Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology John A. Wemmie1, Xiang-ming Zha2, and Michael J. Welsh3 University of Iowa, Roy J. and Lucille A. Carver College of Medicine 1 Department of Psychiatry, Neuroscience Program, and Department of Veterans Affairs Medical Center, Iowa City, IA,USA, john-wemmie@uiowa.edu 2 Department of Internal Medicine and Howard Hughes Medical Institute, Iowa City, IA 52242, USA, xiangming-zha@uiowa.edu 3 Departments of Internal Medicine and Molecular Physiology, and Howard Hughes Medical Institute, Iowa City, IA 52242, USA, michael-welsh@uiowa.edu Abstract. Although brain pH is tightly controlled, it can be more dynamic than commonly appreciated. Physiological fluctuations in extracellular pH provide ample opportunity for protons to influence synaptic signaling. A number of synaptic proteins are modified by extracellular pH. Of these, the acid sensing ion channels (ASICs) are gated by extracellular protons and thus they may be particularly well suited to respond to synaptic pH. Here we review extracellular pH changes that accompany neural activity, the synaptic localization of ASICs, and their known effects on synapse function. Results from manipulating ASICs in mice suggest important roles for synaptic ASICs in behavior and neurological disease. 1 Introduction In general, the brain’s extracellular pH is controlled within a narrow range (1). However, neural activity causes pH alterations that can vary in location and with time. In addition, disease can generate sustained deviations of physiologic pH. Here, we consider some of the changes in brain pH, and some possible molecular targets, focusing particularly on the acid sensing ion channels. 2 Extracellular pH at the Synapse 2.1 Proton Release During Neurotransmission Lowers Synaptic pH Vacuolar-H+ATPases pump protons into synaptic vesicles lowering vesicle pH to ~5.2–5.7 (2–5). The resulting proton gradient energizes neurotransmitter uptake; it may also serve an important signaling role during neurotransmission (Fig. 1a). In addition to free protons, at low pH other molecules in the vesicle are protonated, creating a source of releasable protons. Thus, when synaptic vesicles fuse with the presynaptic membrane, free protons and protonated acids are released into the synaptic cleft (5). At the release site, pH probably falls very quickly, but the J.W. Hell, M.D. Ehlers (eds.), Structural and Functional Organization of the Synapse, DO I : 10.1007/978-0-387-77232-5_22, © Springer Science+Business Media, LLC 2008
662 J.A. Wemmie et al.
reduction is likely to be short lived (milliseconds or less) due to rapid H+ buffering
and diffusion (Fig. 1b). The speed and spatial characteristics of this phenomenon
make it technically challenging to monitor. However, by electrophysiological
methods and by pH sensitive fluorophores, extracellular acidification at the
presynaptic side has been detected (5–8). Based on these measurements, pH in the
retinal ribbon synapse is estimated to drop 0.2–0.6 pH units during
neurotransmission (6, 8). Faster and more sensitive detection strategies may improve
our ability to measure and understand this phenomenon.
As a consequence of synaptic proton release, pH-sensitive voltage-gated Ca2+-
channels (VGCC) are inhibited in the presynaptic membrane of retinal ribbon
synapses (6–8). Other proteins near the synaptic cleft could also be affected. For
example, extracellular acidosis is known to inhibit NMDA receptors (9–12) and
AMPA receptors (13). GABAA-receptors are also pH sensitive; some subunits are
activated (14) and some inhibited by extracellular protons (15). As will be discussed
later (Sections 3 and 4), acid-sensing ion channels (ASICs) in the post-synaptic
membrane are well positioned to respond to rapid acid transients.
2.2 Alkalosis Follows Neurotransmission
Slower changes in extracellular pH follow bursts of synaptic activity. A transient
alkalosis begins ~20 ms after a 100 Hz stimulus train and can last hundreds of
milliseconds (Fig. 1b) (16, 17). This increase in extracellular pH has been detected in
the hippocampus and elsewhere in the brain (16). pH-sensitive microelectrodes in
extracellular fluid detected a rise of ~0.05 pH units (18, 19). However, judging by
the effects on synaptic transmission (18, 19), the magnitude of the alkalosis at the
synapse is probably significantly larger. As described above, additional high-
resolution measurements would help the field. The mechanisms underlying the net
loss of extracellular protons are not clear. Nor is it clear whether the alkalosis is
related to the more rapid acidification that occurs during neurotransmission.
Suspected mechanisms for the alkalosis include glutamate receptor activated H+
transport into cells (17), GABAA-receptor mediated extrusion of HCO3– (17, 20, 21),
and Ca2+- H+ exchange by cell surface ATPase (22–24). Carbonic anhydrase also
plays an important role, since carbonic anhydrase inhibitors magnified the alkaline
transient (25), and adding exogenous carbonic anhydrase to brain slices attenuated
the alkalinization (19).
The alkaline transients can have important physiological consequences. As a
general rule, interstitial alkalosis tends to increase excitability in most central
neurons (26). Because the NMDA receptor is partially blocked by protons at
physiological pH (~7.35), alkalosis would be expected to relieve proton inhibition.
Consistent with this prediction, the alkaline transient boosted NMDA receptor
activation and increased Ca2+ entry (18). As a consequence, depolarization was
prolonged following EPSC trains and excitability was increased (27). The effects of
the alkaline transient on other pH sensitive channels, and on behavior and disease are
not yet clear.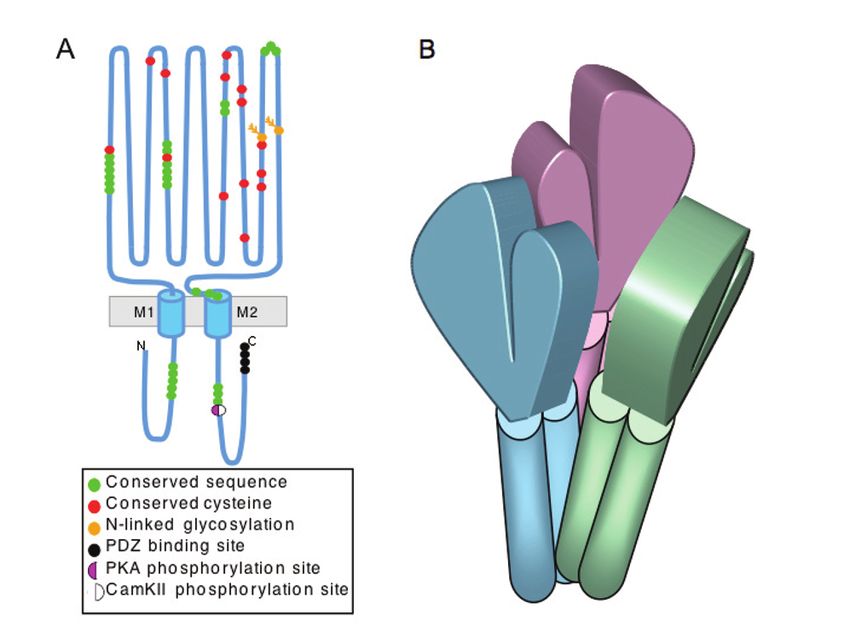
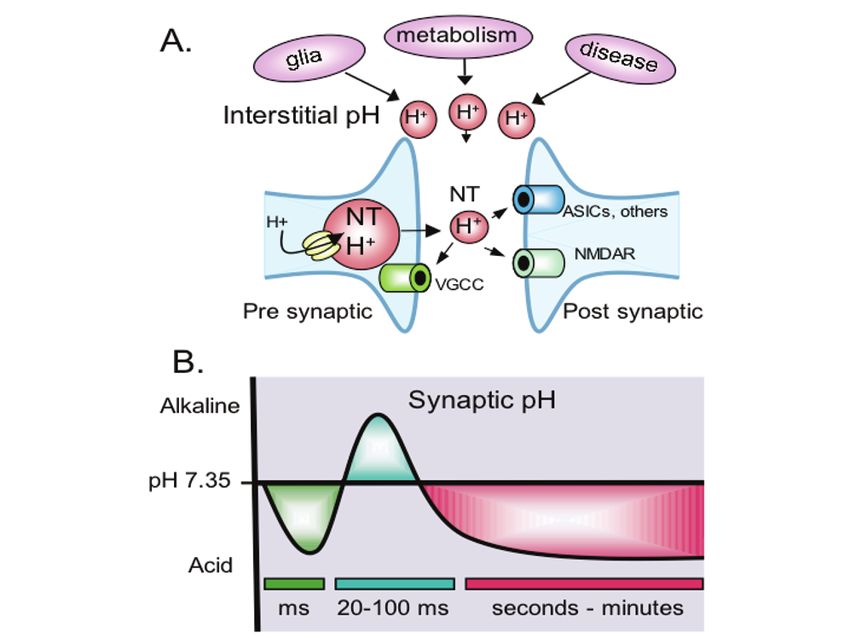
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 663 Fig. 1. Model of pH fluctuations at the synapse. (a) Synaptic vesicles release protons and neurotransmitter (NT) into the synaptic cleft during neurotransmission, which may lower pH in the synaptic cleft and modulate pH sensitive channels and other proteins in the pre- and post-synaptic membrane. Illustrated are voltage-gated Ca2+-channels (VGCC), NMDA receptors, and ASICs, although other pH sensitive proteins may also be present. Glia, metabolism, and disease processes lower interstitial pH and may also affect synaptic pH and physiology. (b) At lease three transient pH fluctuations may occur at the synapse in response to neural activity. Rapid acidification of the cleft (green) may occur with vesicle release and probably lasts milliseconds (ms) or less. A slower alkalosis (blue) has been detected in the interstitial space within tens of milliseconds. An even slower acidosis (red) can follow intense neural activity and last for seconds. Improved techniques are needed to better characterize the degree and duration of these pH fluctuations at the synapse. 2.3 Intense Neural Activity Causes Acidosis On an even slower timescale, intense neural activity produces a wave of acidification (Fig. 1b, red waveform)(17). The acidosis occurs within seconds to minutes of stimulation and can last for minutes or longer. The degree of acidification and its duration appear to depend on the magnitude of neural and metabolic activity. With extreme activity, such as during a seizure, interstitial pH can fall well below 7.0; raising the chance of both synaptic and extra-synaptic consequences. Metabolic mechanisms may contribute to the acidification. Mitochondria take up Ca2+ and extrude H+, which in turn can be transported out of the cell (22–24). Lactate is also produced and extruded from cells (17). Glia move acid across the cell membrane (28, 29), and glial extrusion of lactate may provide an important energy source for active
