the globe. Additionally, many researchers are reluctant to share datasets for fear of losing
credits on new findings. Finally, because of its discrete and high dimensional nature, inappropriate use of genomic data might
lead to sensitive personal information leakage from re-identification or potential phenotype inference to the individuals and
families21, 22 . Genomic data privacy and confidentiality are rising as a great challenge for researchers and the public alike. In the
context of genomic sequence analysis, privacy comes into play when the confidentiality of the data is important: (1) Individual
privacy, (2) Data under embargo, (3) Researcher privacy, (4) Data not shareable under the policy (consent, institutional-level). A
community effort has been made to address privacy issues; for example, iDASH (integrating Data for Analysis, Anonymization,
Sharing)23 has hosted a secure genome analysis competition over the last decade.
Here, we focus on proposing a secure method that performs sequence alignment and inference using Hidden Markov
Models (HMMs)24 . Sequence alignment is the most basic step in genomics analysis methods. The alignment algorithms
compare multiple sequences and find the conserved portions. The alignment can be computed among DNA sequences as well
as other sequences such as amino acid residues of proteins. An alignment of two sequences contains matched columns that
correspond to evolutionarily matching parts and insertions/deletions where sequences have diverged through insertions and
deletions. HMMs are extensively used for modeling and analysis of sequence data because they fit naturally to the discrete
nature of the states and sequential nature of the data since the primary structure of information carrying molecules that originate
from the genome are polymer-based, i.e., composed of "chains" of building blocks such as nucleotides of RNA and DNA and
amino acids of proteins25 .
HMMs use Markov chain-based state space modeling whereby each alignment column is emitted by one of the 3 states,
namely alignment, insertion, and deletion. The model is described by a state transition matrix and an emission matrix. The
state transition matrix contains the probabilities between states, and the emission matrix contains the emission probabilities of
the output values from each state. The process is "hidden" because states are not observed explicitly. We observe only the
output of the HMM that is emitted (i.e., the sequences). Based on the transition and emission probabilities, HMM can be used
to derive the probabilities of the underlying state sequence. The probabilistic model of HMM-based alignment is flexible. For
example, the model can incorporate external information (such as 3D structure information, evolutionary) while performing the
alignment. In this way, HMMs can be used to integrate multiple sources of sequence and other information for probabilistic
inference of alignment. Other applications of HMMs include detection of promoters26 , CpG islands27 , read alignment28 , and for
genotype imputation29 . As such, they are a general class of approaches that are naturally suited to genomic sequence analysis.
We assume that the adversary is an "honest-but-curious" (or semi-honest) entity, that is, the adversary follows the protocol
honestly and provides correct outputs. The protection considerations are centered around the genomic sequence, i.e., only the
genomic sequence is considered confidential. We assume that the model parameters, i.e., transition and emission matrices are
considered publicly available. This assumption is important for reducing the complexity of computations since the model does
not have to be trained. Also, we assume that sequence length is not sensitive. This assumption is reasonable since the sequence
length is not exactly sensitive and depends on the application – read mapping, viral sequence alignment; the sequence lengths
are of common levels and do not generally reveal any information.
We implemented our approach in SHiMMer, a method for secure evaluation of HMMs. For data protection, SHiMMer adopts
Homomorphic Encryption (HE) cryptosystems. In a nutshell, HE enables processing encrypted data directly, without ever
needing to decrypt it. The genomic data is encrypted by the data owner who keeps the private key and it is practically impossible
for an untrusted entity to decrypt the data without this key as the HE cryptosystem is secure against post-quantum attacks. This
enables strong protection of the data and gives complete access control of the data to the owner as the owner can choose to
share the data with anyone that they deem as trusted. Since data is never decrypted in-transit, at-rest, and even in-analysis, the
data is always kept secure during the entire execution of data outsourcing. Even if the encrypted data is stolen, the encrypted
data is indistinguishable from random numbers according to the security protection of HE cryptosystems, and it does not leak
any information about the data. As a consequence, SHiMMer achieves confidential protection of the genomic data and the
inference results against a semi-honest server.
Alternatively, HE runs on commodity hardware, unlike other approaches such as SGX, and does not require additional
communication costs for secure outsourced computation as required by multiparty computation (MPC). This makes HE-
based methods very suitable to be deployed on the cloud where security is hard to maintain but there is virtually unlimited
computational power. However, HE has its own limitations. HE-based frameworks have been deemed impractical since their
inception. Therefore, in comparison to other cryptographically secure methods, such as multiparty computation30 and trusted
execution environments31 , HE-based frameworks have received little attention. Recent theoretical breakthroughs in the HE
2/17literature and a strong community effort32 have rendered HE-based systems practical, and it shows remarkable performance in a
number of applications. Many of these improvements, however, are only beginning to be reflected in practical implementations
and applications of HE algorithms.
Among a few viable HE cryptosystems, the Cheon-Kim-Kim-Song (CKKS) scheme33 has received increasing attention and
has successfully been adopted in various applications such as genomic analysis (e.g., genome-wide association studies34–36
and genotype imputation29 ) and machine learning systems37, 38 since it enables us to perform approximate homomorphic
computation over encrypted real numbers. Despite its versatility, real adoption of real-world problems is yet to be explored
deeply. The main barrier comes from the inherent property of this cryptosystem that it can only evaluate functions of bounded
complexity. In general, the iterative nature of HMM-based inference methods (such as forward-backward39 and Viterbi40 ) leads
to a substantially large circuit depth to be evaluated on HE cryptosystems, so they are not practically amenable to HE-based
evaluations of the models. In addition, they require computations over fractional numbers with sufficient precision.
To address the computational challenges for secure outsourced HMM-based inference, SHiMMer comes up with three
innovations: (1) Compute the HMM-based sequence alignment over encrypted DNA sequences in a HE-friendly manner
by devising a compact one-hot nucleotides encoding method and expressing the update formula of the forward variables as
an arithmetic circuit of low depth, (2) Exploit data parallelization for encrypting DNA sequences, computing the emission
probabilities of alignment symbols, and dealing with an evaluation of a deep circuit, that is, a single ciphertext can be represented
as multiple plaintext values and use the Single Instruction Multiple Data (SIMD) to perform homomorphic operations on these
values in parallel, (3) Propose an effective representation of encrypted data – ciphertext level management to optimize both
time and space for computing the forward variables, and scaling factor management to ensure sufficient precision of decrypted
results obtained by secure approximate computation.
