GENCODE 2021 - Oxford Academic Journals
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
D916–D923 Nucleic Acids Research, 2021, Vol. 49, Database issue Published online 3 December 2020
doi: 10.1093/nar/gkaa1087
GENCODE 2021
Adam Frankish 1 , Mark Diekhans 2 , Irwin Jungreis 3,4 , Julien Lagarde5 ,
Jane E. Loveland 1 , Jonathan M. Mudge1 , Cristina Sisu6,7 , James C. Wright8 ,
Joel Armstrong2 , If Barnes1 , Andrew Berry1 , Alexandra Bignell1 , Carles Boix3,4,9 ,
Silvia Carbonell Sala5 , Fiona Cunningham 1 , Tomás Di Domenico10 , Sarah Donaldson1 ,
Ian T. Fiddes2 , Carlos Garcı́a Girón 1 , Jose Manuel Gonzalez1 , Tiago Grego1 ,
Matthew Hardy1 , Thibaut Hourlier 1 , Kevin L. Howe 1 , Toby Hunt1 , Osagie G. Izuogu1 ,
Rory Johnson 11,12 , Fergal J. Martin 1 , Laura Martı́nez10 , Shamika Mohanan1 ,
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
Paul Muir13,14 , Fabio C. P. Navarro6 , Anne Parker1 , Baikang Pei6 , Fernando Pozo10 , Ferriol
Calvet Riera1 , Magali Ruffier 1 , Bianca M. Schmitt1 , Eloise Stapleton1 ,
Marie-Marthe Suner 1 , Irina Sycheva1 , Barbara Uszczynska-Ratajczak15 , Maxim Y. Wolf16 ,
Jinuri Xu6 , Yucheng T. Yang6,17 , Andrew Yates 1 , Daniel Zerbino 1 , Yan Zhang 6,18 ,
Jyoti S. Choudhary8 , Mark Gerstein6,17,19 , Roderic Guigó5,20 , Tim J. P. Hubbard21 ,
Manolis Kellis3,4 , Benedict Paten2 , Michael L. Tress 10 and Paul Flicek 1,*
1
European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton,
Cambridge CB10 1SD, UK, 2 UC Santa Cruz Genomics Institute, University of California, Santa Cruz, Santa Cruz, CA
95064, USA, 3 MIT Computer Science and Artificial Intelligence Laboratory, 32 Vassar St, Cambridge, MA 02139,
USA, 4 Broad Institute of MIT and Harvard, 415 Main Street, Cambridge, MA 02142, USA, 5 Centre for Genomic
Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, Barcelona, E-08003
Catalonia, Spain, 6 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520,
USA, 7 Department of Bioscience, Brunel University London, Uxbridge UB8 3PH, UK, 8 Functional Proteomics,
Division of Cancer Biology, Institute of Cancer Research, 237 Fulham Road, London SW3 6JB, UK, 9 Computational
and Systems Biology Program, Massachusetts Institute of Technology, Cambridge, MA, USA, 10 Bioinformatics Unit,
Spanish National Cancer Research Centre (CNIO), Madrid, Spain, 11 Department of Medical Oncology, Inselspital,
University Hospital, University of Bern, Bern, Switzerland, 12 Department of Biomedical Research (DBMR), University
of Bern, Bern, Switzerland, 13 Department of Molecular, Cellular & Developmental Biology, Yale University, New
Haven, CT 06520, USA, 14 Systems Biology Institute, Yale University, West Haven, CT 06516, USA, 15 Centre of New
Technologies, University of Warsaw, Warsaw, Poland, 16 Department of Biomedical Informatics at Harvard Medical
School, 10 Shattuck Street, Suite 514, Boston, MA 02115, USA, 17 Program in Computational Biology &