664 J.A. Wemmie et al. neurons (30). Increased metabolic activity also produces more CO2, which can be rapidly hydrated to HCO3– and H+ (31). The physiological consequences of this slower acid transient on synaptic transmission are not clear, although the effects are likely to be mainly inhibitory because prolonged acidosis reduces the excitability of most central neurons (26). 2.4 Neurological Disease can Produce Acidosis Extracellular acidosis is also associated with several neurological diseases. For example, seizures and ischemia are well known to reduce extracellular pH (32–35). Inflammatory diseases, such as multiple sclerosis, and neurodegenerative diseases, such as Huntington’s disease, impair energy metabolism (36, 37). A resulting accumulation of lactate may contribute to acidosis. Interestingly, lactic acid is a particularly potent activator of some ASIC channels (38, 39). Cortical spreading depression, which has been linked to migraine, is also associated with acidosis (32, 40–42). The acidosis associated with these diseases could readily influence synaptic signaling and neuron function. Future challenges include better elucidating the mechanisms underlying the pH alterations, and better understanding of the physiological and pathophysiological consequences of low pH. At the molecular level, pH-sensitive ion channels may mediate, in large part, the physiological effects of acidosis and alkalosis at the synapse. 3 Acid Sensing Ion Channels (ASICs) 3.1 ASICs are Well Suited to Respond to Synaptic pH A number of synaptic channels are sensitive to pH modulation; including glutamate receptors, GABA-receptors, and voltage-gated Ca2+ channels (9–15, 43). These ion channels likely contribute to the physiological effects of fluctuating extracellular pH (9, 44). Unlike these other channels, in which pH modulates the response to a ligand or voltage, extracellular protons directly gate acid sensing ion channels (ASICs). ASICs are members of the DEG/ENaC family located at synapses where they may play an important role in synaptic signaling. They are preferentially permeable to Na+, but to a lesser extent can conduct other cations (Ca2+, K+, Li+, H+). Thus, ASICs may be well suited to mediate effects of acute pH changes at the synapse. 3.2 ASICs are Comprised of Multiple Subunits Five ASIC subunits have been identified in mammalian neurons: ASIC1a, -1b, -2a, - 2b, and -3, (a and b refer to alternatively spliced isoforms). The ASIC subunits combine to form both homomultimeric and heteromultimeric channel complexes (45–47). ASIC2b by itself does not form an acid-activated channel, but modifies currents from other acid-activated subunits (45). The related proteins, ASIC4 and BLINaC, share sequence homology with ASICs, but these mammalian proteins have not been shown to produce or modify acid-activated currents (48, 49), although an ASIC4 homolog in zebrafish is acid-activated (50). The recently determined crystal
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 665 structure of ASIC1a indicates a unique trimeric assembly (Fig. 2b), which suggests that three subunits are required to form a channel (55). Interestingly, previous studies using electrophysiological, biochemical, and fluorescence resonance energy transfer approaches had suggested that 4–9 subunits may combine to form a channel (51–54). Although the explanation for this difference is unknown, understanding the reason for the discrepancy may give the field insight into the structure and function of these channels. Fig. 2. Topology and trimeric assembly of ASICs. (a) ASIC subunits are comprised of two membrane-spanning domains, a large cysteine-rich extracellular domain, and several small conserved amino acid motifs (for recent reviews see (62–64)). Adapted with permission, from (62). (b) Trimeric assembly of ASIC/DEG/ENaC family proteins based on the recently determined crystal structure of chicken ASIC1a minus portions of the amino- and carboxy- termini (55). Individual ASIC1a subunits separated by color. 3.3 Subunit Composition Dictates Channel Properties Homomultimeric ASIC channels vary in activation and desensitization kinetics, and in pH sensitivity (Fig. 3a). For example, one study reported the half-maximal activation (pH0.5) of mouse ASIC subunits as 6.8 (ASIC1a), 6.2 (ASIC1b), 4.9 (ASIC2a), and 6.6 (ASIC3) (46). Slightly different values have also been reported (47), which may to some degree reflect differences between species, for example rat ASIC1a may be less sensitive (pH0.5 = 6.2–5.8) (47, 56) than mouse ASIC1a (46). Properties can also vary considerably when subunits are co-expressed to form heteromultimers. For example,
666 J.A. Wemmie et al. ASIC1a homomultimers are permeable to Ca2+, whereas when ASIC1a heteromultimerizes with ASIC2a the channel loses Ca2+ permeability (57). Also ASIC1a, -2a, and -3 heteromultimers desensitize faster than any of the individual subunits alone (Fig. 3) (46, 47). This result indicates that the properties of heteromultimeric channels are not simply the sum of properties from the individual subunits. Instead, subunits combine to confer new properties on channels. Fig. 3. Representative acid-evoked currents. (a) Currents evoked when indicated subunits were transfected into heterologous (COS-7) cells. Note the effects of subunit combination on rate of desensitization. (b) Acid-evoked current in hippocampal neurons cultured from mice of indicated genotypes. Disrupting ASIC1a eliminated pH 5-evoked current. Adapted from (62) with permission. 3.4 ASIC1a is Critical for Acid-Evoked Currents in the Brain In the brain, ASIC1a, -2a, and -2b are the most prominently expressed subunits (58–61). Of these, ASIC1a plays a critical role. Disrupting the ASIC1a gene eliminates current evoked by lowering extracellular pH to 5.0 in cultured hippocampal, cortical, and amygdala neurons (Fig. 3b). Conversely, disrupting ASIC2 slightly increased pH 5-evoked current (Fig. 3b) (61) (65–67). In addition, an antagonist specific for ASIC1a homomultimers (PcTx1, from tarantula venom) inhibited a significant proportion of the acid-activated current in cultured hippocampal and cortical neurons (60, 67, 68). Thus, much of the acid-activated current in the brain is mediated by ASIC1a homomultimers and ASIC1a-containing heteromultimers. Consequently, relative to the other ASICs, we currently know the most about ASIC1a in the brain.
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 667
3.5 Fluctuating Brain pH Could Activate or Desensitize ASIC1a
ASIC1a homomultimeric channels are activated as pH falls below 7.2, with a pH0.5 of
about 6.8–6.2 (46), (56). This pH sensitivity indicates that ASIC1a is well within the
range of acidosis recorded during both physiological and pathophysiological states.
Because ASIC currents are largely transient and densensitize quickly, some have
considered them best suited to respond to rapid, transient pH fluctuations such as
those thought to occur at the synapse. However, ASICs can also manifest a persistent
current (45, 46, 69), particularly when activated in the presence of modulatory
neuropeptides such as NPFF or FMRFamide (70). Moreover, it is possible that ASIC
activation could produce sustained effects in second messenger pathways, by raising
intracellular Ca2+, for example (57, 67, 71).