We apply SHiMMer to simulated data and demonstrate the feasibility of HE-based pairwise sequence alignment with little
or no change in the inference accuracy. We also present estimates of time and memory requirements for secure HMM evaluation
for different types of HMMs. These can provide general insight into the feasibility of secure HMM evaluations for solving
problems other than sequence alignment.
Results
Scenario and System Model of SHiMMer
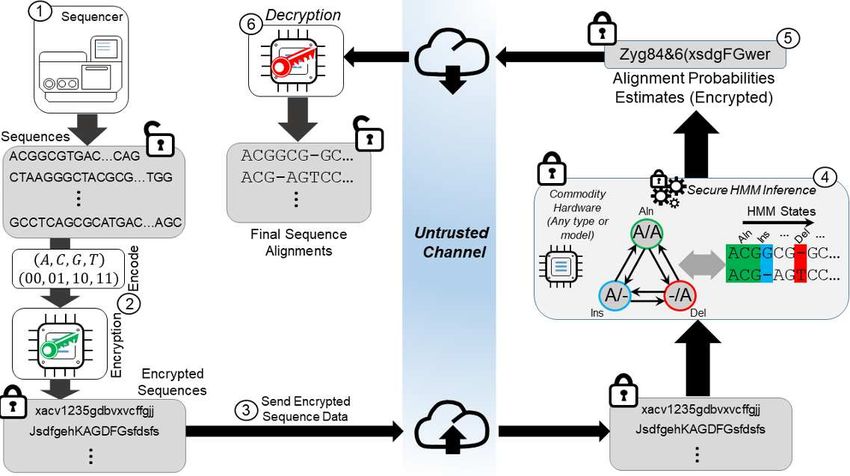
The scenario is summarized in Figure 1. The data owner generates public/private key pairs and encrypts the data using the
public key. After encryption, the encrypted sequence data is sent to the server, which is assumed to be an untrusted entity. The
SHiMMer server evaluates the secure sequence alignment HMM for comparison of sequences in the database. We assume that
the state transition matrix and emission matrix are provided as public data to the server. The encrypted results of sequence
alignment probabilities are returned to the user and can be decrypted using the private key.
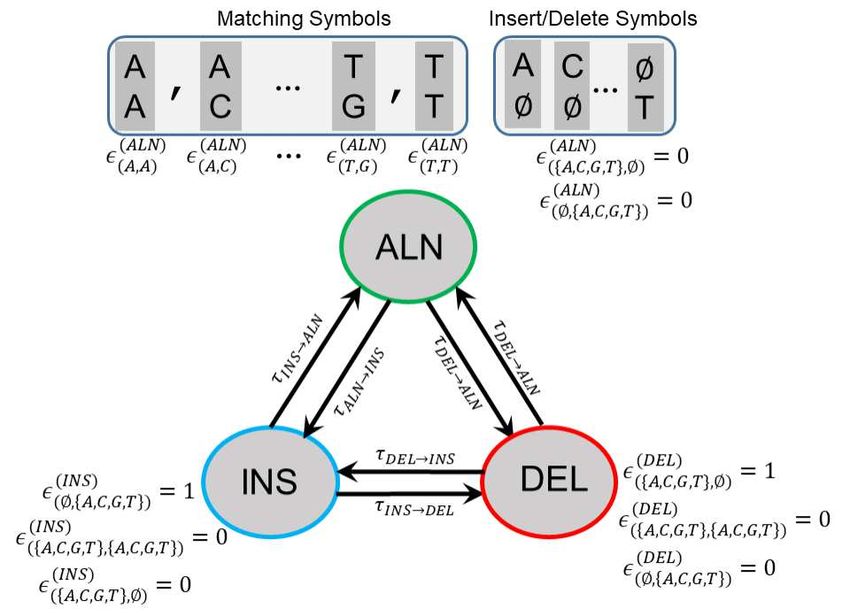
The hidden Markov model states are described in Figure 2. There are 3 states (denoted by ALN, INS, and DEL) that emit
the symbols in a pairwise sequence alignment, wherein each symbol is a 2-element column that makes up a pairwise alignment
from left-to-right. Among the 24 possible alignment symbols (excluding the alignment symbol with two gaps), the 16 symbols
that contain a nucleotide are emitted by the ALN state. The remaining 8 symbols contain a gap and are emitted by INS and
DEL states. State-state transitions probabilities are described by a 3 × 3 transition probability matrix (denoted by τ). For each
state, the emission probabilities of alignment symbols are described by the emission probability vector (ε). SHiMMer utilizes a
compact one-hot-encoding for nucleotide encoding to streamline the computations (See Methods).
Innovation of SHiMMer
In SHiMMer, we first devise a simple and fast encoding/encryption algorithm to support efficient computation of the emission
probabilities of observed alignment states over encrypted SNPs. We convert nucleotides into binary vectors: A 7→ (0, 0),C 7→
(0, 1), G 7→ (1, 0), T 7→ (1, 1). This representation enables us to securely observe the states of DNA sequences over encryption
by bitwise comparison with optimized memory usage for data encryption. Then the HMM-based sequence alignment algorithm
can be expressed as an arithmetic circuit of low depth (see Methods). A ciphertext modulus decreases when applying
homomorphic operations (especially, ciphertext multiplications), and in the end, it becomes too small to get a correct decryption
result. Since the HMM-based inference algorithm is recursive, it requires to refresh low-level ciphertexts after a few levels of
computation, called a bootstrapping transformation. Then it yields a new ciphertext that represents an approximate plaintext
with a larger ciphertext modulus. We address the overhead of bootstrapping operations by exploiting the SIMD parallelism
of the CKKS scheme. To this end, we pack distinct ciphertexts into a single ciphertext by simple slot rotations and perform
parallel bootstrapping over slots at a time. The update of the current 3-state forward variables is resumed by taking as input
refreshed ciphertexts of previous forward variables. SHiMMer is carefully designed to efficiently manage ciphertext levels
and scaling factors during secure computation in order to optimize time/space and guarantee sufficient precision of decrypted
results (Supplementary Notes 1,2).
