Transcriptome Analysis Reveals Key Pathways and Hormone Activities Involved in Early Microtuber Formation of Dioscorea opposita
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Hindawi
BioMed Research International
Volume 2020, Article ID 8057929, 11 pages
https://doi.org/10.1155/2020/8057929
Research Article
Transcriptome Analysis Reveals Key Pathways and Hormone
Activities Involved in Early Microtuber Formation of
Dioscorea opposita
Junhua Li ,1,2,3 Xiting Zhao ,1,2,3 Yahui Dong,1 Shujie Li,1 Jiaojiao Yuan,1 Chenglong Li,1
Xiaoli Zhang,1,2,3 and Mingjun Li 1,2,3
1
College of Life Sciences, Henan Normal University, Xinxiang, Henan 453007, China
2
Engineering Technology Research Center of Nursing and Utilization of Genuine Chinese Crude Drugs of Colleges and Universities in
Henan Province, Xinxiang, Henan 453007, China
3
Engineering Laboratory of Biotechnology for Green Medicinal Plant of Henan Province, Xinxiang, Henan 453007, China
Correspondence should be addressed to Mingjun Li; limingjun2002@263.net
Received 2 November 2019; Accepted 17 February 2020; Published 11 March 2020
Academic Editor: Kui Li
Copyright © 2020 Junhua Li et al. This is an open access article distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Chinese yam (Dioscorea opposita) is an important tuberous crop used for both food and medicine. Despite a long history of
cultivation, the understanding of D. opposita genetics and molecular biology remains scant, which has limited its genetic
improvement. This work presents a de novo transcriptome sequencing analysis of microtuber formation in D. opposita. We
assembled cDNA libraries from different stages during the process of microtuber formation, designated as initial explants (EXP),
axillary bud proliferation after three weeks (BUD), and microtuber visible after four weeks (MTV). More differentially expressed
genes (DEGs) and pathways were identified between BUD vs. EXP than in MTV vs. BUD, indicating that proliferation of the
axillary bud is the key stage of microtuber induction. Gene classification and pathway enrichment analysis showed that
microtuber formation is tightly coordinated with primary metabolism, such as amino acid biosynthesis, ribosomal component
biosynthesis, and starch and sucrose metabolism. The formation of the microtuber is regulated by a variety of plant hormones,
including ABA. Combined with analysis of physiological data, we suggest that ABA positively regulates tuberization in D.
opposita. This study will serve as an empirical foundation for future molecular studies and for the propagation of D. opposita
germplasm in field crops.
1. Introduction has drawn more and more research attentions on its biology,
pathology, and cultivation [1, 2].
Yams (Dioscorea spp.) are a tuberous crop in many tropical Plant diseases, especially virus infections that resulted
and subtropical regions, such as West Africa, East and South from vegetative propagation, are a serious issue for field pro-
Asia, and the Caribbean. Around ten Dioscorea species have duction of D. opposita. The production of virus-free plants
been domesticated, and they are important sources of food through plant tissue culture is widely used in the Dioscorea
and income in these areas. Dioscorea opposita (Chinese genus to avoid virus infections of plant materials [3–5]. How-
yam) is one of the four famous Chinese herbs produced in ever, the virus-free plantlets obtained by this approach are
Huaiqing area, it is also a very popular edible plant and has very fragile and difficult to pack, transport, and transplant.
long been cultivated to promote human health and longevity In addition to plantlets, microtubers are small tubers origi-
through diet, and it is the 2nd most commonly grown tuber- nated from plant tissues in vitro. Microtubers have the poten-
ous crop in China after potato. In recent years, D. opposita tial to be integrated into seed yam programs [6, 7] and are
2 BioMed Research International
particularly convenient for shipping, storage, and exchange
of germplasm. Therefore, the study of microtuber induction
and formation has attracted more and more attention [8, 9].
Tuberization is a highly complex biological process
which is affected by both environmental (such as photope-
riod) and endogenous factors (such as plant growth regula-
tors and plantlet growth stage) [1, 5, 10]. The regulation
mechanism of tuberization is invaluable to devise strategies
to improve tuber yield and quality. Researchers are now
interested in identifying the regulatory molecules related to
the formation of microtubers. The high-throughput capacity
of the next generation of RNA sequencing technology pro-
vides an unprecedented opportunity for genomic exploration
and gene discovery in non-model plant species for which EXP BUD MTV
there is no available reference genome sequence data [11,
12]. RNA-Seq results typically show high levels of reproduc- Figure 1: Microtuber induction and formation stages in D.
ibility for both technical and biological replicates [13, 14]. opposita. Pictures of D. opposita: at the stages of initial explant
During the past decade, the genes and gene networks asso- (EXP), axillary bud proliferation after three weeks (BUD), and
ciated with important potato tuber traits, such as the tuber microtuber visible after four weeks (MTV). The arrow indicates a
formation [15–17], antiviral properties [18–20], and starch microtuber. Bar = 5 mm.
biosynthesis [21–23], have been identified and functionally
characterized using RNA-Seq technology. So far, knowledge
of plant tuberization was mainly obtained from studies in ber formation using a RNAiso for Ploysaccharide-rich Plant
potato. The genomic resources available for D. opposita or Tissue Kit (Takara, Dalian, China). RNA purity was checked
for other members of the Dioscoreaceae family are limited, using a NanoPhotometer® spectrophotometer (IMPLEN,
and the transcriptional changes and molecular mechanisms USA). RNA concentrations were measured using a Qubit®
associated with the developmental process of microtubers RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies,
in D. opposita are still far from being characterized. This USA). RNA integrity was assessed using an RNA Nano 6000
lack of information hampers gene discovery and seriously Assay Kit with an Agilent Bioanalyzer 2100 system (Agilent
hinders the improvement of D. opposita as a commercially Technologies, USA). A total of 3 μg RNA per sample was
important species. used for library construction. For each stage, the RNA sam-
A one-step protocol for induction of microtubers from ples from three individuals were then pooled together in
a single nodal segment was previously established [9], equal amounts to generate one mixed sample. Each stage
which provides a simplified model for the study of microtu- with two replicates, then these six mixed RNA samples were
ber formation in D. opposita. In this study, Illumina RNA- subsequently used in cDNA library construction and Illu-
Seq technology was used to investigate changes in the mina deep sequencing.
transcriptome during microtuber formation in D. opposita. Six cDNA libraries were generated using a NEBNext®
The results of this transcriptomic analysis represent a valu- Ultra™ RNA Library Prep Kit for the Illumina® platform
able bioinformatics resource for further research into the (NEB, USA) following the manufacturer’s recommendations;
genes and gene functions involved in controlling the pro- index codes were added to attribute sequences to each sam-
cess of microtuber formation. ple. Library quality was assessed with an Agilent Bioanalyzer
2100 system. The library preparations were sequenced with
2. Materials and Methods the Illumina Hiseq 2000 platform and paired-end reads were
generated at Beijing Novogene Bioinformatics Biotechnology
2.1. Plant Materials. Microtubers of D. opposita cultivar Tie- Co., Ltd. (Beijing, China). All raw-sequence reads data were
gun were collected from those plants of Henan Province deposited in NCBI Sequence Read Archive (SRA, http://
Engineering Laboratory of Green Medicinal Plant Biotech- www.ncbi.nlm.nih.gov/Traces/sra) with accession number
nology at Henan Normal University in Xinxiang, China. SRP061414.