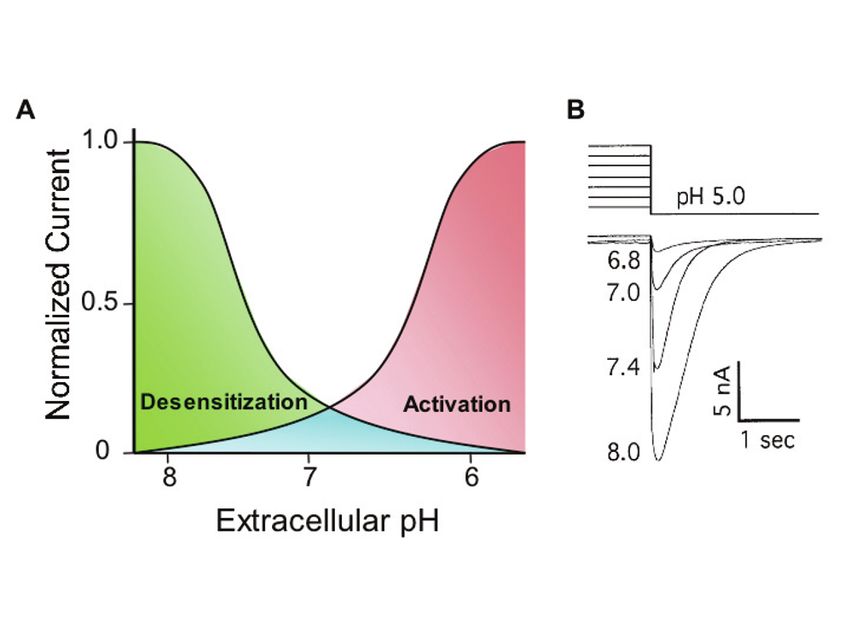
Another way that pH regulates ASICs is by desensitization; sustained exposure to
extracellular protons inactivates the channels. Desensitization begins at pH < 8.0 in
cultured sensory neurons, with half-maximal desensitization occurring at ~pH 7.2
(Fig. 4) (72). Thus, similar to the NMDA receptor, when extracellular pH is held at
Fig. 4. Extracellular pH regulates ASIC activation and steady-state inactivation (desensiti-
zation). (a) Illustration of desensitization and activation sensitivity to extracellular pH in
sensory neurons. Desensitization curve represents the effects of variations in sustained pH on
the subsequent pH 5-evoked current (protocol illustrated in b). Activation curve illustrates
current evoked by stepping from pH 8 to lower pH levels. Depending on the rate of recovery
from inactivation, the overlapping area beneath the two curves (blue) could provide a window
of steady state current; evidence from sensory neurons supports this possibility (68). (b)
Desensitization protocol. Due to channel desensitization, stepping from pH 7 to pH 5 evokes
smaller current than stepping from pH 7.4 to pH 5. Similarly, stepping from pH 8 to pH 5
evokes larger ASIC current. Adapted from (72) with permission.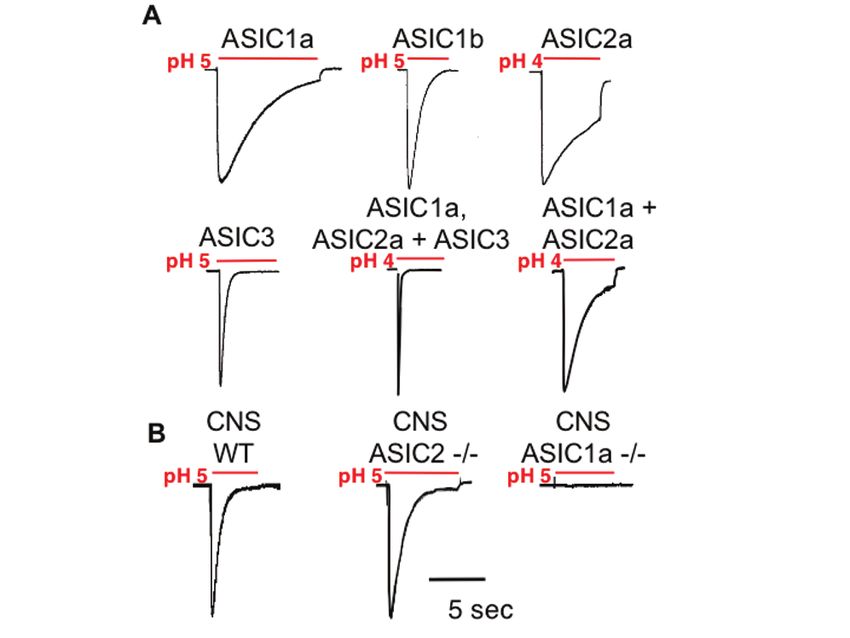
668 J.A. Wemmie et al. 7.35, a significant proportion of ASIC channels are tonically desensitized. As sustained pH becomes more alkaline, desensitization is reduced and transient acidosis evokes greater ASIC current. Similarly, as sustained pH falls, desensitization increases and transient acidosis evokes smaller ASIC current. These dynamic effects of pH raise the possibility that both acid and alkaline pH could regulate ASIC activity in vivo. Moreover, the effects of pH on channel activation and desensitization could generate seemingly paradoxical effects. For example, a generalized alkaline pH might increase the effect of a transient pH drop during neurotransmission. The degree of channel activity may also depend on the speed of onset, magnitude, and duration of the pH change, as well as the presence of modulators in the extracellular milieu (such as FMRFamide (70), Zn2+ (73, 74), lactate (38), arachidonic acid (75) and others (62)). However, because of this complexity, it is difficult to precisely predict the amount of ASIC channel activity in vivo. 4 ASIC1a Modulates Synapse Physiology 4.1 ASIC1a is Localized to Central Synapses Data from multiple experimental approaches place ASIC1a in dendritic spines. By subcellular fractionation, several studies found that ASIC1a is enriched in the synaptosome-containing brain fraction, suggesting a synaptic distribution (65, 76, 77). In cultured neurons, immunohistochemistry also detected endogenous and overexpressed ASIC1a protein in dendritic spines (65, 71, 76, 77). These studies also revealed ASIC1a to be present at puncta along the dendritic shaft and in the cell body (Fig. 5). The improved signal-to-noise ratio obtained from biolistically transfecting ASIC1a into organotypic hippocampal slices revealed a clearer picture of the protein’s distribution (Fig. 5) (71). Using this method, ASIC1a was visualized in almost all dendritic spines, and was clustered in many spine heads. This localization pattern places ASIC1a at the postsynaptic membrane, making it a good candidate for sensing protons there. 4.2 ASIC1a is Not Abundant in Axons Unlike the somatodendritic distribution of ASIC1a, there is controversy about its presence in axons. One study reported ASIC1a immunostaining in axon-like branches of cultured CNS neurons (77). In contrast, others failed to detect endogenous ASIC1a in axons (65). In addition, those studies failed to detect overexpressed ASIC1a in tau-positive branches of cultured neurons. Moreover, no detectable ASIC1a immunofluorescence was seen in axons from biolistically transfected slice neurons co-labeled with an axonal marker, VAMP2/synaptobrevin (71). Together, these data do not rule out the presence of ASIC1a in axons, but suggest it is probably not abundant there. The subcellular distribution of other ASIC subunits in brain neurons remains unclear, although ASIC2 has been suggested to localize to synapses in the cerebellum (78) and the retina (79).