Bioinformatics, Yale University, Bass 432, 266 Whitney Avenue, New Haven, CT 06520, USA, 18 Department of
Biomedical Informatics, College of Medicine, The Ohio State University, Columbus, OH 43210, USA, 19 Department of
Computer Science, Yale University, Bass 432, 266 Whitney Avenue, New Haven, CT 06520, USA, 20 Universitat
Pompeu Fabra (UPF), Barcelona, E-08003 Catalonia, Spain and 21 Department of Medical and Molecular Genetics,
King’s College London, Guys Hospital, Great Maze Pond, London SE1 9RT, UK
Received September 21, 2020; Revised October 21, 2020; Editorial Decision October 22, 2020; Accepted October 24, 2020
ABSTRACT of primary data and bioinformatic tools and analy-
sis generated both within the consortium and ex-
The GENCODE project annotates human and mouse
ternally to support the creation of transcript struc-
genes and transcripts supported by experimental
tures and the determination of their function. Here,
data with high accuracy, providing a foundational re-
we present improvements to our annotation infras-
source that supports genome biology and clinical ge-
tructure, bioinformatics tools, and analysis, and the
nomics. GENCODE annotation processes make use
* To whom correspondence should be addressed. Tel: +44 1223 492581; Fax: +44 1223 494494; Email: flicek@ebi.ac.uk
C The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which
permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.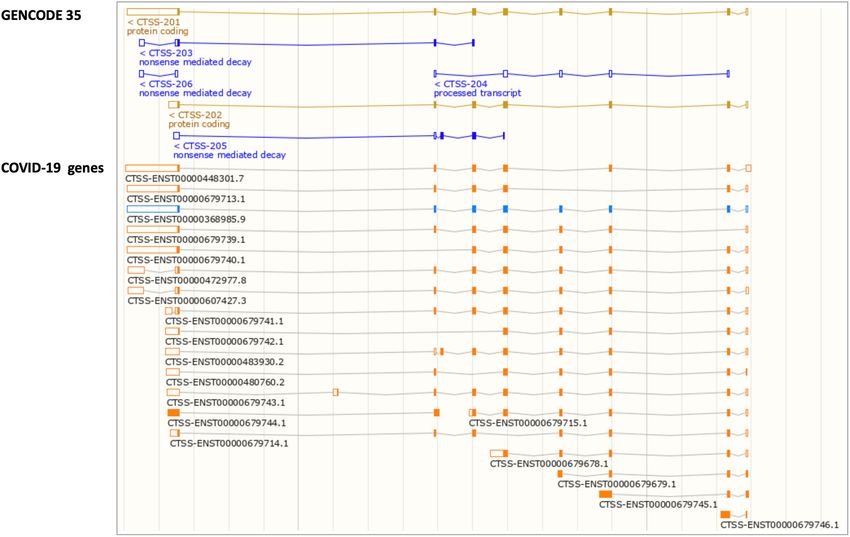
Nucleic Acids Research, 2021, Vol. 49, Database issue D917
advances they support in the annotation of the hu- gene or protein is present in one resource, it will be repre-
man and mouse genomes including: the completion sented in the others or there will be an explanation why not.
of first pass manual annotation for the mouse refer- We are part of the Matched Annotation from NCBI and
ence genome; targeted improvements to the anno- EMBL-EBI (MANE) project to define a single representa-
tation of genes associated with SARS-CoV-2 infec- tive ‘MANE Select’ transcript for all protein-coding genes
and ensure its structure and sequence is identical in both
tion; collaborative projects to achieve convergence
the Ensembl/GENCODE and RefSeq genesets. We anno-
across reference annotation databases for the anno- tated new human protein-coding genes based on improved
tation of human and mouse protein-coding genes; analyses and experimental validation using mass spectrom-
and the first GENCODE manually supervised auto- etry. We have also improved the annotation of lncRNAs via
mated annotation of lncRNAs. Our annotation is ac- the discovery of novel loci and novel transcripts at existing
cessible via Ensembl, the UCSC Genome Browser loci primarily based on incorporating long transcriptomic
and https://www.gencodegenes.org. sequence data generated using the CLS protocol (5).