3/17We implemented the SHiMMer protocol with Lattigo version 2.2, which includes implementations of the bootstrappable
CKKS. We use the default parameter set in Lattigo which provides at least 128-bit of security level according to the LWE-
estimator41 . We refer to Methods for further technical and implementation details of our system.
Time and Memory Requirements
To demonstrate the practicability and scalability of our system, we performed a detailed analysis of the running time and
memory requirements of secure HMM-based inference with various lengths of DNA sequences. We divided the alignment
process into four steps such as key generation, encryption, secure evaluation, and decryption. And then, we measured the
running time and the peak memory requirements for each step. Our experiments were conducted on a machine with an Intel
Platinum 8268 2.9GHz CPU featuring 16 cores and 192GB of main memory.
The SHiMMer protocol takes around 23.019 seconds to generate all the required cryptographic keys for secure computation
together with bootstrapping operations: (1) 0.169 seconds to generate the public/private key pairs, (2) 1.247 seconds to generate
the relinearization key (used for ciphertext multiplications), (3) 9.616 seconds to generate rotation keys (used for ciphertext
rotations), (4) 11.988 seconds to generate bootstrapping keys.
Figure 3a shows the detailed time requirement for the encryption step, which exhibits a linear scaling with the increasing
number of sizes of DNA sequences. The packed-nucleotides encryption method is 1.3-1.5 times faster than the single-nucleotide
encryption method. The packed implementation also reduces the memory usage, using 20.9%-26.8% less space (see Figure 3b).
The secure evaluation step consists of three procedures: (1) For thread-safe multi-threading, it first copies the memory pools
of the evaluation and bootstrapping structures to be used concurrently on homomorphic computations. (2) Then it computes
the forward variables over encrypted DNA sequences (3) It performs bootstrapping operations if needed (e.g., a ciphertext
level is too small to be used for homomorphic computation). Figure 3c shows the detailed time requirements for each step
and the aggregated time. All steps exhibited a linear scaling with the number of sum of sequence lengths (|S1 | + |S2 |). The
most time-consuming step in homomorphic evaluation is the bootstrapping procedures. Decryption, the last step, took less
than 85 milliseconds to decrypt a single ciphertext of the total probability of sequence alignment. For the alignment of two
sequences each of which is 100 nucleotides long, the total running time will be less than 13 minutes. Figure 3d shows the
memory requirements of SHiMMer, which compute the peak memory required for an arrangement of the thread pools and
secure evaluation. We note that the underlying HE cryptosystem should generate all the required keys regardless of DNA
lengths, which is around 6.004 GB in size. The memory usage during secure evaluation scales linearly with the size of the input
DNA sequences.
Figure 4 shows the run time and memory requirements of SHiMMer with constraints on the maximum separation (d)
on the sequence indices on the computations, i.e., cutting edges of the alignment space (see Figure 5). We used d =
⌈0.25 × max(|S1 |, |S2 |)⌉ as the constraint parameter. Although there are no perceptible changes in the run time and memory
usage for encryption compared with the full-space computation method (Figure 4a, 4b), we get a speedup of 1.5-1.8 times
for variable computations over the constrained implementation (Figure 4c). Likewise, memory requirements increase with
increasing sequence lengths. But the increase is much lower, and the constrained variant uses 25.8%-43.1% less space
(Figure 4d). These results indicate that constraints on the alignment space can be effective for decreasing time and memory
requirements.
Accuracy of HMM-based Inference
We estimated the secure HMM evaluation accuracy. This is necessary because HE operations in CKKS are performed
approximately, that is, noise is introduced for enabling real number computation. The HMM inference of the forward variables
exhibits substantial dependency on each other in the recursive computations. For this reason, we believe that this is a challenging
case for accuracy comparison and a good test case scenario for the precision effect from the secure computation. As an absolute
metric of accuracy, we computed the total absolute difference between the decrypted forward variable and the forward variable
for the whole sequence:
(Plain) (Secure)
∆α (S1 , S2 ) = ∑ |α|S (s) − α|S |,|S | (s)|, (1)
1 |,|S2 | 1 2
s
(Plain) (Secure)
where α|S |,|S | (s) denotes the forward variable at the positions |S1 | and |S2 | with the state s in the clear, and α|S |,|S | (s) is the
1 2 1 2
corresponding forward variable from the secure computation. Figure 6a shows the binary logarithm of total absolute difference
(log2 (∆α (S1 , S2 )) of the full and constrained space computation methods. We observed that the error decreases with sequence
length since forward variables become smaller with increasing sequence lengths. The constrained space computation method
has a slightly smaller error than the full-space computation method because the forward variables are smaller by its constraints
on the sequence indices (i.e., by setting some boundary variables as zeros). The error, however, is very small in comparison to
the absolute magnitude of the forward variable. This result provides evidence that the forward variable and associated values
4/17(Plain)
can be estimated without much precision loss. For the sake of the brevity, let PPlain (S1 , S2 ) = ∑s α|S |,|S | (s) be the probability
1 2
of sequences obtained by unencrypted computation. Figure 6b plots the ratio between the total absolute difference ∆α (S1 , S2 )
and the probability of sequences PPlain (S1 , S2 ). We observed that the relative error in encrypted forward variables with respect
to the true values increases with sequence length. This is because HMM-based inference requires a larger circuit depth to
compute forward variables as sequence lengths increase. As a result, it gives rise to larger computational errors from secure
computation. The full-space computation and constrained space computation result in almost the same relative errors. This is
in line with the theoretical noise estimation of the CKKS scheme that the noise is determined by the depth of a circuit to be
evaluated. In practice, two computation methods have the same depth requirement for secure evaluation. The error between the
cleartext forward variable and securely computed variable is less than 4 × 10−5 when the sequence lengths are less than 41.
This indicates that for the sequence lengths, the inference of posterior alignment probabilities will be impacted at most up to
4 × 10−5 by secure computation. For alignment of two sequences each of which is 100 nucleotides long, the error term will be
approximately 10−4 .
Discussion
Data encryption is currently one of the few methods that are recognized at the legislative level for data protection with clear
guarantees on security, including collaborations between countries that have different privacy regulations, such as HIPAA in the
United States and the GDPR in the European Union. Therefore, there is great promise in the development and adoption of
cryptographically secure methods that are based on multiparty computation and HE. Although HE has long been deemed as a
theoretical formalism, theoretical progress in the last decade has enabled substantial improvements in the time and memory
requirements of these secure methods.