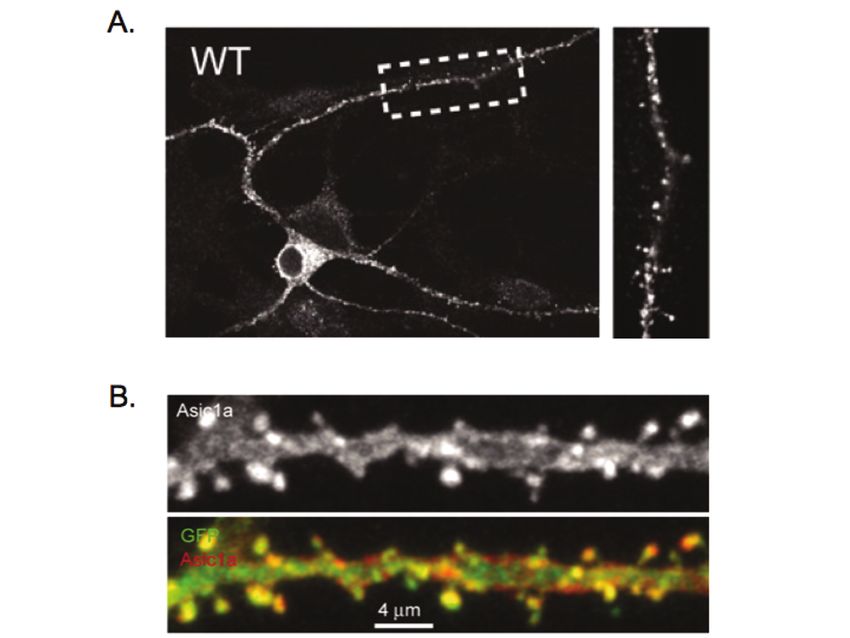
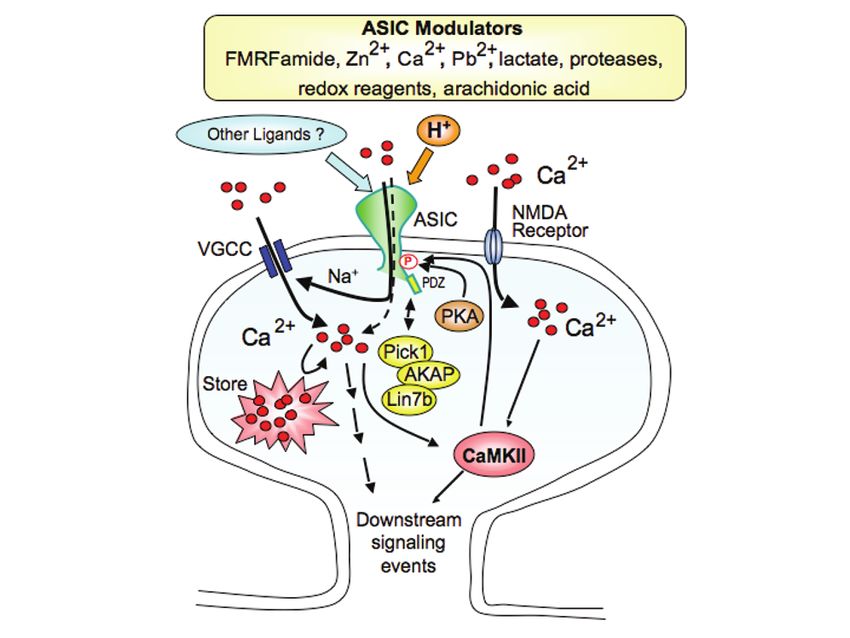
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 669 Fig. 5. ASIC1a is localized to dendritic spines. (a) Immunofluorescence (IF) of endogenous ASIC1a in dissociated cortical neurons. Shown is a cortical neuron from a wild-type culture. ASIC1a IF is visible in the cell body and dendrites. Little or no fluorescence was observed in cultures from ASIC1a knockout mice (71). Right is the high-magnification view of the boxed area. (b) IF of ASIC1a in transfected hippocampal slice neurons. Shown is an enlarged view of an apical dendrite of a CA1 pyramidal neuron. Top panel shows ASIC1a IF. Bottom panel is the merged image of ASIC1a (red) and eGFP (green). In both endogenous staining and in hippocampal slices, ASIC1a IF presents in a clustered pattern in spines and dendrites. Reproduced with permission from (71). 4.3 ASIC1a Associates with Post-Synaptic Scaffolding Proteins Consistent with the speculation that ASIC1a functions in the postsynaptic membrane, ASIC1a associates with at least two synaptic scaffolding proteins, PICK1 and AKAP150 (Fig. 6) (80–82). Both PICK1 and AKAP150 interact with ASIC1a biochemically and colocalize with ASIC1a in neurons. In addition to these interactions, ASIC1a co-localizes with PSD-95 in dendritic spines (65, 76). This observation is intriguing, although there is currently no biochemical evidence for a direct interaction between ASIC1a and PSD-95. However, other ASIC family members interact with PSD-95 (83), raising the possibility of an indirect association between ASIC1a and PSD-95. Functionally, ASIC1a associates with several other synaptic signaling molecules including calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA) and NMDA receptors (71, 82, 84, 85). In mice, disrupting ASIC1a reduced CaMKII phosphorylation, and overexpressing ASIC1a
670 J.A. Wemmie et al. increased CaMKII phosphorylation (71). Similarly, CaMKII-dependent phosphory- lation affected ASIC1a function; in response to NMDA receptor activation, CaMKII phosphorylated the intracellular C-terminus of human ASIC1a at serines 478 and 479, which potentiated proton-elicited current and acid-induced neuron damage (84). Hippocampal slice recordings also suggest a functional interaction between ASIC1a and the NMDA receptor (Section 4.5). PKA also phosphorylates mouse ASIC1a at serine 478 and disrupts its interaction and co-localization with PICK1 in heterologous cells (85). This event is likely to be functionally important, because PKA binding with AKAP150 reduced ASIC1a current in heterologous cells and in cultured neurons (82). Together, these studies implicate ASIC1a in synaptic signaling networks. Fig. 6. Model of ASICs and interacting molecules at the synapse. ASICs are activated by protons, possibly from pre-synaptic release or other sources. Additional molecules can modify acid-evoked ASIC currents, suggesting the possibility of other ligands not yet identified (62). Upon activation, ASIC1a increases intracellular calcium [Ca2+]i, largely through voltage-gated calcium channels (VGCCs) and the release of intracellular Ca2+ stores (71). The resulting rise in [Ca2+]i initiates downstream signaling events, including CaMKII activation. ASIC1a channel activity may be regulated by interactions with CaMKII, PKA, AKAP150, calcineurin, PICK1 and possibly other PDZ-domain proteins (81–85). 4.4 ASIC1a Confers Acid Sensitivity to Dendritic Spines The presence of ASIC1a at dendritic spines raised the question about its physiological effects there. To explore this question, one study tested whether
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 671
ASIC1a influenced the intracellular Ca2+ concentration ([Ca2+]i) at dendritic spines
by transfecting a ratiometric Ca2+ reporter, cameleon YC3.60, into hippocampal slices
(71). Acute acid application induced [Ca2+]i transients in wild-type spines (Fig. 7).
Knocking down ASIC1a with siRNA attenuated the number of acid-sensitive spines.
Conversely ASIC1a overexpression increased the number of acid-sensitive spines.
The Ca2+ signal was also observed in the dendritic shaft and the cell body. These data
combined with the ASIC1a localization indicate that ASIC1a confers acid sensitivity
to dendritic spines and that ASIC1a can function as a postsynaptic proton receptor.
ASIC1a homomultimeric channels conduct both Na+ and Ca2+, thus channel
activation could increase [Ca2+]i directly, or indirectly through voltage-gated Ca2+
channels (VGCC) and by releasing from intracellular Ca2+ stores. In heterologous
cells, Ca2+ influx through ASIC1a is responsible for acid-induced increase in [Ca2+]i
(57). In contrast, in hippocampal slices, direct Ca2+ influx through ASIC1a made
only a small contribution; most of the [Ca2+]i increase came from VGCCs and from
intracellular stores (71). These observations illustrate the multiple mechanisms by
which ASIC1a may influence [Ca2+]i at synapses.