Plants were then grown in a growth chamber in the liquid
MS medium containing 60 g L-1sucrose under a 16 h light/8 h
dark photoperiod with a light intensity of 38 μm sec-1 m-2 at 2.3. De Novo Transcriptome Assembly. Raw data (raw reads)
22-25°C [9]. Three different stages of microtuber formation in the fastq format were initially processed with in-house perl
are shown in Figure 1. All samples were dissected from the scripts. In this step, clean data (clean reads) were obtained by
plants, immediately frozen with liquid nitrogen, and then removing reads containing adapters, reads containing ploy-
were put at -80°C. N, and low-quality reads from raw data. At the same time,
the Q20 and Q30 values, the GC-content, and the sequence
2.2. RNA Extraction and cDNA Library Preparation for duplication level of the clean data were calculated. All of
Transcriptome Sequencing. Total RNA used for RT-qPCR the downstream analyses were based on the high-quality data
analysis was extracted from tissues of three stages of microtu- prepared via these initial processing procedures.BioMed Research International 3
2.4. Sequence Annotation and Classification. For annotation, 3. Results
the sequences were searched against the NCBI NR protein
database (https://www.ncbi.nlm.nih.gov/protein/) using the 3.1. Illumina Sequencing and the Assembly of Sequence Reads.
BlastX algorithm, with a cut-off E value of 1e-5. Gene Ontol- In a previous study, a simplified method for in vitro induction
ogy (GO) terms were extracted from the annotation of high- of microtubers from D. opposita was developed, by which
score BLAST matches in the NCBI NR protein database microtubers can be induced from almost all of the internodes
using Blast2GO v2.5 [24] and then sorted for the GO catego- of single nodal segments in MS medium under rotary shaking
ries using in-house perl scripts. Functional annotation of the conditions [9]. The hormone-free induction conditions and
proteome was carried out by a BlastP homology search the highly reproducible results made this method a model sys-
against the NCBI eukaryotic Ortholog Groups (KOG) data- tem for studies of the molecular mechanisms behind the pro-
base. KEGG pathway annotations were performed using cess of microtuber formation, such as differences in gene
Blastall software. expression and transcriptome profile. In order to perform an
expression profiling study, here we divided the process of
2.5. Differential Expression Analysis. After the D. opposita microtuber formation into the following three stages based
transcriptome was assembled, counting of alignments was on appearance changes of the leaf-axil region and the microtu-
performed using the RSEM package [25]. Differential ber emergence (Figure 1): initial explant (EXP), axillary bud
expression analysis of two conditions/groups was performed proliferation after three weeks (BUD), and microtuber visible
using the DESeq R package [26, 27]. The DESeq R package after four weeks (MTV). Based on the present study and our
provides statistical routines for determining differential previous work [9], the starting control material, microtuber
expression in digital gene expression data using a model induction, and microtuber formation are thought to be well-
based on a negative binomial distribution. The P value sets represented by materials from the above three stages.
the threshold for the differential gene expression tests. The Total RNA from these stages was extracted; cDNA librar-
resulting P values were adjusted using Benjamini and Hoch- ies for each developmental stage (EXP, BUD, and MTV) were
berg’s approach for controlling the false discovery rate constructed and sequenced. After eliminating primer and
(FDR) [28]. Genes with an adjusted P value < 0.05 deter- adapter sequences and filtering out the low-quality reads,
mined by DESeq were classified as differentially expressed. we pooled all the high-quality Illumina reads from the three
The sequences of the DEGs were Blast (BlastX) against the different stage libraries. We obtained 50,072,432 and
genome of a related species to obtain the Predicted 53,666,222, 43,944,164 and 57,705,804, and 53,460,778 and
Protein-Protein Interactions (PPI) of these DEGs. The PPI 65,554,462 qualified Illumina reads, respectively, for the
of the DEGs were visualized in Cytoscape [29]. We used two EXP samples, the two BUD samples, and the two MTV
KOBAS software to test the statistical enrichment of differ- samples, giving rise to 5.01G and 5.36G, 4.39G and 5.78G,
entially expressed genes in KEGG pathways [30]. and 5.34G and 6.56G total bases, respectively (Table S1).
Using Trinity software, we then combined all the reads
2.6. Real-Time Quantitative PCR to Validate the RNA-Seq to form a transcriptome database for D. opposita [34].
Data. Nine DEGs were chosen for validation using real- We identified 181,047 transcripts, with total residues of
time qPCR. The primers, designed with Primer Premier 234,740,027 bp. The average length of each transcript was
5.0 software, are detailed in Table S8. Total RNA was 1,297 bp; the shortest sequence was 201 bp in length and
extracted from tissues at each stage using a TaKaRa the longest sequence was 18,739 bp in length (Table S1).
MiniBEST Plant RNA Extraction Kit (TaKaRa, Japan) and The sequence length distribution of the unigenes and
reverse-transcribed into cDNA using HiScript® Q Select RT transcripts is shown in Figure S1. The unigenes have a
SuperMix for qPCR (Vazyme, USA). Real-time qPCR was length of less than 301 bp (21,687), representing the highest
performed on an ABI 7500 Real-Time PCR system (Applied proportion (30.23%), followed by the 301-500 bp sequences
Biosystems, USA). The level of expression was calculated (17,465, 24.35%) and 501-1000 bp sequences (15,314,
with normalization using Actin as a reference [31]. A 21.35%); there were 45,419 transcripts (25.09%) in the size
comparative Ct method (2–ΔΔCt) of relative quantification range of 1001-2000 bp, 39,546 (21.84%) at >2000 bp, and
was used to analyze the real-time quantitative PCR data 37,413 (20.66%) at 501-1000 bp.