The HMM inference in the secure setting of HE-based encryption enables researcher and user privacy whereby the genomic
sequences are under complete control of the owner. This way, large databases can be restructured such that sequences are
encrypted and large-scale inferences from the databases can be performed while sequences are being compared to each
other. Future work is necessary for managing these complex datasets to integrate user permissions so that any request can be
streamlined for secure analysis. This is necessary since data owners do not share keys. These can also be maintained by a
trusted entity such as the NIH, which can allow access to the datasets by centrally managing private keys.
Our methods can be extended to different problems where HMMs such as multiple sequence alignment wherein multiple
sequences are compared to each other, e.g., sequences from different viral species. In another direction, the framework can be
adapted to build secure dynamic programming-based algorithms where sensitive data are processed42 . However, there is still
work needed to adapt our method to these large-scale problems in sequential data modeling and analysis. In parallel, these
methods can also be used in other fields such as speech recognition43 .
Unlike some other reports that indicate accuracy as a limitation of HE-based schemes, we observed that the accuracy of
secure HMM evaluation is high and practically the same as inference in the clear, as our results show the secure evaluation
step incurs a normalized error term less than 10−4 . Our approach can be re-parameterized for applications that require higher
precision although this may incur additional time/memory costs. As input sequence (and alignment) lengths are increased, the
time requirement increases quadratically. This is expected since HMM inference is an intensely recursive process and exhibits
high dependency. Some of the heuristic approaches that are used in plaintext HMM inference, such as "corner-cutting" can be
adopted by secure methods to decrease the dependencies and the time/memory usage requirements.
Methods
HMM-based Sequence Alignment
Alignment Hidden Markov Model (HMM) is a 3-state generative model that emits aligned sequences. The different states
correspond to the three different modes of alignment emissions that correspond to (1) Emission of one nucleotide from each
sequence (ALN state), (2) Emission of one nucleotide in the 1st sequence and a gap in the second sequence (INS state), (3)
Emission of one nucleotide from the 2nd sequence and a gap in the first sequence (DEL state).
As generative models, HMMs are particularly useful since they can be used to build probability distributions on the emitted
alignments. From this aspect, they can be treated simply as Markov chains where only the dependence on consecutive alignment
states is necessary to model the alignment and sequence. Given the sequences and alignments, Markov chains can be efficiently
used to estimate the probability of the sequences and alignments simultaneously:
(s )
P(S10/ , S20/ , a|τ, ε) = ∏ P(εS1ki ,S2l · τsi−1 →si ), (2)
(k,l,s)∈a
where S10/ and S20/ are the aligned sequences include the gap symbols (0)
/ that are used in the emissions of INS and DEL states. a
is the state sequence of the alignment; for example a = (ALN, INS, INS, ALN, . . .). In principle, a is redundant because the
5/17alignment state sequence can be inferred from S10/ and S20/ . But we include these to be consistent with previous literature, and we
include these for clarity of presentation. Of note, the lengths of S10/ and S20/ are equal and are bound in terms of lengths of S1 and
S2 : max(|S1 |, |S2 |) ≤ |S10/ | = |S20/ | ≤ (|S1 | + |S2 |).
In Equation (2), the sequences include the gap symbol. However, the gap symbols are not observed (or hidden) from a
sequencing experiment. Clearly, these symbols are not meaningful for a single sequence because the alignment information
is, by definition, computed relative to the other sequences in the alignment. In addition, ALN states may emit alignment
columns that contain non-matching nucleotides. Therefore, given two sequences that we would like to align, the positions of
gap symbols and non-matching nucleotide positions must be inferred. Given two DNA sequences, we can use the distribution to
infer the most likely alignment and probability of alignment of two specific nucleotide positions of the sequences. Given only
the nucleotide sequences, the alignment states underlying the sequences are unknown and must be inferred using an analytical
model. For this, HMMs are used to define probability space over the possible set of states that would that we use to infer the
probability distributions of underlying alignment states. The total probability of sequences is:
P(S1 , S2 |τ, ε) = ∑ P(S10/ , S20/ , a|τ, ε), (3)
a∈A
where S1 , S2 are the measured nucleotide sequences, S10/ and S20/ are the gap-inclusive alignment symbol sequences with the gap
symbol 0/ in concordance with a. Also, τ and ε are state transition and alignment column emission probabilities. In Equation (3),
the summation extends over all possible alignment of two sequences where all gap positions (for INS and DEL states) and
all mismatching positions (for ALN states) are enumerated. This summation is not tractable, as the number of alignments
increases exponentially with the length of sequences42 . This summation can be efficiently estimated using forward algorithm
that quadratically increases with the length of sequences, O(|S1 | · |S2 |), given the number of states is a constant. For efficient
computation, a forward variable αi, j (s) at the positions i and j with the state s is used:
αi, j (s) = ∑ P(S1,[1,i] , S2,[1, j] , ψi, j = s|a, τ, ε), (4)
a∈A
where ψi, j denotes the emitting state at the positions i and j. Here, S1,[1,i] is the subsequence of nucleotides (S1,1 , S1,2 , . . . , S1,i )
and S2,[1, j] is defined as a subsequence of S2 .
A recursive estimation formula is used to compute the forward variable:
(s)
αi, j (s) = ∑ αi−δ1 (s), j−δ2 (s) (s′ ) · τs′ →s · εS ′ ,S ′ , (5)
i j
s′ ∈ALN,INS,DEL
where δ1 (s) returns an index update for the first sequence given that the current state is s:
(
1 if s ∈ {ALN, INS},
δ1 (s) = (6)
0 otherwise,
and δ2 (s) is defined similarly:
(
1 if s ∈ {ALN, DEL},
δ2 (s) = (7)
0 otherwise.
Finally, Si′ and S j′ are the symbols that are emitted state s. For INS and DEL states, these include the gap symbol. For ALN
state, these are the corresponding nucleotides at i and j from the DNA sequences. This recursive formula makes use of the
state-space dependency between transitions and decomposes emission and state transitions. Thereby, the probabilities can be
computed starting from the smallest subsequences while the subsequence length is grown.