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
INTRODUCTION GENE ANNOTATION INFRASTRUCTURE
GENCODE produces widely-used reference genome an- We have made several key improvements to our processes
notation of protein-coding and non-coding loci including and tools used for manual gene annotation.
alternatively spliced transcripts and pseudogenes for the The Ensembl/GENCODE geneset is a merge of the man-
human and mouse genomes and makes these annotations ual gene annotation created by the Ensembl-HAVANA
freely available for the benefit of biomedical research and team (methods and validation described in 6–8) and the
genome interpretation. The GENCODE consortium devel- automated annotation produced by the Ensembl Geneb-
ops, maintains and improves targeted tools, analysis and uild team (9,10). Historically, these data were produced sep-
primary transcriptomic and proteomic data in support of arately and stored in independent and structurally differ-
gene and transcript annotation. These resources support ent databases before being merged into a single set for re-
updates to genes in all functional classes or biotypes, includ- lease. To speed data release and reduce complexity, we have
ing (i) the discovery of new features such as novel protein- now moved all manual annotation and computational an-
coding genes and long non-coding RNA (lncRNA) genes; notation into a single database for human (and another for
(ii) the extension of existing annotation including the identi- mouse). In addition to continuing the support of manual
fication of novel alternatively spliced transcripts at protein- annotation, this transition allows manual annotators to di-
coding and lncRNA loci and (iii) the continuous critical rectly ‘bless’, update or remove computationally annotated
reappraisal of existing annotation that may result in re- models. Most significantly, new genes and transcripts re-
moval or reclassification of protein-coding genes that lack leased early via the GENCODE update trackhub will be
evidence of protein-coding potential given all data now assigned their Ensembl (ENSX) formatted stable IDs at
available. GENCODE defines genes in terms of their tran- their creation, having previously been given an interim ID
scriptional and functional overlap. The functional informa- (OTTX format).
tion implicit in the CDS of protein-coding gene supports Long-read transcriptomic sequencing methods includ-
decision making and provides high confidence in the inter- ing those from Pacific Biosciences (PacBio) and Oxford
pretation of protein-coding genes. For lncRNAs, the lack Nanopore Technologies (ONT) produce data volumes that
of analogous knowledge makes representation of complex require change to our manual annotation process. In re-
lncRNA loci difficult and we are working with lncRNA sponse, we developed the TAGENE pipeline to support
community and other reference annotation databases to im- greater automation of transcript model creation based on
prove their annotation. long-read datasets generated both within GENCODE and
Among other achievements, over the last two years we by other groups. TAGENE implements filtering and merg-
have developed a manually supervised automated annota- ing of long transcriptomic datasets before clustering puta-
tion pipeline and an annotation triage tool to leverage the tive transcripts into loci (both existing and novel) and ap-
volume of data generated by current transcriptomics experi- plying further filters based on other transcriptomic datasets,
ments while ensuring that the resulting annotated transcript including RNA-seq supported introns and existing GEN-
models maintain the quality of expert human annotation. CODE annotation (Figure 1). The clustering and final filter-
We have completed the first pass manual annotation of the ing steps are applied following multiple iterations of manual
mouse reference genome based on experiences on complet- review until a point is reached where the false positive rate
ing the human annotation in 2013 and have used whole for the addition of spurious models is
D918 Nucleic Acids Research, 2021, Vol. 49, Database issue
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
Figure 1. Schematic of the TAGENE workflow to add long transcriptomic data to GENCODE annotation. Points in the workflow where manual review
is applied are indicated.
ated by the TAGENE pipeline. Kestrel is complementary notation for all chromosomes. LncRNAs continue to show
to our set of high quality annotation tools in Zmap, Blixem the largest increases in number, particularly in human where
and Dotter, which were initially developed for the clone-by- our efforts have been concentrated.