[32, 33]. All quantitative PCR analyses for each gene used
three biological replicates. 3.2. Real-Time Quantitative PCR Validation of RNA-Seq
Results. To validate the RNA-Seq results, nine DEGs were
2.7. ABA Treatment. To investigate the influence of ABA on selected and RT-qPCR assays were performed. The expres-
microtuber formation, single nodal segments were cultured sion patterns of these genes at each of the three stages of
under the same conditions that we developed previously microtuber formation are shown in Figure S2. Although the
[9] in medium containing 1 μM ABA or 0.5 mM sodium calculated fold changes showed slightly varied, most genes
tungstate (ST). For each treatment, three independent measured by real-time qPCR were consistent with the RNA-
experiments were conducted and each repetition contained Seq results. These experiments confirmed the accuracy of the
36 single nodal segments. Values represent means ± SE of RNA-Seq results reported in this study.
three independent measurements. Student’s t-test was used
to evaluate the differences between every treatment and 3.3. Sequence Annotation. Since there is no reference sequence
the control. data available for directly assembling the transcriptome of4 BioMed Research International
100 21510
10 2151
Number of genes
Percent of genes
1 215.1
0.1 21.51
Cell killing
Rhythmic process
Growth
Biological adhesion
Immune system process
Locomotion
Positive regulation of biological process
Negative regulation of biological process
Reproductive process
Reproduction
Developmental process
Multicellular organismal process
Multi-organism process
Signaling
Cellular component organization or biogenesis
Establishment of localization
Localization
Regulation of biological process
Biological regulation
Single-organism process
Metabolic process
Cellular process
Extracellular matrix
Virion
Extracellular region
Membrane-enclosed lumen
Membrane
Macromolecular complex
Enzyme regulator activity
Molecular transducer activity
Structural molecule activity
Nucleic acid binding transcription factor activity
Transporter activity
Catalytic activity
Binding
Response to stimulus
Extracellular matrix part
Extracellular region part
Virion part
Membrane part
Organelle part
Organelle
Cell
Channel regulator activity
Antioxidant activity
Protein binding transcription factor activity
Receptor activity
Cell part
Biological process
Cellular component
Molecular function
Figure 2: Gene Ontology (GO) assignments for the microtuber induction and formation transcriptome of D. opposita. Results are
summarized under three main GO categories: biological process, cellular component, and molecular function. The left y-axis indicates the
percentage of a specific subcategory of genes in each main category. The right y-axis indicates the number (count) of genes.
D. opposita, sequences obtained were searched against the categories and 48 subcategories. Of the 181,047 assembled
NCBI nonredundant (NR) database using BlastX [35] with a transcripts, 21,514 were successfully annotated with GO
cut-off E value at 10-5. A total of 23,310 (32.49%) sequences assignments; some of these belonged to one or more of the
showed significant similarity to known proteins in the NR three categories (Figure 2). The “biological process” categories
database. Among them, 13.89% demonstrated 80%-100% sim- included cellular process (13,117, 60.97%), metabolic process
ilarity as reported in the BlastX results, and 38.21% showed (12,416, 57.71%), single-organism process (6,549, 30.44%),
60%-80% of RNA sequence similarity (Figure S3A). After a biological regulation (4,328, 20.12%), regulation of biological
closer look of the specific origins of the matched sequences, process (3,977, 18.49%), and other categories (18,347,
we found that 13.53% of the sequences matched to the 85.28%). The major proportion of the “cellular component”
transcripts of Vitis vinifera, which was followed by Oryza categories included cell (8,582, 39.89%), cell part (8,556,
sativa (13.05%) and Arabidopsis (11.19%) (Figure S3B). 39.77%), organelle (6,478, 30.11%), macromolecular complex
(4,464, 20.75%), membrane (4,209, 19.56%), organelle part
3.4. GO, KOG, and KEGG Classification. To address the gene (3,672, 17.07%), and membrane part (3,579, 16.64%). The
product properties, we adopted Gene Ontology (GO) terms top three most abundant subcategories of the “molecular func-
using Blast2GO v2.5 [24], which annotates high-score tion” category were binding (12,455, 57.89%), catalytic activity
BLAST matches to sequences in the NCBI NR protein data- (10,407, 48.37%), and transporter activity (1,602, 7.45%).
base. Genes of D. opposita were classified into biological Identification of orthologous groups is useful for genome
process, cellular component, and molecular function GO annotation. The annotated sequences were subjected to aBioMed Research International 5
20 [A] RNA processing and modification
[B] Chromatin structure and dynamics
[J] Translation, ribosomal structure and biogenesis
[K] Transcription
[L] Replication, recombination and repair
[D] Cell cycle control, cell division, chromosome partitioning
15 [M] Cell wall/membrane/envelope biogenesis
[N] Cell motility
[O] Posttranslational modification, protein turnover, chaperones
[T] Signal transduction mechanisms
Percent (%)
[U] Intracellular trafficking, secretion, and vesicular transport
[V] Defense mechanisms
10 [W] Extracellular structures
[Y] Nuclear structure
[Z] Cytoskeleton
[C] Energy production and conversion
[E] Amino acid transport and metabolism
[F] Nucleotide transport and metabolism
[G] Carbohydrate transport and metabolism
5 [H] Coenzyme transport and metabolism
[I] Lipid transport and metabolism
[P] Inorganic ion transport and metabolism
[Q] Secondary metabolites biosynthesis, transport and catabolism
[R] General function prediction only
[X]Unamed protein
0 [S] Function unknown
A B J K L D M N O T U V W Y Z C E F G H I P Q R X S
Function class
Figure 3: KOG functional classification for the microtuber induction and formation transcriptome of D. opposita. From a total of 181,047 de
novo assembled transcripts, 9,404 transcripts with significant homologies in the KOG database (E value ≤ 1e − 3) were classified into 26 KOG
categories.
search against the eukaryotic Ortholog Groups (KOG) (EXP, BUD, and MTV) onto our assembled transcriptome
database [36], for functional prediction and classification. database. The expression of each gene was calculated using
Based on sequence homology, 9,404 unique sequences were the number of reads that mapped onto a given transcript.
assigned a KOG functional classification. These sequences The DEG Venn diagram of the comparisons is depicted in
were classified into 26 KOG categories involved in cellular Figure 5(a). It shows 1,804 and 923 genes are specific differ-
process, signal transduction, metabolism, and other pro- entially expressed in BUD vs. EXP and MTV vs. EXP com-
cesses (Figure 3). The most common category was the non- parisons during microtuber formation, respectively.