One of the challenges around computation is that there is a strong dependency, which limits the potential of parallelization
of computations: The value of αi, j (s) depends on all of the forward variable values for smaller subsequences, i.e., αi′ , j′ (s)
for i′ < i, j′ < j. Thus, all of the smaller subsequences need to be computed before αi, j (s) can be computed. While it is
necessary to loop over all 2-tuples (i, j) in a growing fashion, the order of computations can be selected arbitrarily as long as
the dependency conditions are not violated.
Constraints on Alignment Space
The full computation of forward variable requires a full 2-dimensional computation over all (i, j) for 1 ≤ i ≤ |S1 |, 1 ≤ j ≤ |S2 |.
This can be prohibitive for alignment of long sequences. Numerous approaches have been proposed for "cutting-corners"
6/17of alignments whereby the difference on the sequence indices are constrained42, 44 . Figure 5 illustrates this constraint. The
constraint can be implemented into forward variable computation by setting the constraint on difference between nucleotide
indices on two sequences, that is,
(
αi, j (s) if |i − j| < d,
α̃i, j (s) = (8)
0 otherwise.
The motivation for this constraint is the assumption that the alignments of subsequences do not deviate substantially in the
alignment (i.e., |i − j| < d). In other words, alignments do not include long runs of insertions or deletions. In addition, this
constraint is biologically plausible since the constraint excludes biologically uninformative alignments such as the alignment
where one sequence is emitted by an insertion (or deletion) state.
Homomorphic Encryption Cryptosystem
Homomorphic encryption allows one to perform arithmetic operations on encrypted data and receive an encrypted result
corresponding to the result of operations performed in plaintext. It enables us to outsource computation on encrypted data
in an untrusted cloud environment while mitigating privacy risks by allowing all computation to be done in an encrypted
manner. Among a few viable solutions, the CKKS cryptosystem can be considered as one of the promising privacy-preserving
outsourcing protocols. A ciphertext has an inherent error for security, and this error is fused with a real message as a ciphertext
in the CKKS scheme. In practice, a message is scaled by a predetermined factor before encryption to ensure the correctness of
the decryption. As ciphertext multiplication operations bring about an increased scaling factor of the messages, the built-in
rescaling operation on encrypted data is used for rounding off the least significant digits over encryption as in plain fixed-
point computation. This technique leads to precision adjustment to get rid of accumulated extra digits after homomorphic
computation, thereby enabling us to control the magnitude of messages. However, when a ciphertext modulus becomes too
small after a number of multiplications, the correctness of decryption cannot be guaranteed. To address this challenge, Cheon
et al.45 presented a bootstrapping procedure that refreshes low-level ciphertexts, resulting in a new ciphertext that encrypts
an approximate message with a larger ciphertext modulus. But this operation is still computationally intensive for practical
use46, 47 .
Homomorphic Encryption Notation. Mult(c1 , c2 ) indicates a homomorphic multiplication between ciphertexts c1 and c2 .
Sqr(ct) denotes a homomorphic squaring operation of a ciphertext ct. Rot(ct; ρ) denotes a homomorphic rotation operation of
a ciphertext ct by an amount of ρ to the left. MultPlain(ct, pt) indicates a homomorphic multiplication between a ciphertext ct
and a plaintext pt (a value or a vector).
HE-Friendly Reformulation of HMM-based Sequence Alignment
For the sake of brevity, we identify three different modes of alignment emissions into a set S = {1, 2, 3} as follows: ALN 7→ 1,
INS 7→ 2, DEL 7→ 3. We abuse the notation by writing τs′ s instead of τs′ →s . Then the forward variable computation proceeds as
follows:
1 1
• Initialize the forward variable α0,0 (s) = |S| = 3 for s ∈ S.
• For each 1 ≤ i ≤ |S1 |, 1 ≤ j ≤ |S2 |, compute the forward variable as follows:
! !
(ALN)
• αi, j (1) = ∑ αi−1, j−1 (s) · τs1 · ε(x ,y )
i j
= ∑ αi−1, j−1 (s) · τs1 · εi j , (9)
s∈S s∈S
!
(INS)
• αi, j (2) = ∑ αi−1, j (s) · τs2 · ε(x ,0)
/
i
= ∑ αi−1, j (s) · τs2 , (10)
s∈S s∈S
!
(DEL)
• αi, j (3) = ∑ αi, j−1 (s) · τs3 · ε(0,y
/ j)
= ∑ αi, j−1 (s) · τs3 , (11)
s∈S s∈S
where we denote by εi j be the emission probability of the nucleotides xi and y j from each sequence at the ALN state.
• Obtain the total probability of sequences Pr(S1 , S2 |τ, ε) = ∑s∈S α|S1 |,|S2 | (s).
7/17Privacy-Preserving Alignment of Genomic Sequences
We convert each nucleotide to a binary vector representation: A 7→ (0, 0),C 7→ (0, 1), G 7→ (1, 0), T 7→ (1, 1). In the following,
we identify nucleotides with their binary vector representations. Then, for each entry in the DNA genomic sequence, we
can compute two ciphertexts, one for the first entry and the one for the second entry. To reduce the encryption time and
minimize the size of encrypted data, we use the SIMD technique for encrypting a binary vector as a single ciphertext. Given a
predetermined scaling factor of ∆, each binary vector is multiplied by the factor and converted to a ciphertext. We provide a
detailed explanation of how to set the input scaling factors and encryption levels in Supplementary Notes 1,2.
Given two nucleotides xi , y j ∈ {A,C, G, T }, the emission probability εi j at the positions i and j with respect to the ALN
state is defined as follows:
(
ε if xi = y j ,
εi j = (12)
ε ′ otherwise.
Then it can be expressed as an arithmetic circuit:
εi j = ε ′ · (1 − di j ) + ε · di j , (13)
where xi = (xi1 , xi2 ), y j = (y j1 , y j2 ), and di, j = ((xi1 − y j1 )2 − 1) · ((xi2 − y j2 )2 − 1). Note that di j is 1 if and only if xi = y j ;
otherwise it is 0.