clone annotation approach used for the first pass annota-
tion of the human and mouse reference genomes. Kestrel’s
streamlined functionality is often all that is required to an- PROTEIN-CODING GENES
swer emerging manual annotation questions and thus faster In response to the SARS-CoV-2 pandemic, we have ap-
than our traditional workflow. plied our annotation resources to human genes with poten-
tial links to viral infection and COVID-19 disease primarily
by investigating whether existing annotation for these genes
GENE ANNOTATION UPDATES
can be improved. Our list of genes for reannotation comes
The GENCODE consortium has improved and extended from several sources including recently published drug re-
the annotation of the human and mouse reference genomes purposing studies identifying host proteins associated with
and makes the annotation publicly available (see Table 1 for other related coronaviruses (14) and human proteins found
annotation statistics from the most recent GENCODE re- to physically associate with SARS-CoV-2 viral proteins in
leases). the cell (15). We also included genes curated by UniProt (4)
Since June 2018, ∼37 000 genes (∼32 000 human and and the Human Cell Atlas project (16) as well as interferon-
5000 mouse) and ∼63 000 transcripts (∼55 000 human and stimulated genes with known antiviral activity (17). These
∼8000 mouse) have either been created or updated in the efforts added previously unannotated alternatively-spliced
GENCODE geneset (see Table 2 for a breakdown of new transcript models and updated existing GENCODE tran-
and updated genes and transcripts by functional biotype). script models, in particular ‘partial’ models that were in-
During this period we have completed the first pass annota- complete at their 5 and/or 3 ends that could be extended
tion of the mouse reference genome and conducted a num- to full length. All annotation takes advantage of long tran-
ber of tightly focussed annotation projects including the hu- scriptomic datasets and RNAseq data that was unavailable
man and mouse olfactory receptor repertoire (12) and a re- at the time of initial annotation. To date we have updated
annotation of developmental and epileptic encephalopathy- the annotation for 280 genes, adding ∼3700 novel tran-
associated genes (13). scripts and updating a further ∼850.
Although a number of protein-coding genes in both hu- GENCODE has been actively collaborating with other
man and mouse have been added, removed or had their reference annotation databases to try to achieve conver-
biotype changed over the past two years, the total number gence on the annotation of protein-coding genes in hu-
of genes is stable. Similarly, the number of pseudogenes of man and mouse. The MANE project aims to create a sin-
protein-coding genes is broadly stable for human, although gle agreed transcript for every human protein-coding gene
our ability to better identify unitary pseudogenes has led that has a 100% match for sequence and structure (splic-
to an increase in this specific class. In mouse, an increase ing, UTR and CDS) in both the Ensembl/GENCODE and
in pseudogene count reflects the completion of manual an- RefSeq (3) annotation sets. The project is driven by two in-Nucleic Acids Research, 2021, Vol. 49, Database issue D919
Table 1. Total numbers of genes and transcripts in the GENCODE 35 (Human) and GENCODE M25 (Mouse) releases by gene functional biotype
Protein-coding LncRNA Pseudogene sRNA IG/TR
Human GENCODE 35 Genes 19954 17957 14767 7569 645
Transcripts 154580 48684 18664 7569 666
Mouse GENCODE M25 Genes 21859 13197 13741 6108 700
Transcripts 102241 18856 14522 6108 864
Table 2. Numbers of genes and transcripts that have been added to or updated in GENCODE Human and Mouse annotation since June 2018
Human Mouse
New Updated New and updated New Updated New and updated
Protein-coding 131 17995 18126 845 1584 2429
Genes LncRNA 1965 7678 9643 670 282 952
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
Pseudogene 75 4152 4227 676 266 942
Total 2171 29825 31996 2191 2132 4323
Protein-coding 11334 21406 32740 4323 968 5291
Transcripts LncRNA 19042 2807 21849 1171 73 1244
Pseudogene 247 259 506 794 137 931
Total 30623 24472 55095 6288 1178 7466
dependent pipelines, one from each centre, followed by ex- genes, novel pseudogenes, and novel coding sequence (21).