specific category of “general function prediction only” There were 5,136, 4,297, and 143 genes differentially
(1,859, 19.77%), followed by “posttranslational modification, expressed (P < 0:05) between sample pairs of BUD-EXP,
protein turnover, and chaperones” (1,230, 13.08%), “signal MTV-EXP, and MTV-BUD, respectively. The expression
transduction mechanisms” (843, 8.96%), “translation, ribo- patterns of the differentially expressed genes (DEGs) from
somal structure, and biogenesis” (580, 6.17%), “secondary the EXP vs. BUD are depicted in Figure 5(b); there were
metabolite biosynthesis, transport, and catabolism” (566, 2,943 genes that are shown to be 10-fold greater and 2,193
6.02%), and “intracellular trafficking, secretion, and vesicular genes that are shown to be 9-fold weaker in BUD vs. EXP.
transport” (555, 5.90%). Among other categories represented The expression patterns of DEGs from the MTV vs. EXP
were “translation” (529, 5.63%) and “carbohydrate transport comparison are depicted in Figure 5(b); there were 2,593
and metabolism” (496, 5.27%). The least well-represented genes that are shown to be 9-fold greater and 1704 genes that
categories were “cell motility” (3, 0.03%) and “unnamed pro- are shown to be 8-fold weaker in MTV vs. EXP.
tein” (1, 0.01%). To further investigate genes that were highly correlated
In order to identify biochemical pathways, we mapped with microtuber formation, we looked into the 20 most
the annotated sequences onto the KEGG (Kyoto Encyclope- upregulated and the 20 most downregulated genes selected
dia of Genes and Genomes) database [30], which is an alter- from the BUD vs. EXP, MTV vs. EXP, and MTV vs. BUD
native approach to predict and assign gene function by comparisons. Some of these genes showed matches to
emphasizing biochemical pathways (Figure 4). A total of sequences in the NCBI NR database, such as fructose-bispho-
7,278 annotated genes were assigned to 262 KEGG pathways. sphatealdolase, chlorophyll a/b-binding protein type III pre-
The maps with the highest unigene representation were car- cursor, protein LHY-like, glycosyltransferase GT14A05, and
bohydrate metabolism (750, 10.31%), followed by signal Chain A, Banana Lectin (Table S2-S4).
transduction (636, 8.74%) and translation (622, 8.55%). The
pathways with the highest representation were carbon 3.6. Pathway Enrichment Analysis of DEGs. In order to
metabolism (315, 4.33%), biosynthesis of amino acids (277, explore the functions of the DEGs in greater depth, the
3.81%), ribosome (251, 3.45%), and plant hormone signal KEGG Orthology-Based Annotation System (KOBAS)
transduction (223, 3.06%). These pathway assignments can [37, 38] was used. Among these pathways, the terms that
provide valuable information for the investigations focused were found to be significantly differently expressed (cor-
on specific biochemical and developmental processes. rected P value ≦ 0.05) in both the BUD vs. EXP and the
MTV vs. EXP comparisons were involved in “glycolysis/-
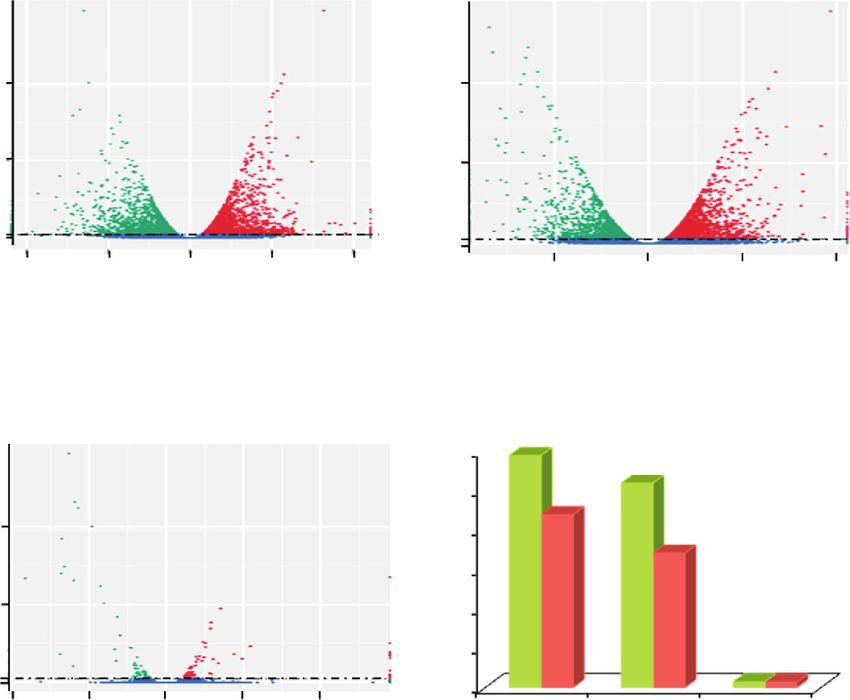
3.5. Differentially Expressed Genes (DEGs) during Microtuber gluconeogenesis” and the “citrate cycle” (TCA cycle). We
Formation. We mapped the Illumina reads from each stage noticed a high percentage of significantly DEGs of the6 BioMed Research International
KEGG classification
Sensory system 26
Nervous system 199
Immune system 196
Excretory system 60
Environmental adaptation 278 E
Endocrine system 272
Digestive system 129
Development 39
Circulatory system 77
Xenobiotics biodegradation and metabolism 117
Overview 524
Nucleotide metabolism 191
Metabolism of terpenoids and polyketides 220
Metabolism of other amino acids 206
Metabolism of cofactors and vitamins 213 D
Lipid metabolism 382
Glycan biosynthesis and metabolism 102
Energy metabolism 580
Carbohydrate metabolism 750
Biosynthesis of other secondary metabolites 304
Amino acid metabolism 540
Translation 622
Transcription 243 C
Replication and repair 150
Folding, sorting and degradation 498
Signaling molecules and interaction 4 B
Signal transduction 636
Membrane transport 76
Transport and catabolism 372
Cell motility 77 A
Cell growth and death 224
Cell communication 91
0 2 4 6 8 10 12 14
Percent of genes (%)
Figure 4: KEGG functional classification for the microtuber induction and formation transcriptome of D. opposita. From a total of 181,047 de
novo assembled transcripts, 7,278 transcripts with significant homologies in the KEGG database were classified into 5 KEGG categories. A:
cellular processes; B: environmental information processing; C: genetic information processing; D: metabolism; and E: organismal systems.
biosynthesis of secondary metabolites, microbial metabo- that almost all of the key genes in these pathways were found
lism in diverse environments, carbon metabolism, and among the DEGs, including nineteen starch and sucrose met-
ribosome pathways (see Discussion). We found that the abolic genes in the BUD vs. EXP comparison and sixteen
only pathway enriched in MTV vs. BUD comparison was genes in the MTV vs. BUD comparison (Table S6). We also
the photosynthesis-antenna protein pathway, which means found that there were no DEGs in the starch or sucrose
DEGs of BUD vs. EXP and MTV vs. EXP comparisons metabolism pathways in the MTV vs. BUD comparison.