For 1 ≤ i ≤ |S1 | and 1 ≤ j ≤ |S2 |, we first compute ct.xyi j = Sqr(Enc(xi ) − Enc(y j )) − 1, which represents the numbers of
((xi1 − y j1 )2 − 1) and ((xi2 − y j2 )2 − 1) at the first and second entries, respectively. Then an encryption of di j can be computed
by evaluating
ct.di j = Mult(ct.xyi j , Rot(ct.xyi j ; 1)). (14)
From Equation (13), an encryption of the emission probability εi j is obtained by
ct.εi j = MultPlain(1 − ct.di j , ε ′ ) + MultPlain(ct.di j , ε). (15)
Let ct.αi j (s) be a ciphertext of the forward variable of two sequences at the positions i and j with the state s. Then the forward
algorithm can proceed as follows:
• ct.αi, j (1) = Mult ∑s∈S MultPlain(ct.αi−1, j−1 (s), τs1 ), ct.εi j ,
• ct.αi, j (2) = ∑s∈S MultPlain(ct.αi−1, j (s), τs2 ),
• ct.αi, j (3) = ∑s∈S MultPlain(ct.αi, j−1 (s) · τs3 ).
Ciphertext Levels Management
A freshly encrypted ciphertext of the CKKS scheme is represented as a pair of polynomials in ZQ [X]/(X N + 1) where L is set as
the maximum multiplication level to be supported by the HE cryptosystem and Q is a product of (L + 1) pairwise co-primes qi .
If a ciphertext modulus is over ZQℓ for Qℓ = ∏0≤i≤ℓ qi , we say that the ciphertext is at level ℓ. Indeed, multiplication operations
bring about decreased ciphertext modulus. By the update formula of the forward variables, we have the followings:
• lvl(ct.αi, j (1)) = mins∈S {lvl(ct.αi−1, j−1 (s))} − 2,
• lvl(ct.αi, j (2)) = mins∈S {lvl(ct.αi−1, j (s))} − 1,
• lvl(ct.αi, j (3)) = mins∈S {lvl(ct.αi, j−1 (s))} − 1,
where lvl(ct) denotes the level of the ciphertext ct. This implies that ciphertexts on the same anti-diagonal have the same
ciphertext level (see Supplementary Figure S1). So, we keep on updating the forward variable until a ciphertext level reaches
one. When such a situation has arisen, we apply the bootstrapping operation of Cheon et al.48 to refresh low-level ciphertexts
and repeatedly perform these procedures until we get ciphertexts of the forward variables for the whole sequence. We provide a
detailed explanation of ciphertext level management in Supplementary Note 1.
Scaling Factors Management
In the CKKS scheme, a real number is multiplied by a scaling factor before encryption to ensure that the encoded values ensure
sufficient precision. Assume that an encryption of the emission probability εi j has a scaling factor of ∆ε when i = 1 or j = 1;
otherwise, it is scaled by the factor of qt when the encryption ct.εi j is at level t. Using mathematical induction, it can be shown
that an encryption of αi, j (s) is scaled by ∆ε for 1 ≤ i ≤ |S1 |, 1 ≤ j ≤ |S2 |. Thus, we set the encryption levels of input DNA
sequences to satisfy this assumption, so that the resulting ciphertext of the total probability of sequences has a sufficiently large
precision to get a meaningful result. We provide detailed proof of scaling factors management in Supplementary Note 2.
8/17Code availability
The source code implementation of secure evaluation of the developed approaches are available to download at https://
drive.google.com/drive/folders/1s5N7TCR4iUVtitTUucfPw7y6Et3WSAws?usp=sharing. The source
code will be made publicly available upon publication.
Acknowledgements
The work of M.K. was supported by the Settlement Research Fund (No. 1.200109.01) of UNIST (Ulsan National Institute of
Science & Technology) and National Research Foundation of Korea (NRF) Grant funded by the Korea Government (MSIT)
under Grant 2021R1C1C1010173. X.J. is CPRIT Scholar in Cancer Research (RR180012), and he was supported in part by
Christopher Sarofim Family Professorship, UT Stars award, UTHealth startup, the National Institute of Health (NIH) under
award number R13HG009072 and R01AG066749-S1.
Author Contributions
All authors designed the secure alignment scenario and developed the methods. M.K. and A.H. implemented the software and
conducted the benchmarking experiments. All authors wrote the manuscript.
Competing Interests
The authors declare that they have no competing financial interests.
References
1. Ng, P. C. & Kirkness, E. F. Whole genome sequencing. In Genetic variation, 215–226 (Springer, 2010).
2. Shendure, J. et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017).
3. Chisholm, J., Caulfield, M., Parker, M., Davies, J. & Palin, M. Briefing genomics england and the 100K genome project.
Genomics Engl (2013).
4. Consortium, T. . G. P. A global reference for human genetic variation. Nature 526, 68–74, DOI: 10.1038/nature15393
(2015).
5. Schwarze, K., Buchanan, J., Taylor, J. C. & Wordsworth, S. Are whole-exome and whole-genome sequencing approaches
cost-effective? A systematic review of the literature. Genet. Medicine 20, 1122–1130 (2018).
6. Allen, H. L. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature
467, 832–838 (2010).
7. Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
8. Gibson, G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 (2012).
9. Agarwala, V. et al. Evaluating empirical bounds on complex disease genetic architecture. Nat. genetics 45, 1418 (2013).
10. Chen, J., Harmanci, A. S. & Harmanci, A. O. Detecting and annotating rare variants. Encycl. Bioinforma. Comput. Biol.
388–399 (2019).
11. Cooper, J. D. et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat.
genetics 40, 1399 (2008).
12. Russell, J. J. et al. Non-model model organisms. BMC Biol. 15, 1–31, DOI: 10.1186/s12915-017-0391-5 (2017).
13. Rehm, H. L. Evolving health care through personal genomics. Nat. Rev. Genet. 18, 259 (2017).
14. Home - gene - ncbi. https://www.ncbi.nlm.nih.gov/gene/.
15. Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob.
Challenges 1, 33–46, DOI: 10.1002/gch2.1018 (2017).
16. Sboner, A., Mu, X., Greenbaum, D., Auerbach, R. K. & Gerstein, M. B. The real cost of sequencing: higher than you
think! Genome Biol. 12, 125, DOI: 10.1186/gb-2011-12-8-125 (2011).