tensive investigation and discussion by expert human anno- We have automated our process to generate updated lists of
tators where the pipelines do not agree. The latest release PhyloCSF Candidate Coding Regions (PCCRs), which are
of MANE v0.91, gives an overall coverage of 84% of all then examined by manual annotators. In human, PCCRs
protein-coding genes. are part of the standard annotation workflow. In mouse, a
We have been working extensively to improve the interop- targeted review of unannotated PCCRs analogous to that
erability of the existing annotations with UniProt. Genome previously undertaken in human has led to the identifica-
Integration with FuncTion and Sequence (GIFTS) is a joint tion of 64 novel protein-coding genes, 376 novel coding
project between Ensembl and the EMBL-EBI component exons in preexisting protein-coding genes, and 202 pseu-
of the UniProt project and is currently available for human dogenes including 56 unitary pseudogenes. PhyloCSF has
and mouse proteins https://www.ebi.ac.uk/gifts/. GIFTS also been used to identify candidate ribosomal stop codon
calculates mappings and pairwise alignments between En- readthrough events in human and mouse (22,23). Follow-
sembl transcripts that have a protein translation with their ing manual review of these and several others identified ex-
corresponding UniProt protein entries. Unmapped UniProt perimentally, 14 and 11 genes with stop codon readthrough
proteins are investigated by annotators from both teams events have been annotated in human and mouse, respec-
and edited where necessary. We have investigated 1044 tively (Figure 2).
unmapped human (716) and mouse (328) proteins from GENCODE annotation utilises proteomics data to
UniProt and identified cases where the GENCODE anno- supplement transcriptomic and evolutionary evidence of
tation needs to be updated (2 human, 49 mouse), and pro- protein-coding functionality and we have continued to both
teins that appear invalid in their putative genomic context generate experimental MS data and use publicly available
(640 human, 54 mouse). data sets to aid the identification and annotation of protein-
We continue to analyse publications external to the GEN- coding genes. Our data generation focus is on elements of
CODE consortium reporting additional protein-coding the proteome that are missed by standard proteomics ap-
genes in the light of GENCODE criteria. For example, we proaches including the use of 155 novel synthetic peptides
examined the novel protein-coding genes reported in the targeting distinct and unique peptides mapping to putative
CHESS gene annotation set (18), adding five protein-coding coding genes, newly discovered protein coding genes that re-
genes, 16 pseudogenes and 37 lncRNAs. A recent survey of quire validation, and pseudogenes that have shown strong
heart ORFs (19), has so far resulted in the annotation of 12 peptide evidence in previous experiments. These peptides
additional human protein-coding genes. are compiled into a reference spectral library, which is used
GENCODE annotation makes substantial use of com- to validate their existence in our experimental proteomics
parative genomics to help identify regions on the genome data and large public MS datasets. For example transcrip-
with protein-coding potential. For example, we have used tomic, conservation, and ribosome profiling data combined
Cactus to create a 600-way vertebrate whole genome align- with experimental peptide evidence supported the discovery
ment incorporating data from the 200 Mammals and Bird and validation of an alternate protein isoform originating
10K projects as the basis of a single base-pair resolution from a non-ATG start site in the gene POLG (24), and high-
map of evolutionary selection (20). We will directly use lighted a novel class of unannotated protein-coding features
these alignments within the PhyloCSF phylogenetic analy- that are now under active investigation.
sis tool (1). The PhyloCSF pipeline has also been run on the To support the automated analysis of proteomics data for
each new release of the human and mouse genome annota- genome annotation we collaborated with the PRIDE (25)
tions to facilitate the discovery of additional novel coding proteomics repository at EMBL-EBI to build a reprocess-D920 Nucleic Acids Research, 2021, Vol. 49, Database issue
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
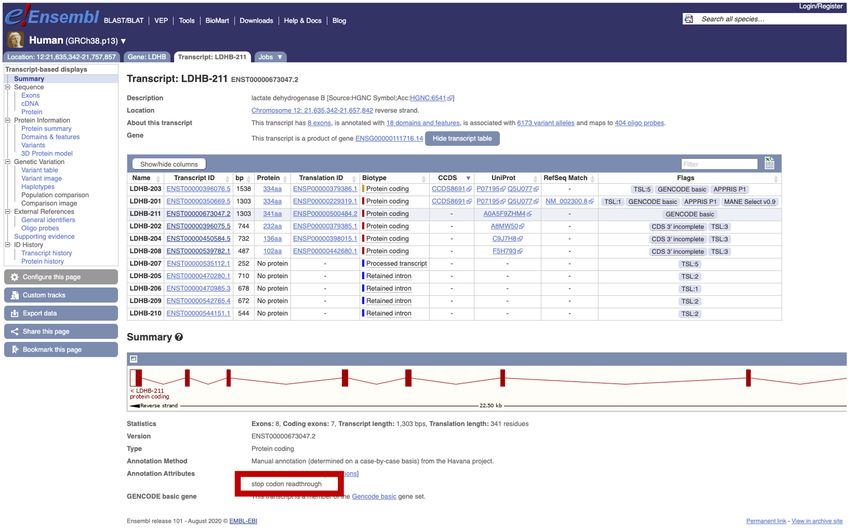
Figure 2. Screenshot from the Ensembl genome browser of the transcript view page for the gene LDHB, which contains a transcript (ENST00000673047,
LDHB-211) with an annotated stop-codon readthrough event. The location of the annotation attribute flagging the stop-codon readthrough is highlighted
by the red box.