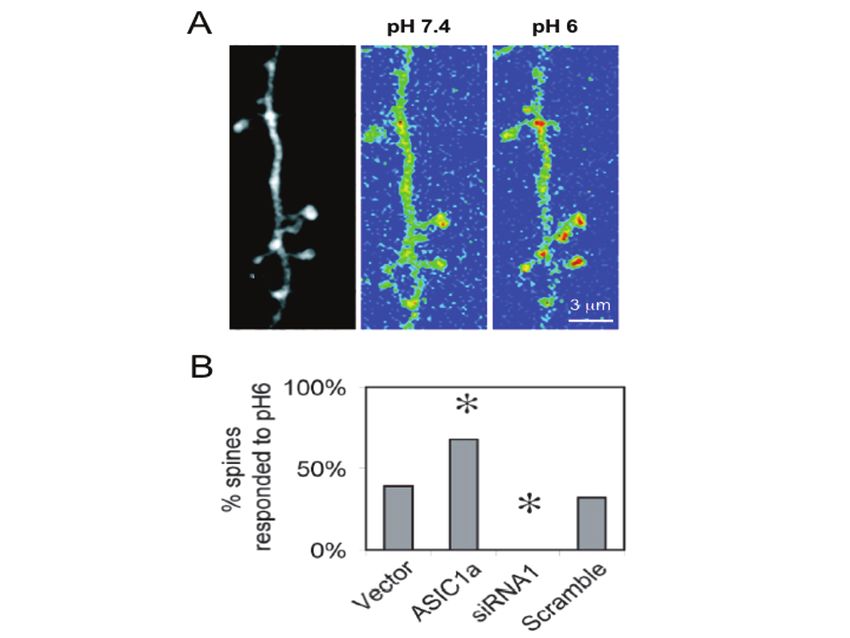
Fig. 7. ASIC1a confers pH sensitivity to dendritic spines. (a) Image on left shows a segment
of an apical dendrite from a neuron cotransfected with ASIC1a and cameleon. Two images on
the right show the YFP/CFP fluorescence ratio (blue indicates a low ratio and red indicates a
high ratio) obtained at baseline (pH 7.4) and during acid stimulation (pH 6). Note that pH 6
increased Ca2+ levels in most spines. (b) Acid-induced calcium increase in spines was
ASIC1a-dependent. ASIC1a overexpression increased while ASIC1a siRNA reduced the % of
spines responding to acid stimulation. Adapted from (71) with permission.672 J.A. Wemmie et al. 4.5 Disrupting ASIC1a Impairs Synaptic Transmission and Plasticity The pH changes at the synapse, the subcellular distribution of ASIC1a, and its effects on dendritic [Ca2+], hinted that ASIC1a may play an important role in synaptic physiology and plasticity. Results from ASIC1a knockout mice support this possibility. An analysis of synaptic function in hippocampal slices found that long term potentiation (LTP) evoked by Schaffer collateral fiber stimulation, was impaired by the loss of ASIC1a (65). High frequency stimulation (HFS-100 Hz) produced potentiation in wild-type mice lasting beyond 40 minutes. However, within this time frame, potentiation of excitatory post-synaptic potentials (EPSP) decayed to baseline in ASIC1a knockout slices. Prior to LTP induction, paired-pulse facilitation and single evoked EPSPs were normal, suggesting ASIC1a disruption did not impair presynaptic vesicle release. However, loss of ASIC1a attenuated the summation of EPSPs during the HFS. Interestingly, the NMDA receptor antagonist D-APV inhibited EPSP summation in wild-type slices but not in ASIC1a-null slices suggesting that the loss of ASIC1a impaired NMDA-receptor function. These data support the possible connection between ASIC1a and the NMDA receptor, however the mechanism for the suggested crosstalk remains uncertain. 4.6 ASICs at Retinal Synapses Glutamate release at retinal ribbon synapses is accompanied by acidosis, raising the possibility that ASICs could affect neurotransmission in the retina. Recent studies investigated this possibility and found ASIC1a protein located in the retina near the synaptic cleft in bipolar cells. ASIC1a-antisense RNA and PcTx1 significantly decreased a- and b-electroretinogram measurements, supporting the idea that ASIC1a contributes to synaptic signaling in the retina (86). While ASIC1a inhibition decreased electroretinogram waves, knocking out ASIC2 increased the a- and b- electroretinogram waves and caused light-induced retinal degeneration (79). Thus, disrupting ASIC2 produced an effect essentially opposite to that of disrupting ASIC1a. Further studies are needed to better understand the roles of ASIC1a and ASIC2 at the retinal synapse. However, the retina may be an excellent location for studying synaptic ASICs because of large size of retinal synapses and the proton release recorded there (6–8). 4.7 Model of ASIC1a Function at the Synapse The available data support a model of ASIC1a at the synapse (Figs. 1 and 6). In the post-synaptic membrane, ASICs seem well positioned to respond to protons released from presynaptic neurotransmitter (NT)-containing vesicles and also from other sources. In this model, various extracellular modulators and intracellular ASIC- interacting proteins might influence the response, which would be expected to depolarize the membrane potential and raise intracellular Ca2+ concentration, perhaps influencing other receptors and signaling proteins. An important prediction from this model is that ASIC currents will be activated during neurotransmission. However, thus far, studies in brain slices (65) and cultured neurons (77) have not detected
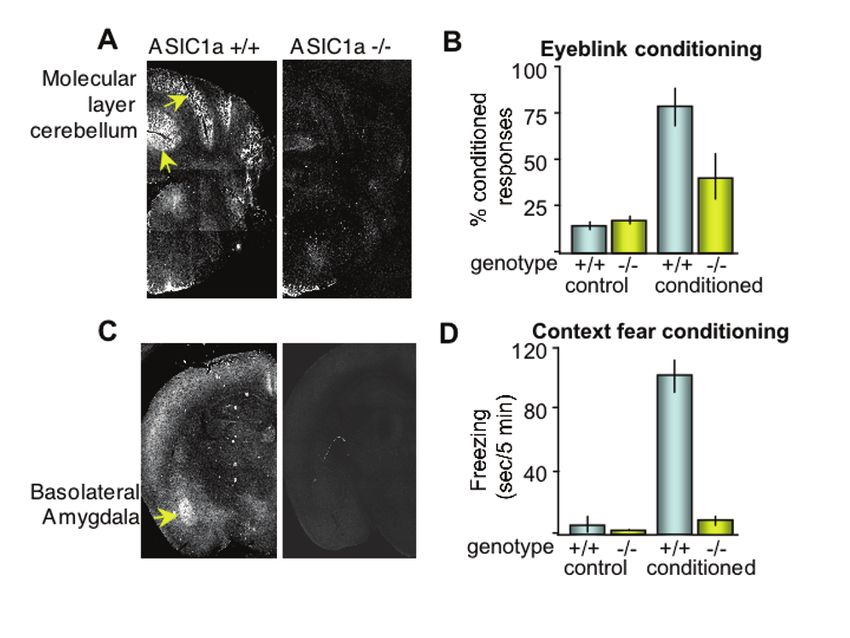
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 673 them. There are many possible explanations for these negative results, but they hint that the story could be more complex. Perhaps the appropriate experimental conditions have not been identified. Interestingly however, a recent study discovered spontaneous ASIC activation in HEK293 cells (87). Presumably, protons released by vesicles evoked the ASIC current, because blocking vesicle acidification with bafilomycin prevented the spontaneous currents. This finding represents a potentially important step in identifying how endogenous ASICs may be activated. 4.8 ASIC1a Affects Synaptic Spine Density Regardless of how ASIC1a acts at the synapse, evidence for some potentially important consequences has been obtained. For example, it was recently found that knocking down ASIC1a expression with RNAi in hippocampal slice neurons reduced the number of dendritic spines (71). An ASIC1a dominant-negative construct had a similar effect. In contrast, overexpressing ASIC1a in CA1 neurons had the opposite effect and increased the number of dendritic spines. Possibly, the ability of ASIC1a to affect spine number is related to its effects on synaptic [Ca2+] (57, 67, 71, 84) and LTP (65), although the precise mechanisms have not yet been established. Nevertheless, these observations suggest ASICs may influence spine remodeling in disease. Unlike the effects of acutely changing ASIC1a expression, disrupting ASIC1a throughout development in the knockout mice did not change spine number, suggesting developmental mechanisms might compensate for some effects of ASIC1a disruption (71). Together, these functional effects on synaptic physiology suggest that genetically or pharmacologically altering ASIC1a might have important physiological and behavioral consequences. 5 ASIC1a Regulates Behavior and May Contribute to Neurological disease 5.1 Disrupting ASIC1a Impaired Cerebellum-Dependent Learning The location of ASIC1a in mouse brain provided important clues about the potential contribution of the channel to behavior. By immunohistochemistry and protein blotting, it was found that ASIC1a protein was enriched in gray matter and in brain regions with high synaptic density (65, 66, 88). One region where ASIC1a was abundant was the molecular layer of the cerebellar cortex (Fig. 8a) (65, 66). In addition, Purkinje cells which populate this layer of the cerebellum have large acid- evoked currents (75). Consistent with a role in cerebellum-dependent learning, the loss of ASIC1a significantly impaired classical delay eyeblink conditioning (Fig. 8b) (65). Mice normally learn that a tone predicts a periorbital shock and acquire the ability to defensively blink before the shock occurs (i.e. a conditioned response). After 10 days of training, wild-type mice produced conditioned responses in more than 80% of trials, whereas the ASIC1a-null mice produced a conditioned response in only 40% of trials. Disrupting ASIC1a did not alter shock sensitivity (65), hearing (88), or the blink reflex (65), suggesting a learning-related deficit. Although the
674 J.A. Wemmie et al. mechanism has not been determined, the loss of ASIC1a may have impaired plasticity in the cerebellar cortex. Fig. 8 . Localization of ASIC1a in brain structures underlying Pavlovian conditioning. (a) Immunofluorescent labeling of ASIC1a protein in ASIC1a+/+ relative to ASIC1a–/– mice revealed abundant ASIC1a expression in the molecular layer of the cerebellar cortex (arrowheads). (b) Consistent with a role in cerebellum-dependent learning, loss of ASIC1a significantly impaired delay eyeblink conditioning. When the tone and shock were not paired (control), neither group developed conditioned responses. (c) In the forebrain, ASIC1a immunolabeling was particularly abundant in the basolateral amygdala of wild-type mice (arrowheads). (d) Suggesting a role in amygdala-dependent learning, ASIC1a–/– disruption severely impaired single-shock context fear conditioning. Twenty-four hours after receiving a single 0.5 mA footshock, wild-type mice froze when returned to the training chamber. In contrast, ASIC1a-null mice froze very little. Mice not receiving a footshock during training (control) froze little or none during testing. Panels (a), (b), and (c) reproduced with permission from (65, 66, 88). 5.2 ASIC1a Contributes to Pavlovian Fear Conditioning Outside of the cerebellum, ASIC1a is particularly abundant in structures that underlie fear behaviors including the amygdala complex, bed nucleus of the stria terminalis, lateral hypothalamus, habenula, cingulate cortex, and periacqueductal gray (56, 59, 66, 77, 88, 89). In the amygdala, multiple nuclei express ASIC1a including the lateral, central, and medial nuclei, although ASIC1a immuno-labeling was especially pronounced in the basolateral amygdala (88, 89).