17. Heather, J. M. & Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 107, 1–8 (2016).
18. Ozercan, H. I., Ileri, A. M., Ayday, E. & Alkan, C. Realizing the potential of blockchain technologies in genomics. Genome
Res. 28, 1255–1263, DOI: 10.1101/gr.207464.116 (2018).
9/1719. Starr, D. Forensics gone wrong: When dna snares the innocent. Science DOI: 10.1126/science.aaf4160 (2016).
20. Lunshof, J. E., Chadwick, R., Vorhaus, D. B. & Church, G. M. From genetic privacy to open consent. Nat. reviews. Genet.
9, 406–411, DOI: 10.1038/nrg2360 (2008).
21. Harmanci, A. & Gerstein, M. Quantification of private information leakage from phenotype-genotype data: linking attacks.
Nat. methods 13, 251–256 (2016).
22. Harmanci, A. & Gerstein, M. Analysis of sensitive information leakage in functional genomics signal profiles through
genomic deletions. Nat. communications 9, 1–10 (2018).
23. iDASH (integrating Data for Analysis, Anonymization, Sharing) genome privacy competition. http://www.
humangenomeprivacy.org/.
24. Rabiner, L. R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 77,
257–286, DOI: 10.1109/5.18626 (1989).
25. Yoon, B.-J. Hidden Markov models and their applications in biological sequence analysis. Curr. genomics 10, 402–415
(2009).
26. Won, K.-J., Sandelin, A., Marstrand, T. T. & Krogh, A. Modeling promoter grammars with evolving hidden Markov
models. Bioinformatics 24, 1669–1675, DOI: 10.1093/bioinformatics/btn254 (2008).
27. Wu, H., Caffo, B., Jaffee, H. A., Irizarry, R. A. & Feinberg, A. P. Redefining cpg islands using hidden Markov models.
Biostatistics 11, 499–514, DOI: 10.1093/biostatistics/kxq005 (2010).
28. Canzar, S. & Salzberg, S. L. Short read mapping: An algorithmic tour. vol. 105, 436–458, DOI: 10.1109/JPROC.2015.
2455551 (Institute of Electrical and Electronics Engineers Inc., 2017).
29. Kim, M. et al. Ultrafast homomorphic encryption models enable secure outsourcing of genotype imputation. Cell Syst.
DOI: 10.1016/j.cels.2021.07.010 (2021).
30. Cho, H., Wu, D. J. & Berger, B. Secure genome-wide association analysis using multiparty computation. Nat. biotechnology
36, 547–551 (2018).
31. Kockan, C. et al. Sketching algorithms for genomic data analysis and querying in a secure enclave. Nat. Methods 17,
295–301 (2020).
32. Homomorphic encryption standardization (HES). https://homomorphicencryption.org. HES.
33. Cheon, J. H., Kim, A., Kim, M. & Song, Y. Homomorphic encryption for arithmetic of approximate numbers. In
International Conference on the Theory and Application of Cryptology and Information Security, 409–437 (Springer,
2017).
34. Kim, M., Song, Y., Li, B. & Micciancio, D. Semi-parallel logistic regression for GWAS on encrypted data. BMC Med.
Genomics 13, 1–13 (2020).
35. Blatt, M., Gusev, A., Polyakov, Y. & Goldwasser, S. Secure large-scale genome-wide association studies using homomor-
phic encryption. Proc. Natl. Acad. Sci. 117, 11608–11613 (2020).
36. Froelicher, D. et al. Truly privacy-preserving federated analytics for precision medicine with multiparty homomorphic
encryption. bioRxiv (2021).
37. Jiang, X., Kim, M., Lauter, K. & Song, Y. Secure outsourced matrix computation and application to neural networks. In
Proceedings of the 2018 ACM SIGSAC Conference on Computer and Communications Security, 1209–1222 (ACM, 2018).
38. Kim, M., Song, Y., Wang, S., Xia, Y. & Jiang, X. Secure logistic regression based on homomorphic encryption: design and
evaluation. JMIR medical informatics 6 (2018).
39. Bahl, L. R., Cocke, J., Jelinek, F. & Raviv, J. Optimal decoding of linear codes for minimizing symbol error rate. IEEE
Transactions on Inf. Theory 20, 284–287, DOI: 10.1109/TIT.1974.1055186 (1974).
40. Forney, G. D. The viterbi algorithm. Proc. IEEE 61, 268–278, DOI: 10.1109/PROC.1973.9030 (1973).
41. Albrecht, M. R., Player, R. & Scott, S. On the concrete hardness of learning with errors. J. Math. Cryptol. 9, 169–203
(2015).
42. Durbin, R., Eddy, S., Krogh, A. & Mitchison, G. Biological sequence analysis: Probabilistic models of proteins and nucleic
acids (Cambridge University Press., 1998).
43. Pathak, M., Rane, S., Sun, W. & Raj, B. Privacy preserving probabilistic inference with hidden Markov models. In 2011
IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), 5868–5871 (IEEE, 2011).
10/1744. Harmanci AO, M. D., Sharma G. Efficient pairwise RNA structure prediction using probabilistic alignment constraints in
dynalign. BMC Bioinforma. 8, 130 (2007).
45. Cheon, J. H., Han, K., Kim, A., Kim, M. & Song, Y. Bootstrapping for approximate homomorphic encryption. In Annual
International Conference on the Theory and Applications of Cryptographic Techniques, 360–384 (Springer, 2018).
46. Chen, H., Chillotti, I. & Song, Y. Improved bootstrapping for approximate homomorphic encryption. In Annual
International Conference on the Theory and Applications of Cryptographic Techniques, 34–54 (Springer, 2019).
47. Bossuat, J.-P., Mouchet, C., Troncoso-Pastoriza, J. & Hubaux, J.-P. Efficient bootstrapping for approximate homomorphic
encryption with non-sparse keys. In Annual International Conference on the Theory and Applications of Cryptographic
Techniques, 587–617 (Springer, 2021).
48. Cheon, J. H., Han, K., Kim, A., Kim, M. & Song, Y. A full RNS variant of approximate homomorphic encryption. In
Selected Areas in Cryptography – SAC 2018, 347–368 (Springer, 2018).