may play a more important role during microtuber forma-
tion (Table 1). Then, the KEGG enrichment on the specific 3.7. Differentially Expressed Genes Involved in Plant Hormone
DEGs of BUD vs. EXP and MTV vs. EXP comparisons was Signaling Pathways during Microtuber Formation. A group of
conducted. We found that the genes involved in biosynthesis genes involved in plant hormone activities were upregulated
of amino acids, followed by carbon metabolism and phenyl- at BUD and MTV compared to EXP (Table S7). Those genes
propanoid biosynthesis pathways, were significantly enriched include ARF, GH3, AHP, ARR, PP2C, JAZ, and TGA. Among
in BUD, suggesting these pathways represent key metabolic the upregulated genes, the ethylene-responsive genes EIN3
processes during microtuber induction. Our data also sug- and CTR1 were significantly upregulated at BUD while the
gested that ribosome, photosynthesis and the metabolism of ABA-responsive gene PP2C was significantly upregulated at
amino sugar, and nucleotide sugar play roles in microtuber MTV. These results indicate that the formation of the
formation (Table S5). microtuber is coordinated and regulated by a variety of
Tubers are starch-rich storage organs, in which large hormones, such as ethylene and ABA.
amounts of starch are deposited. In potato, studies showed
that expression of the sucrose synthase gene and genes 3.8. ABA Is a Positive Regulator of Microtuber Formation.
encoding enzymes involved in starch biosynthesis were Transcriptional profiling showed that microtuber formation
increased during tuber formation [39–41]. Microtubers of in D. opposita is associated with altered transcription state
D. opposita likely share the same fundamental mechanism of PP2C, which has both positive and negative roles in ABA
of its development as do potato and other tuberous plants. signaling [42]. We therefore carried out further studies to
Given this hypothesis, we examined gene activities of the elucidate the role of ABA in microtuber formation. We tested
starch and sucrose metabolism pathways. Our data showed the influence of ABA and ST (an ABA inhibitor) onBioMed Research International 7
BUD vs. EXP MTV vs. EXP
3274
1804 923
39
19 61
24
MTV vs. BUD
(a)
BUD vs. EXP MTV vs. EXP
46
-log10 (padj)
-log10 (padj)
66
33 23
1.3 1.3
0 0
–10 –5 0 5 10 –5 0 5 10
log2 (fold change) log2 (fold change)
Different expressed genes (5136) Different expressed genes (4297)
Up-regulated: 2943 Up-regulated: 2593
Down-regulated: 2193 Down-regulated: 1704
MTV vs. BUD
2943
3000
2593
2500 2193
Number of DEGs
-log10 (padj)
56 2000 1704
1500
28 1000
500 73 70
1.3
0 0
–10 –5 0 5 10 BUD vs. EXP MTV vs. EXP MTV vs. BUD
log2 (fold change)
Up-regulated genes
Different expressed genes (143)
Down-regulated genes
Up-regulated: 73
Down-regulated: 70
(b)
Figure 5: Differentially expressed genes (DEGs) in the microtuber formation-stage comparisons. (a) DEG Venn diagram. The sum of the
numbers in each large circle represents the total number of DEGs of the comparative combination, and the circle overlapping part
represents what the DEG combination has in common. (b) Whole-study overview of log-fold changes in gene expression in comparisons
BUD vs. EXP (upper left), MTV vs. EXP (upper right), and MTV vs. BUD (down left). The x-axis indicates the log-fold changes between
the two samples. The y-axis indicates the absolute expression levels (–log10 (Padj)). The number of up- or downregulated genes in BUD
vs. EXP, MTV vs. EXP, and MTV vs. BUD are shown in the down-right panel.8 BioMed Research International
Table 1: Significantly enriched pathways identified using the KOBAS database based on DEGs identified during microtuber formation.
BUD vs. EXP MTV vs. EXP MTV vs. BUD
Pathway Corrected Corrected Corrected
Pathway Bn Nt P value Nt P value Nt P value
ID P value P value P value
Photosynthesis-antenna proteins ko00196 39 7 3.36E-10 5.94E-08
Flavonoid biosynthesis ko00941 81 35 5.07E-07 8.97E-05
Ribosome ko03010 251 104 0 0
Glycolysis/gluconeogenesis ko00010 145 51 3.27E-06 0.000289 45 6.34E-06 0.000561
Phenylalanine metabolism ko00360 110 41 5.76E-06 0.00034
Citrate cycle (TCA cycle) ko00020 73 27 0.000252 0.008928 24 0.000341 0.020113
Phenylpropanoid biosynthesis ko00940 163 50 0.000236 0.008928
Microbial metabolism in diverse
ko01120 418 107 0.000518 0.01477
environments
Flavone and flavonol biosynthesis ko00944 27 13 0.000584 0.01477
Carbon metabolism ko01200 296 79 0.000741 0.016394
Biosynthesis of secondary
ko01110 1104 250 0.001075 0.019019
metabolites
Butanoate metabolism ko00650 22 11 0.001043 0.019019
Steroid biosynthesis ko00100 27 12 0.000511 0.022627
DNA replication ko03030 37 14 0.001257 0.044508
Note: Bn (background number) indicates the total number of transcripts present in each pathway. Nt (number of transcripts) indicates the number of DEGs in
each pathway. Pathways enrichment analysis with corrected P value ≤ 0.05 were included.
120
results indicate that ABA had a positive role in regulating
100 microtuber formation.
Visible microtubers (%)
80
4. Discussion
60 4.1. Transcriptomic Study Provides a Basis for Further
⁎ Research of Microtuber Formation in D. opposita. D. opposita
40
⁎
is an important tuberous crop for both food and medicine.
20 However, a limited genetic resource, including genomic
information, is available for this species, which has limited
0 its genetic improvement. In this study, we generated cDNA
15 30
Days sequence data, comprising long sequences of good quality,
which will facilitate subsequent and more detailed studies.