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 675
The distribution of ASIC1a in fear circuit structures and the effects of ASIC1a on
synaptic function, led to the question of whether ASIC1a influences Pavlovian fear
conditioning. Wild-type and ASIC1a-null mice were tested in their ability to learn
the association between a fear-producing footshock and either the environmental
context or an audible tone (66). Disrupting ASIC1a significantly impaired both
context fear conditioning (Fig. 8d) and tone conditioning (66). Conversely,
overexpressing ASIC1a in the mouse brain increased fear conditioning (76). The
effects were not due to sensory dysfunction because both hearing (88) and footshock
sensitivity (66) were unaffected by ASIC1a gene disruption.
5.3 Interrupting ASIC1a Reduces Unconditioned Fear
Effects of ASIC1a on unconditioned fear responses have also been tested (67). The
loss of ASIC1a reduced unconditioned fear in the open field test, during acoustic
startle, and in response to predator odor. Curiously, the loss of ASIC1a did not
reduce undonditioned fear in the elevated plus maze, suggesting that this fear
behavior is not ASIC1a-dependent (65). The effects of ASIC1a disruption on
unconditioned fear are probably not due to developmental abnormalities, because
acutely inhibiting ASIC1a in the brain post-developmentally with PcTx1 venom
reduced TMT-evoked fear in wild-type mice, but not in ASIC1a-null mice (67).
The observation that ASIC1a contributes to fear learning, suggests that ASIC1a
contributes to plasticity in the fear circuit. In addition, the finding that ASIC1a
affects unconditioned fear suggests that ASIC1a can regulate fear circuit activity
independent of long-lasting plasticity. These effects of ASIC1a resemble those of
AMPA and NMDA receptors, the inhibition of which reduces both fear learning and
the expression of learned fear. However, further studies are needed to better
undestand how ASIC1a affects neurotransmission in the amygdala and elsewhere in
the fear circuit. Nevertheless, the ability of ASIC1a inhibition to reduce multiple fear
behaviors, raises the possibility that ASIC1a antagonists could reduce anxiety in
humans and might benefit other psychiatric diseases. Anti-anxiety medications with
novel mechanisms of action could offer advantages over current medications, which
can cause a number of unwanted side effects.
5.4 Ischemic Stroke
Central acidosis exacerbates neurodegenerative diseases including ischemic stroke.
Because Ca2+ overload causes toxicity in the ischemic brain, and because ASIC1a
activation raises intracellular Ca2+, it was hypothesized that ASIC1a activation might
increase ischemic cell death. Enhancement of ASIC1a function by activation of
NMDA receptors (84), lack of oxygen or glucose (67), or accumulation of lactate or
arachidonic acid (75) might further exacerbate ASIC1a-mediated neurotoxicity. A
possible role for ASIC1a in ischemic toxicity was initially supported by experiments
in cells heterologously expressing ASIC1a and in cultured hippocampal neurons
(57). Neurons lacking ASIC1a and cells treated with amiloride or PcTx1 resisted
acidosis-induced injury (57, 67). Moreover, PcTx1 diminished the effects of NMDA-
induced cell death (67). Establishing a strong case for a role in ischemic disease,676 J.A. Wemmie et al. disrupting ASIC1a in mice reduced infarct volume by 60% following experimental stroke due to middle cerebral artery (MCA) occlusion (67). Interestingly, pH remains low for hours after a stroke, thus opening a window of therapeutic opportunity. Even administering PcTx1 five hours after MCA occlusion reduced infarct volume by >50% (90). These encouraging results endorse ASIC1a inhibitors as a potential new treatment for stroke. 5.5 Central Pain Regulation In peripheral neurons, ASICs contribute to acid-evoked nociception (reviewed in (62)). Recently, ASICs in the central nervous system (CNS) were also implicated in pain control (91). Inhibiting ASIC1a in the CNS by injecting PcTx1 or ASIC1a anti- sense RNA into the cerebrospinal fluid reduced inflammatory and neuropathic pain. The analgesic effects were linked to elevated met-enkephalin levels. The possible relationship between ASIC1a function at the synapse and met-enkephalin expression is not yet clear, although it is interesting to speculate that blocking ASIC1a could attenuate previously observed anti-opiod effects of FMRFamide related peptides (92). Thexse observations suggest a promising opportunity for targeting ASICs in pain control. 6 Concluding Remarks The identification of ASIC1a as synaptic proton receptors and an increasing appreciation of pH dynamics in the CNS suggest new possibilities for proton mediated cellular signaling. Although much remains to be learned about the ASICs, the available data indicate an intriguing role in synapse function. Perhaps like a neurotransmitter, protons cross the synapse to activate or modulate ASICs and other proteins. In addition, at the cell surface and at synapses, ASICs may be readily accessible to pharmacological modification, suggesting fertile ground for novel drug discovery. Studies in mice point to possible therapeutic benefits of ASIC1a antagonists in psychiatric disease, neurodegeneration, and pain. Acknowledgements We thank Theresa Mayhew and Ashley Small for assistance preparing the manuscript, and Kelsey Coulter for help preparing the figures. We also thank Drs. Chris Benson, Candice Askwith, and Mikael Schnizler for examples of electrophysiological traces. JW is supported by a Department of Veteran’s Affairs Advanced Career Development Award, a NARSAD young investigator award, and a beginning grant in aid award from the American Heart Association. XZ is an Associ- ate and MJW is an Investigator of the Howard Hughes Medical Institute.
Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 677
References
1. Kaila, K. and B.R. Ransom, Concept of pH and its importance in neurobiology, in pH
and Brain Function, K. Kaila and B.R. Ransom, Editors. 1998, Wiley-Liss, Inc. p. 1–10.
2. Morel, N., Neurotransmitter release: the dark side of the vacuolar-H+ATPase. Biol.
Cell., 2003. 95(7): p. 453–457.
3. Fuldner, H.H. and H. Stadler, 31P-NMR analysis of synaptic vesicles. Status of ATP and
internal pH. Eur. J. Biochem., 1982. 121(3): p. 519–524.
4. Michaelson, D.M. and I. Angel, Determination of delta pH in cholinergic synaptic
vesicles: its effect on storage and release of acetylcholine. Life Sci., 1980. 27(1): p.
39–44.
5. Miesenbock, G., D.A. De Angelis, and J.E. Rothman, Visualizing secretion and synaptic
transmission with pH-sensitive green fluorescent proteins. Nature, 1998. 394: p.
192–195.
6. DeVries, S.H., Exocytosed protons feedback to suppress the Ca(2+) current in
mammalian cone photoreceptors. Neuron, 2001. 32(6): p. 1107–1117.
7. Vessey, J.P., et al., Proton-mediated feedback inhibition of presynaptic calcium channels
at the cone photoreceptor synapse. J. Neurosci., 2005. 25(16): p. 4108–4117.
8. Palmer, M.J., et al., Synaptic cleft acidification and modulation of short-term depression
by exocytosed protons in retinal bipolar cells. J. Neurosci., 2003. 23(36): p.
11332–11341.
9. Traynelis, S.F., pH modulation of ligand-gated ion channels, in pH and Brain Function,
K. Kaila and B.R. Ransom, Editors. 1998, Wiley-Liss, Inc.
10. Traynelis, S.F. and S.G. Cull-Candy, Proton inhibition of N-methyl-D-aspartate
receptors in cerebellar neurons. Nature, 1990. 345(6273): p. 347–350.