11/17Figures
Figure 1
Figure 1. Illustration of the secure HMM-based sequence alignment scenario. The raw DNA sequences are generated by a
DNA sequencer (Step 1). The data owner generates public/private key pairs and encrypts the query sequences using the public
key (Step 2). The encrypted genomic sequences are sent to the semi-honest server (Step 3). The SHiMMer server performs a
secure HMM-based sequence alignment using the HMM parameters (Step 4). The encrypted results of the sequence alignment
probabilities are returned to the user (Step 5). The data owner decrypts the sequence alignment probabilities using the private
key (Step 6).
12/17Figure 2
Figure 2. Illustration of the 3-State HMM used for pairwise sequence alignment. The states are denoted by alignment (ALN),
insertion (INS), and deletion (DEL). The state transition probability from the state s′ to the state s is denoted by τs′ →s . The
(s)
emission probabilities of alignment symbols with respect to the state s are denoted by εS′ ,S′′ , where S′ and S′′ are the symbols
that are emitted state s.
13/17Figure 3
(a) Run time (b) Peak memory usage
5
Packed Packed 4.59
3.5 Single Single 4.2
4 3.81
3.49
3 3.36
3.17
2.97
Gigabytes
3
Seconds
2.78 2.78
2.5
2.2
2
2
1.5 1
1 0
(13, 13) (20, 20) (27, 27) (34, 34) (41, 41) (13, 13) (20, 20) (27, 27) (34, 34) (41, 41)
Sequence lengths (|S1 |, |S2 |) Sequence lengths (|S1 |, |S2 |)
(c) Run time (d) Peak memory usage
350
Thread-pools 317.4 30 Thread-pools 27.94
300 Variable Comp. Evaluation
Bootstrap 25 24.01
253.8
250 Total 21.36
20
Seconds
Gigabytes
18.84
200 180.1
188.5
15.94
150 144.1 15 13.31
123.7 11.89
115.4
99.9
92.9
100 10 9.04
68.5 7.62
57.3 57.8 7.04
47.7
50 32.3
17.9
9.9 11.7 16.9 18.3 5
7.1
(13, 13) (20, 20) (27, 27) (34, 34) (41, 41) (13, 13) (20, 20) (27, 27) (34, 34) (41, 41)
Sequence lengths (|S1 |, |S2 |) Sequence lengths (|S1 |, |S2 |)
Figure 3. Detailed run time and memory requirements for secure HMM-based sequence alignment. The implementation
exploits multiple cores when available. The complete execution of secure computation is divided into four steps: key
generation, encryption, evaluation, and decryption. (a),(b): Running time and peak memory usage for encryption with the
packed-nucleotides implementation and single-nucleotide implementation over different sequence lengths. (c) Running time for
homomorphic evaluation with different sequence lengths. It consists of three procedures: thread-pools (copy the memory pools
of the evaluation and bootstrapping structures to be used concurrently on homomorphic computation), variable compute
(update the forward variables over encrypted inputs), and bootstrap (perform bootstrapping operations when needed). The
aggregated time is also shown. (d) Peak memory usage during the evaluation process.
14/17Figure 4
(a) Run time (b) Peak memory usage
5
Packed Packed 4.6
3.5
Single Single 4.1
4 3.7
3 3.36
3.3
3.11
Gigabytes
2.91
3
Seconds
2.71
2.5 2.6
2.13
2
2
1.5 1
0
(13, 13) (20, 20) (27, 27) (34, 34) (41, 41) (13, 13) (20, 20) (27, 27) (34, 34) (41, 41)
Sequence lengths (|S1 |, |S2 |) Sequence lengths (|S1 |, |S2 |)
(c) Run time (d) Peak memory usage
20
Thread-pools 244.5 Thread-pools
250
Variable Compute Evaluation
16.5
Bootstrap 199.1 15.5
15.9
200 Total 15
163.6
Seconds
Gigabytes
12.4 12.5
150 134.6
128.8
10
100 82.7
92.4
76.3 7.2 7.2 7.3
65.7
6
53.5
50 40.7 37.8
5 4.2
26
20.517.6
2.1 2.2 4.5 4.6 4.6
(13, 13) (20, 20) (27, 27) (34, 34) (41, 41) (13, 13) (20, 20) (27, 27) (34, 34) (41, 41)
Sequence lengths (|S1 |, |S2 |) Sequence lengths (|S1 |, |S2 |)
Figure 4. Detailed run time and memory requirements for secure HMM-based sequence alignment using cutting-corners of
alignments. We take into account 2-tuples (i, j)’s with |i − j| ≤ d with the constraint parameter d = ⌈0.25 × max(|S1 |, |S2 |)⌉.
(a),(b): Running time and peak memory usage for encryption with packed-nucleotides implementation and single-nucleotide
implementation over different sequence lengths. (c) Running time for evaluation with different sequence lengths. (d) Peak
memory usage during the evaluation process.
15/17Figure 5
Figure 5. Illustration of constraints on alignment. The square represents 2-tuples (i, j)’s that are required for full-space
computation. The red triangles illustrate the corners that correspond to |i − j| > d, for d that constrains the total number of INS
and DEL states. The 2-tuples that are in the red triangles will not be taken into account in the constrained inference.
16/17Figure 6
(a) Absolute error (b) Relative error
·10−5
F-space F-space
C-space 4 C-space
−24
∆α (S1 , S2 )/P(Plain) (S1 , S2 )
3.5
−25 3
log2 (∆α (S1 , S2 ))
−26 2.5
2
−27
1.5
−28 1
−29 0.5
0
(13, 13) (20, 20) (27, 27) (34, 34) (41, 41) (13, 13) (20, 20) (27, 27) (34, 34) (41, 41)
Length of DNA sequences (|S1 |, |S2 |) Length of DNA sequences (|S1 |, |S2 |)
Figure 6. Accuracy comparison of secure HMM-based inference with plain HMM-based inference of the full-space
computation (denoted by F-space) and constrained space computation (C-space) on the alignment space. (a) The binary
logarithm of the total absolute difference ∆α (S1 , S2 ). (b) The ratio between the total absolute difference ∆α (S1 , S2 ) and the total
probability of sequences alignment from plain computation P(Plain) (S1 , S2 ).
17/17Supplementary Files
This is a list of supplementary les associated with this preprint. Click to download.
supplementary.pdfYou can also read

















