CK ST
ABA The analysis included sample cDNA libraries from three dif-
ferent stages of microtuber formation. We identified a total of
Figure 6: The influence of exogenous ABA on the rates of 71,739 unigenes, and the sequences were annotated against
microtuber formation. ST: sodium tungstate. Data are presented as the NCBI NR database using BlastX. By using Illumina’s dig-
means ± SD. Asterisks indicate significance (∗ P < 0:01 versus ital gene expression platform, we investigated differential
control, Student’s t-test). gene expression during the formation of D. opposita microtu-
bers and analyzed the expression of unigenes in the context
microtuber formation. The formation of microtubers could of the pathways of the KEGG. We found biosynthesis of
be observed on 17.6% of single nodal segments on day 15 amino acids and ribosome may represent key metabolic
in medium with 1 μM ABA, which is obviously earlier than processes during microtuber induction and formation,
that of the control (Figure 6). In contrast, the percentage of respectively. We identified biosynthesis of amino acids and
microtuber formation was reduced three times compared ribosome pathway genes that were differentially expressed
with the control on day 30, when microtubers were formed during microtuber induction and formation. We also found
in most single nodal segments of the control and the ABA- that key genes in the metabolism of starch and sucrose were
treat group (Figure 6). In addition, the endogenous levels of differentially expressed during the various stages of microtu-
ABA were measured in different stages of microtuber forma- ber formation. To our knowledge, this is the first comprehen-
tion in our previous study [43], in which the ABA contents sive transcriptomic study of D. opposita to identify genes and
showed a positive association with the process of microtuber pathways that are differentially expressed during microtuber
formation, which reached its maximum level at the MTV formation; the results are foundational for functional geno-
stage, and maintained at a high level thereafter [43]. These mic studies in this species.BioMed Research International 9
For gene annotation, the sequences were searched against 4.3. Potential Roles of Starch Formation and Hormone
the NCBI NR database. Because of the limited genetic infor- Activities in Microtuber Formation. As storage organs, large
mation available, no matches were obtained for as many as amounts of starch are deposited in tubers. Our data showed
48,429 (67.51%) of the sequences. Some of these sequences that key genes in starch and sucrose metabolic pathways are
may represent novel genes in D. opposita, suggesting that associated with early microtuber induction. In our previous
some unique processes and pathways might be involved with study, microtubers could be easily induced in a high sugar
microtuber formation in D. opposita. Interestingly, the medium [9]. It is conceivable that the availability of starch
annotated sequences of D. opposita shared relatively high or one of its precursors acts as a signal, to initiate the mor-
homology with Vitis vinifera proteins (Figure S3). In spite phological typical changes not only for tuberization but also
of the large proportion of sequences that showed no for the activation of the “tuber-specific” genes and early
matches, a large number of transcripts were nevertheless microtuber induction.
assigned to a wide range of GO and KOG classifications, Transcriptome data indicated that microtuber formation
which indicated that our transcriptome data represented a is regulated by a variety of hormones, such as ethylene and
broad diversity of transcripts in D. opposita. KEGG functional ABA, since multiple genes involved in ethylene and ABA
classification identified many genes associated with carbon activities were upregulated at BUD and MTV (Table S7).
metabolism, including genes of glycolysis/gluconeogenesis, The results are consistent with our previous study, in which
the citrate cycle (TCA cycle), fructose and mannose the ABA content increased during microtuber formation
metabolism, galactose metabolism, pentose biosynthesis, [43]. We showed that exogenous ABA and its inhibitor
and inositol phosphate metabolism. Additionally, we significantly influenced microtuber formation and concluded
identified genes related to starch and sucrose metabolism. that ABA plays a positive role in this process. It is well
Starch composition is one of the most economically known that ABA functions directly in plant responses to
important characteristics of D. opposita, since tubers and stress (e.g., [46]); therefore, in D. opposita, ABA might be an
fully matured microtubers are a rich source of energy, and internal signal integrator through which plants can sensitively
starch constitutes most of the dry weight of the microtubers. perceive incoming stresses and regulate their physiology
accordingly (such as the production of microtubers). In
4.2. DEGs Revealed Pathways Enriched during Microtuber summary, our results not only advance our understanding of
Formation. Although the analysis of DEGs during microtu- early tuberization but also are potentially valuable for future
ber formation has been performed using microarray technol- mechanism studies, e.g., roles of other plant hormones in
ogy in potato [16], the availability of data relating to the microtuber formation of D. opposita.
transcriptomes of microtuber formation at different stages
in D. opposita remains vacant. Here, the analysis of DEGs 5. Conclusions
during microtuber formation has been performed using Illu-
mina RNA-Seq method microarray in D. opposita, and a The transcriptome sequencing analysis of microtuber forma-
number of DEGs were identified, which showed differential tion indicates that the proliferation of axillary bud is the key
expression patterns in the following comparisons: BUD vs. stage of microtuber induction. DEGs involved in starch and
EXP, MTV vs. EXP, and BUD vs. MTV. sucrose metabolism during microtuber formation were iden-
Analysis of DEGs showed that there was only one pathway tified. We suggest that ABA positively regulates tuberization
enrichment in BUD vs. MTV comparison. Therefore, DEGs of in D. opposita.
BUD vs. EXP and MTV vs. EXP comparisons may play an
important role during microtuber formation (Table S5). Abbreviations
Through analyzing the KEGG enrichment data on the DEGs
of BUD vs. EXP and MTV vs. EXP comparisons, we found BUD: Axillary bud proliferation
that the biosynthesis of amino acids, carbon metabolism, EXP: Initial explants
and phenylpropanoid biosynthesis is significantly differently GO: Gene Ontology
expressed (corrected P value ≦ 0.05) in the BUD vs. EXP KOG: EuKaryotic Ortholog Groups
and speculated that the biosynthesis of amino acids, followed KEGG: Kyoto Encyclopedia of Genes and Genomes
by carbon metabolism and phenylpropanoid biosynthesis, MTV: Microtuber visible after four weeks
may play key regulatory roles during microtuber induction NR: Nonredundant
of D. opposita. Our results are consistent with those of DEGs: Differentially expressed genes
Roessner-Tunali et al. [44], who linked sucrose levels to the KOBAS: KEGG Orthology-Based Annotation System.
regulation of de novo amino acid synthesis at the
transcriptional level. A possible explanation for these results Data Availability
may be an elevated requirement for free amino acid levels to
maintain cellular osmoticum during tuber induction [45]. The datasets supporting the results of this article are
The ribosome pathway appears to play an important role included in the article and the supplemental files accessible
during microtuber formation of D. opposita. In addition, through the journal website. The raw sequencing datasets
photosynthesis and metabolism of amino sugars and nucle- supporting the results of this article were deposited in
otide sugars also appear to have a supporting function in the NCBI SRA repository [http://www.ncbi.nlm.nih.gov/
these processes. sra?term = SRP061414].10 BioMed Research International
Conflicts of Interest and D. praehensilis,” Plant Cell Tissue and Organ Culture,
vol. 41, no. 3, pp. 229–235, 1995.
The authors declare that they have no competing interests. [5] P. O. Ovono, C. Kevers, and J. Dommes, “Axillary prolifera-
tion and tuberisation of Dioscorea cayenensis–D. rotundata
Authors’ Contributions complex,” Plant Cell Tissue and Organ Culture, vol. 91, no. 2,
pp. 107–114, 2007.