11. Vyklicky, L.J., V. Vlachova, and J. Krusek, The effect of external pH changes on
responses to excitatory amino acids in mouse hippocampal neurones. J. Physiol. (Lond),
1990. 430: p. 497–517.
12. Tang, C.M., M. Dichter, and M. Morad, Modulation of the N-methyl-D-aspartate
channel by extracellular H+. Proc. Natl. Acad. Sci. U. S. A., 1990. 87(16): p. 6445–6449.
13. Ihle, E.C. and D.K. Patneau, Modulation of ∂-amino-3-hydroxy-5-methyl-4-
isoxazolepropionic acid receptor desensitization by extracellular protons. Mol.
Pharmacol., 2000. 58(6): p. 1204–1212.
14. Pasternack, M., S. Smirnov, and K. Kaila, Proton modulation of functionally distinct
GABAA receptors in acutely isolated pyramidal neurons of rat hippocampus.
Neuropharmacology, 1996. 35(9–10): p. 1279–1288.
15. Huang, R.Q., Z. Chen, and G.H. Dillon, Molecular basis for modulation of recombinant
alpha1beta2gamma2 GABAA receptors by protons. J. Neurophysiol., 2004. 92(2): p.
883–894.
16. Chesler, M., Regulation and modulation of pH in the brain. Physiol. Rev., 2003. 83(4): p.
1183–1221.
17. Kaila, K. and M. Chesler, Activity-evoked changes in extracellular pH, in pH and Brain
Function, K. Kaila and B.R. Ransom, Editors. 1998, Wiley-Liss, Inc. p. 309.
18. Fedirko, N., et al., Regulation of postsynaptic Ca2+ influx in hippocampal CA1
pyramidal neurons via extracellular carbonic anhydrase. J. Neurosci., 2007. 27(5): p.
1167–1175.
19. Makani, S. and M. Chesler, Endogenous alkaline transients boost postsynaptic NMDA
receptor responses in hippocampal CA1 pyramidal neurons. J. Neurosci., 2007. 27(28):
p. 7438–7446.
20. Kaila, K. and J. Voipio, Postsynaptic fall in intracellular pH induced by GABA-activated
bicarbonate conductance. Nature, 1987. 330(6144): p. 163–165.678 J.A. Wemmie et al.
21. Kaila, K., J. Saarikoski, and J. Voipio, Mechanism of action of GABA on intracellular
pH and on surface pH in crayfish muscle fibres. J. Physiol., 1990. 427: p. 241–260.
22. Schwiening, C.J., H.J. Kennedy, and R.C. Thomas, Calcium-hydrogen exchange by the
plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc. R. Soc. Lond.
Biol. Sci., 1993. 253(1338): p. 285–289.
23. Schwiening, C.J. and R.C. Thomas, pH consequences of calcium regulation, in pH and
Brain Function, K. Kaila and B. Ransom, Editors. 1998, Wiley-Liss, Inc. p. 277.
24. Thomas, R.C., Proton channels in snail neurones. Does calcium entry mimic the effects
of proton influx? Ann. N. Y. Acad. Sci., 1989. 574: p. 287–293.
25. Chen, J.C. and M. Chesler, pH transients evoked by excitatory synaptic transmission are
increased by inhibition of extracellular carbonic anhydrase. Proc. Natl. Acad. Sci. U. S.
A., 1992. 89(16): p. 7786–7790.
26. Somjen, G.G. and G.C. Tombaugh, pH modulation of neuronal excitability and central
nervous system functions, in pH and Brain Function, K. Kaila and B.R. Ransom,
Editors. 1998, Wiley-Leiss Inc: New York. p. 373–393.
27. Taira, T., et al., Relative contributions of excitatory and inhibitory neuronal activity to
alkaline transients evoked by stimulation of Schaffer collaterals in the rat hippocampal
slice. J. Neurophysiol., 1995. 74(2): p. 643–649.
28. Chesler, M. and R.P. Kraig, Intracellular pH of astrocytes increases rapidly with cortical
stimulation. Am. J. Physiol., 1987. 253(4 Pt 2): p. R666–R6670.
29. Chesler, M. and R.P. Kraig, Intracellular pH transients of mammalian astrocytes. J.
Neurosci., 1989. 9(6): p. 2011–2019.
30. Pellerin, L., et al., Activity-dependent regulation of energy metabolism by astrocytes: An
update. Glia, 2007. 55(12): p. 1251–1262.
31. Voipio, J. and K. Kaila, Interstitial PCO2 and pH in rat hippocampal slices measured by
means of a novel fast CO2/H(+)-sensitive microelectrode based on a PVC-gelled
membrane. Pflugers Arch., 1993. 423(3–4): p. 193–201.
32. Somjen, G.G., Acidification of interstitial fluid in hippocampal formation caused by
seizures and by spreading depression. Brain Res., 1984. 311(1): p. 186–188.
33. Siesjo, B.K., et al., Extra- and intracellular pH in the brain during seizures and in the
recovery period following the arrest of seizure activity. J. Cereb. Blood Flow Metab.,
1985. 5(1): p. 47–57.
34. Siesjo, B.K., Acidosis and ischemic brain damage. Neurochem. Pathol., 1988. 9: p.
31–88.
35. Wang, R.I.H. and S. R.R., PH of cerebral cortex during induced convulsions. J
Neurophysiol., 1955. 18(2): p. 130–137.
36. Waxman, S.G., Axonal conduction and injury in multiple sclerosis: the role of sodium
channels. Nat. Rev. Neurosci., 2006. 7(12): p. 932–941.
37. Browne, S.E. and M.F. Beal, The energetics of Huntington's disease. Neurochem. Res.,
2004. 29(3): p. 531–546.
38. Immke, D.C. and E.W. McCleskey, Lactate enhances the acid-sensing Na+ channel on
ischemia-sensing neurons. Nat. Neurosci., 2001. 4(9): p. 869–870.
39. Immke, D.C. and E.W. McCleskey, Protons open acid-sensing ion channels by
catalyzing relief of Ca2+ blockade. Neuron, 2003. 37(1): p. 75–84.
40. Parsons, A.A. and P.J. Strijbos, The neuronal versus vascular hypothesis of migraine and
cortical spreading depression. Curr. Opin. Pharmacol., 2003. 3(1): p. 73–77.
41. Sanchez-Del-Rio, M., U. Reuter, and M.A. Moskowitz, New insights into migraine
pathophysiology. Curr. Opin. Neurol., 2006. 19(3): p. 294–298.
42. Csiba, L., W. Paschen, and G. Mies, Regional changes in tissue pH and glucose content
during cortical spreading depression in rat brain. Brain Res., 1985. 336(1): p. 167–170.Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 679
43. Tombaugh, G.C. and G.G. Somjen, Effects of extracellular pH on voltage-gated Na+, K+
and Ca2+ currents in isolated rat CA1 neurons. J. Physiol., 1996. 493(Pt 3): p.
719–732.
44. Tombaugh, G.C. and G.G. Somjen, pH modulation of voltage-gated ion channels, in pH
and Brain Function, K. Kaila and B.R. Ransom, Editors. 1998, Wiley-Liss, Inc. p. 395.
45. Lingueglia, E., et al., A modulatory subunit of acid sensing ion channels in brain and
dorsal root ganglion cells. J. Biol. Chem., 1997. 272(47): p. 29778–29783.
46. Benson, C.J., et al., Heteromultimerics of DEG/ENaC subunits form H+-gated channels
in mouse sensory neurons. Proc. Natl. Acad. Sci. U. S. A., 2002. 99(4): p. 2338–2343.
47. Hesselager, M., D.B. Timmermann, and P.K. Ahring, pH-dependency and
desensitization kinetics of heterologously expressed combinations of ASIC subunits. J.
Biol. Chem., 2004. 279(12): p. 11006–11015.
48. Akopian, A.N., et al., A new member of the acid-sensing ion channel family.
Neuroreport, 2000. 11(10): p. 2217–2222.
49. Sakai, H., et al., Cloning and functional expression of a novel degenerin-like Na+
channel gene in mammals. J. Physiol. (Lond), 1999. 519(2): p. 323–333.
50. Paukert, M., et al., A family of acid-sensing ion channels from the zebrafish: widespread
expression in the central nervous system suggests a conserved role in neuronal
communication. J. Biol. Chem., 2004. 279(18): p. 18783–18791.
51. Gao, Y., et al., Fluorescence resonance energy transfer analysis of subunit assembly of
the ASIC channel. Biochem. Biophys. Res. Commun., 2007. 359(1): p. 143–150.
52. Snyder, P.M., et al., Electrophysiological and biochemical evicence that DEG/ENaC
cation channels are composed of nine subunits. J. Biol. Chem., 1998. 273(2): p. 681–684.
53. Firsov, D., et al., The heterotetrameric architecture of the epithelial sodium channel
(ENaC). EMBO J., 1998. 17(2): p. 344–352.
54. Eskandari, S., et al., Number of subunits comprising the epithelial sodium channel. J.
Biol. Chem., 1999. 274(38): p. 27281–27286.
55. Jasti, J., et al., Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH.
Nature, 2007. 449(7160): p. 316–323.
56. Waldmann, R., et al., A proton-gated cation channel involved in acid-sensing. Nature,
1997. 386: p. 173–177.
57. Yermolaieva, O., et al., Extracellular acidosis increases neuronal cell calcium by
activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U. S. A., 2004. 101(17): p.
6752–6757.
58. Waldmann, R., Proton-gated cation channels–neuronal acid sensors in the central and
peripheral nervous system. Adv. Exp. Med. Biol., 2001. 502: p. 293–304.
59. García-Añoveros, J., et al., BNaC1 and BNaC2 constitute a new family of human
neuronal sodium channels related to degenerins and epithelial sodium channels. Proc.
Natl. Acad. Sci. U. S. A., 1997. 94: p. 1459–1464.
60. Baron, A., R. Waldmann, and M. Lazdunski, ASIC-like, proton-activated currents in rat
hippocampal neurons. J. Physiol., 2002. 539(Pt 2): p. 485–494.
61. Askwith, C.C., et al., ASIC2 modulates ASIC1 H+-activated currents in hippocampal
neurons. J. Biol. Chem., 2003. 279(18): p. 18296–18305.
62. Wemmie, J.A., M.P. Price, and M.J. Welsh, Acid-sensing ion channels: advances,
questions and therapeutic opportunities. Trends Neurosci., 2006. 29(10): p. 578–586.
63. Krishtal, O., The ASICs: signaling molecules? Modulators? Trends Neurosci., 2003.
26(9): p. 477–483.
64. Lingueglia, E., E. Deval, and M. Lazdunski, FMRFamide-gated sodium channel and
ASIC channels: a new class of ionotropic receptors for FMRFamide and related peptides.
Peptides, 2006. 27(5): p. 1138–1152.680 J.A. Wemmie et al.
65. Wemmie, J.A., et al., The acid-activated ion channel ASIC contributes to synaptic
plasticity, learning, and memory. Neuron, 2002. 34: p. 463–477.
66. Wemmie, J.A., et al., Acid-sensing ion channel 1 is localized in brain regions with high
synaptic density and contributes to fear conditioning. J. Neurosci., 2003. 23(13):
p. 5496–5502.
67. Xiong, Z.G., et al., Neuroprotection in ischemia: blocking calcium-permeable acid-
sensing ion channels. Cell, 2004. 118(6): p. 687–698.
68. Coryell, M.W., Ziemann, A.E., Westmoreland, P.J., Haenfler, J.M., Kurjakovic, Z., Zha,
X.M., Price, M., Schinzler, M.K., Wemmie, J.A. Targeting ASIC1a reduces innate fear and
alters neuronal acitivity in the fear circuit. Biol. Psychiatry., 2007. 62 (10): P. 1140–1148.
69. Yagi, J., et al., Sustained currents through ASIC3 ion channels at the modest pH changes
that occur during myocardial ischemia. Circ. Res., 2006. 99(5): p. 501–509.
70. Askwith, C.C., et al., Neuropeptide FF and FMRFamide potentiate acid-evoked currents
from sensory neurons and proton-gated DEG/ENaC channels. Neuron, 2000. 26: p.
133–141.
71. Zha, X.-M., J.A. Wemmie, and M.J. Welsh, ASIC1a is a postsynaptic proton receptor
that influences the density of dendritic spines. Proc. Natl. Acad. Sci. U. S. A., 2006.
103(44): p. 16556–16561.
72. Benson, C.J., S.P. Eckert, and E.W. McCleskey, Acid-evoked currents in cardiac sensory
neurons: a possible mediator of myocardial ischemic sensation. Circ. Res., 1999. 84(8):
p. 921–928.
73. Chu, X.P., et al., Subunit-dependent high-affinity zinc inhibition of acid-sensing ion
channels. J. Neurosci., 2004. 24(40): p. 8678–8689.
74. Baron, A., et al., Zn2+ and H+, coactivators of acid sensing ion channels (ASIC). J. Biol.
Chem., 2001. 276: p. 35361–35367.
75. Allen, N.J. and D. Attwell, Modulation of ASIC channels in rat cerebellar Purkinje
neurons by ischaemia-related signals. J. Physiol., 2002. 543(2): p. 521–529.
76. Wemmie, J., Coryell M, Askwith C, Lamani E, Leonard S, Sigmund C, Welsh M,
Overexpression of acid-sensing ion channel 1a in transgenic mice increases fear-related
behavior. Proc. Natl. Acad. Sci. U. S. A., 2004. 101(10): p. 3621–3626.
77. Alvarez de la Rosa, D., et al., Distribution, subcellular localization and ontogeny of
ASIC1 in the mammalian central nervous system. J. Physiol., 2003. 546(Pt 1): p. 77–87.
78. Jovov, B., et al., Immunolocalization of the acid-sensing ion channel 2a in the rat
cerebellum. Histochem. Cell Biol., 2003. 119(6): p. 437.
79. Ettaiche, M., et al., Acid-sensing ion channel 2 is important for retinal function and
protects against light-induced retinal degeneration. J. Neurosci., 2004. 24(5): p.
1005–1012.
80. Hruska-Hageman, A.M., et al., Interaction of the synaptic protein PICK1 (protein
interacting with C kinase 1) with the non-voltage gated sodium channels BNC1 (brain
Na+ channel 1) and ASIC (acid-sensing ion channel). Biochem. J., 2002. 361(Pt 3): p.
443–450.
81. Duggan, A., J. Garcia-Anoveros, and D.P. Corey, The PDZ domain protein PICK1 and
the sodium channel BNaC1 interact and localize at mechanosensory terminals of dorsal
root ganglion neurons and dendrites of central neurons. J. Biol. Chem., 2002. 277(7): p.
5203–5208.
82. Chai, S., et al., A Kinase-anchoring Protein 150 and Calcineurin Are Involved in
Regulation of Acid-sensing Ion Channels ASIC1a and ASIC2a. J. Biol. Chem., 2007.
282(31): p. 22668–22677.
83. Hruska-Hageman, A.M., et al., PSD-95 and Lin-7b interact with acid-sensing ion
channel-3 and have opposite effects on H+- gated current. J. Biol. Chem., 2004. 279(45):
p. 46962–46968.Acid-Sensing Ion Channels (ASICs) and pH in Synapse Physiology 681
84. Gao, J., et al., Coupling between NMDA receptor and acid-sensing ion channel
contributes to ischemic neuronal death. Neuron, 2005. 48(4): p. 635–646.
85. Leonard, A.S., et al., cAMP-dependent protein kinase phosphorylation of the acid-
sensing ion channel-1 regulates its binding to the protein interacting with C-kinase-1.
Proc. Natl. Acad. Sci. U. S. A., 2003. 100(4): p. 2029–2034.
86. Ettaiche, M., et al., Silencing acid-sensing ion channel 1a alters cone-mediated retinal
function. J. Neurosci., 2006. 26(21): p. 5800–5809.
87. Lalo, U., et al., Spontaneous autocrine release of protons activates ASIC-mediated
currents in HEK293 cells. J. Cell. Physiol., 2007. 212(2): p. 473–480.
88. Coryell, M.W., et al., Targeting ASIC1a Reduces Innate Fear and Alters Neuronal
Activity in the Fear Circuit. Biol. Psychiatry, 2007: p. Epub ahead of Print.
89. Olson, T.H., et al., An acid sensing ion channel (ASIC) localizes to small primary
afferent neurons in rats. Neuron, 1998. 9: p. 1109–1113.
90. Pignataro, G., R.P. Simon, and Z.G. Xiong, Prolonged activation of ASIC1a and the time
window for neuroprotection in cerebral ischaemia. Brain, 2007. 130(Pt 1): p. 151–158.
91. Mazzuca, M., et al., A tarantula peptide against pain via ASIC1a channels and opioid
mechanisms. Nat. Neurosci., 2007. 10(8): p. 943–945.
92. Roumy, M. and J.M. Zajac, Neuropeptide FF, pain and analgesia. Eur. J. Pharmacol.,
1998. 345: p. 1–11.You can also read

















