JL, XZ, and ML contributed to the project design, data [6] M. Balogun, I. Fawole, S. Ng, N. Ng, H. Shiwachi, and
analysis, and manuscript writing. YD, SL, JY, CL, and XZ H. Kikuno, “Interaction among cultural factors in microtuber-
performed the sample collection, analyzed the data, and ization of white yam (Dioscorea rotundata),” Tropical Science,
drafted the manuscript. All authors have read and approved vol. 46, no. 1, pp. 55–59, 2006.
the final manuscript. Junhua Li and Xiting Zhao contributed [7] F.-Q. Chen, Y. Fu, D.-L. Wang, X. Gao, and L. Wang, “The
equally to this work. effect of plant growth regulators and sucrose on the micropro-
pagation and microtuberization of Dioscorea nipponica
Makino,” Journal of Plant Growth Regulation, vol. 26, no. 1,
Acknowledgments pp. 38–45, 2007.
We are grateful to Dr. Qingxiang Yang for reading the man- [8] A. E. Uduebo, “Effect of external supply of growth substances
on axillary proliferation and development in Dioscorea bulbi-
uscript. This research was supported by the National Nature
fera,” Annals of Botany, vol. 35, no. 1, pp. 159–163, 1971.
Science Foundation of China (81274019 and 31670317) and
the Plan for Scientific Innovation Talent of Henan Province [9] M. Li, J. Li, Y. I. P. E. N. G. Wang et al., “A simple method for
microtuber production in Dioscorea opposita using single
(114200510013).
nodal segments,” Pakistan Journal of Botany, vol. 47,
pp. 665–668, 2015.
Supplementary Materials [10] P. O. Ovono, C. Kevers, and J. Dommes, “Effects of reducing
sugar concentration on in vitro tuber formation and sprouting
Figure S1: sequence-length distributions of unigenes and in yam (Dioscorea cayenensis–D. rotundata complex),” Plant
transcripts assembled from the Illumina reads. Figure S2: Cell Tissue and Organ Culture (PCTOC), vol. 99, no. 1,
verification of RNA-Seq results by qRT-PCR. Figure S3: pp. 55–59, 2009.
result summary for sequence-homology searching against [11] Z. Wang, M. Gerstein, and M. Snyder, “RNA-Seq: a revolu-
the NCBI NR database. Table S1: summary of Illumina tionary tool for transcriptomics,” Nature Reviews Genetics,
transcriptome sequencing for microtuber formation of D. vol. 10, no. 1, pp. 57–63, 2009.
opposita. Table S2: description of the 20 most up- and down- [12] J. A. Ward, L. Ponnala, and C. A. Weber, “Strategies for tran-
regulated genes in the BUD vs. EXP comparison. Table S3: scriptome analysis in nonmodel plants,” American Journal of
description of the 20 most up- and downregulated genes in Botany, vol. 99, no. 2, pp. 267–276, 2012.
the MTV vs. EXP comparison. Table S4: description of the [13] N. Cloonan and S. M. Grimmond, “Transcriptome content
20 most up- and downregulated genes in the MTV vs. BUD and dynamics at single-nucleotide resolution,” Genome Biol-
comparison. Table S5: KEGG enriched pathways of specific- ogy, vol. 9, no. 9, p. 234, 2008.
ity DEGs identified during microtuber induction and forma- [14] U. Nagalakshmi, Z. Wang, K. Waern et al., “The transcrip-
tion. Table S6: differentially expressed genes related to starch tional landscape of the yeast genome defined by RNA sequenc-
and sucrose metabolism during microtuber formation. Table ing,” Science, vol. 320, no. 5881, pp. 1344–1349, 2008.
S7: differentially expressed genes related to plant hormone [15] W. L. Morris, R. D. Hancock, L. J. M. Ducreux et al., “Day
signal transduction pathways during microtuber formation. length dependent restructuring of the leaf transcriptome and
Table S8: the primers used for the analysis of gene expression metabolome in potato genotypes with contrasting tuberization
by qRT-PCR. (Supplementary Materials) phenotypes,” Plant, Cell & Environment, vol. 37, no. 6,
pp. 1351–1363, 2014.
References [16] J. Shan, W. Song, J. Zhou et al., “Transcriptome analysis
reveals novel genes potentially involved in photoperiodic
[1] M. Li, J. Li, W. Liu et al., “A protocol forin vitroproduction of tuberization in potato,” Genomics, vol. 102, no. 4, pp. 388–
microtubers in Chinese yam (Dioscorea opposita),” Bioscience, 396, 2013.
Biotechnology, and Biochemistry, vol. 78, no. 6, pp. 1005–1009, [17] J. P. van Dijk, K. Cankar, P. J. M. Hendriksen et al., “The iden-
2014. tification and interpretation of differences in the transcrip-
[2] M. Cheng, D. Wang, and H. Peng, “Evolution and change of tomes of organically and conventionally grown potato
the species quality and authentic producing areas of Dioscorea tubers,” Journal of Agricultural and Food Chemistry, vol. 60,
opposita Thunb,” Chinese Journal of Medical History, vol. 44, no. 9, pp. 2090–2101, 2012.
no. 2, pp. 81–84, 2014. [18] S. D’Ippólito, M. L. Martín, M. F. Salcedo et al., “Tran-
[3] M. Borges, W. Ceiro, S. Meneses et al., “Regeneration and mul- scriptome profiling of Fusarium solani f. sp. eumartii-infected
tiplication of Dioscorea alata germplasm maintained in vitro,” potato tubers provides evidence of an inducible defense
Plant Cell, Tissue and Organ Culture, vol. 76, no. 1, pp. 87–90, response,” Physiological and Molecular Plant Pathology,
2004. vol. 75, no. 1-2, pp. 3–12, 2010.
[4] B. Malaurie, O. Pungu, and M.-F. Trouslot, “Effect of growth [19] M. W. Dees, E. Lysøe, M. B. Brurberg, P. Somervuo,
regulators concentrations on morphological development of M. Almvik, and J. P. T. Valkonen, “Global gene expression in
meristem-tips in Dioscorea cayenensis-D. rotundata complex the common scab pathogen, Streptomyces scabies, exposed toBioMed Research International 11
potato microtubers,” Annals of Applied Biology, vol. 165, no. 1, [36] R. L. Tatusov, N. D. Fedorova, J. D. Jackson et al., “The COG
pp. 43–52, 2014. database: an updated version includes eukaryotes,” BMC Bio-
[20] L. Gao, Z. J. Tu, B. P. Millett, and J. M. Bradeen, “Insights into informatics, vol. 4, no. 1, p. 41, 2003.
organ-specific pathogen defense responses in plants: RNA-seq [37] X. Mao, T. Cai, J. G. Olyarchuk, and L. Wei, “Automated
analysis of potato tuber-Phytophthora infestans interactions,” genome annotation and pathway identification using the
BMC Genomics, vol. 14, no. 1, p. 340, 2013. KEGG Orthology (KO) as a controlled vocabulary,” Bioinfor-
[21] S. J. Ferreira, M. Senning, S. Sonnewald, P. M. Keßling, matics, vol. 21, no. 19, pp. 3787–3793, 2005.
R. Goldstein, and U. Sonnewald, “Comparative transcriptome [38] W. C. Dunlap, A. Starcevic, D. Baranasic et al., “KEGG
analysis coupled to X-ray CT reveals sucrose supply and orthology-based annotation of the predicted proteome of
growth velocity as major determinants of potato tuber starch Acropora digitifera: ZoophyteBase—an open access and
biosynthesis,” BMC Genomics, vol. 11, no. 1, p. 93, 2010. searchable database of a coral genome,” BMC Genomics,
[22] K. P. Kaminski, A. H. Petersen, M. Sønderkær et al., “Tran- vol. 14, no. 1, p. 509, 2013.
scriptome analysis suggests that starch synthesis may proceed [39] D. M. Beckles, J. Craig, and A. M. Smith, “ADP-glucose
via multiple metabolic routes in high yielding potato culti- pyrophosphorylase is located in the plastid in developing
vars,” PLoS One, vol. 7, no. 12, article e51248, 2012. tomato fruit,” Plant Physiology, vol. 126, no. 1, pp. 261–266,
[23] J. K. Van Harsselaar, J. Lorenz, M. Senning, U. Sonnewald, and 2001.
S. Sonnewald, “Genome-wide analysis of starch metabolism [40] E. Ewing and P. Struik, “Tuber formation in potato: induction,
genes in potato (Solanum tuberosum L.),” BMC Genomics, initiation and growth,” Horticultural Reviews, vol. 14, p. 197,
vol. 18, no. 1, p. 37, 2017. 1992.
[24] S. Götz, J. M. García-Gómez, J. Terol et al., “High-throughput [41] R. G. F. Visser, D. Vreugdenhil, T. Hendriks, and E. Jacobsen,
functional annotation and data mining with the Blast2GO “Gene expression and carbohydrate content during stolon to
suite,” Nucleic Acids Research, vol. 36, no. 10, pp. 3420–3435, tuber transition in potatoes (Solanum tuberosum),” Physiolo-
2008. gia Plantarum, vol. 90, no. 2, pp. 285–292, 1994.
[25] B. Li and C. N. Dewey, “RSEM: accurate transcript quantifica- [42] Q. Yang, K. Liu, X. Niu et al., “Genome-wide Identification of
tion from RNA-Seq data with or without a reference genome,” PP2C Genes and Their Expression Profiling in Response to
BMC Bioinformatics, vol. 12, no. 1, 2011. Drought and Cold Stresses in Medicago truncatula,” Scientific
[26] S. Anders and W. Huber, “Differential expression analysis for Reports, vol. 8, no. 1, p. 12841, 2018.
sequence count data,” Genome Biology, vol. 11, no. 10, [43] M. Li, S. Liu, W. Liu, J. Li, X. Zhang, and X. Zhao, “Physiolog-
p. R106, 2010. ical and biochemical changes in Dioscorea opposita during the
[27] L. Wang, Z. Feng, X. Wang, X. Wang, and X. Zhang, “DEGseq: process of microtuber formation,” Plant Physiology Journal,
an R package for identifying differentially expressed genes vol. 53, pp. 807–814, 2017.
from RNA-seq data,” Bioinformatics, vol. 26, no. 1, pp. 136– [44] U. Roessner-Tunali, E. Urbanczyk-Wochniak, T. Czechowski,
138, 2010. A. Kolbe, L. Willmitzer, and A. R. Fernie, “De novo amino acid
[28] Y. Benjamini and D. Yekutieli, “The control of the false discov- biosynthesis in potato tubers is regulated by sucrose levels,”
ery rate in multiple testing under dependency,” Annals of Sta- Plant Physiology, vol. 133, no. 2, pp. 683–692, 2003.
tistics, vol. 29, pp. 1165–1188, 2001.
[45] A. R. Fernie, A. Tiessen, M. Stitt, L. Willmitzer, and
[29] T. van Parys, I. Melckenbeeck, M. Houbraken et al., “A Cytos- P. Geigenberger, “Altered metabolic fluxes result from shifts
cape app for motif enumeration with ISMAGS,” Bioinformat- in metabolite levels in sucrose phosphorylase-expressing
ics, vol. 33, no. 3, pp. btw626–btw463, 2017. potato tubers,” Plant, Cell & Environment, vol. 25, no. 10,
[30] M. Kanehisa and S. Goto, “KEGG: Kyoto Encyclopedia of pp. 1219–1232, 2002.
Genes and Genomes,” Nucleic Acids Research, vol. 28, no. 1, [46] Z. Zhang, J. Li, H. Liu, K. Chong, and Y. Xu, “Roles of
pp. 27–30, 2000. ubiquitination-mediated protein degradation in plant
[31] X. Zhao, X. Zhang, X. Guo et al., “Identification and validation responses to abiotic stresses,” Environmental and Experimen-
of reference genes for qRT-PCR studies of gene expression in tal Botany, vol. 114, pp. 92–103, 2015.
Dioscorea opposita,” BioMed Research International,
vol. 2016, Article ID 3089584, 13 pages, 2016.
[32] S. Guo, S. Dai, P. K. Singh et al., “A membrane-bound NAC-
like transcription factor OsNTL5 represses the flowering in
Oryza sativa,” Frontiers in Plant Science, vol. 9, pp. 555–555,
2018.
[33] H. Liu, S. Guo, M. Lu et al., “Biosynthesis of DHGA12 and its
roles in Arabidopsis seedling establishment,” Nature Commu-
nications, vol. 10, no. 1, p. 1768, 2019.
[34] M. G. Grabherr, B. J. Haas, M. Yassour et al., “Full-length tran-
scriptome assembly from RNA-Seq data without a reference
genome,” Nature Biotechnology, vol. 29, no. 7, pp. 644–652,
2011.
[35] S. F. Altschul, T. L. Madden, A. A. Schäffer et al., “Gapped
BLAST and PSI-BLAST: a new generation of protein database
search programs,” Nucleic Acids Research, vol. 25, no. 17,
pp. 3389–3402, 1997.You can also read

















