ing and peptide-to-genome mapping pipeline for public pro- in both human and mouse. Focusing primarily on unan-
teomics. notated regions such as GWAS sites, putative enhancers,
Finally, we developed a pipeline based on UniProt (4), and non-GENCODE lncRNA catalogs (e.g. miTranscrip-
APPRIS (26), PhyloCSF (1), Ensembl gene trees (10), tome (28), NONCODE (29), FANTOM CAT (30)). In to-
RNA-seq, MS and variation data to identify annotated tal we have produced more than 36 million ONT reads
protein-coding genes with weak or no support. This method and 2 million PacBio Sequel (PBS) reads identifying thou-
enables us to scrutinise currently annotated protein-coding sands of potential novel loci (∼1600 in Human, ∼4500 in
genes in the human and mouse gene set for misclassified mouse) in currently unannotated genomic regions for re-
gene models. To date we have flagged as potential non- view and inclusion in the Ensembl/GENCODE geneset.
coding genes more than 2475 human and 1807 mouse genes Long transcriptomic sequence data produced within GEN-
that were annotated as protein-coding. These are then re- CODE and from public data archives has been run through
viewed in an iterative and ongoing process by expert manual our TAGENE workflow and the results of this first set
annotators and retained, removed or reclassified based on of analysis released to the public in GENCODE 31 (June
their current supporting evidence. To date, ∼1000 human 2019). These initial results have already made a signifi-
protein-coding genes have been reviewed and 119 removed cant difference to the coverage of lncRNAs in GENCODE,
or reclassified. A complementary approach has also been with the addition of 1711 novel loci and 17 858 transcripts,
developed to identify missing and partially complete gene an 11% and 60% increase compared to the previous release
models in the human genome and submit to manual review. respectively.
LncRNAS PSEUDOGENES
We have made improvements to the Capture Long Se- Our pseudogene annotation has benefited from the analy-
quencing (CLS) lab protocol (5), including a 5 cap se- sis of new datasets. For example, using RNA-seq datasets
lection step (‘CapTrap’) (27), which increases the propor- from ENTEx-pseudogene expression in various human tis-
tion of sequenced full-length transcripts and the use of sues we have developed a computational framework to ac-
Spiked-in RNA Variant Control Mixes (SIRVs). Applying curately quantify the expression level of pseudogenes, and
CLS, we have generated long transcriptomic data target- identify actively transcribed pseudogenes in each tissue. We
ing a variety of suspected lncRNA-producing genomic loci have also used our pseudogene annotation in 16 closely re-Nucleic Acids Research, 2021, Vol. 49, Database issue D921
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
Figure 3. A screenshot from the Ensembl genome browser of the location view for the CTSS gene. The Comprehensive annotation from GENCODE 35 is
shown in the upper panel and the updated annotation in the COVID-19 genes trackhub is shown in the lower panel. Transcript models that are unchanged
with respect to release Ensembl 101 are coloured blue, whereas new models or pre-existing models that have been modified are shown in orange.
lated mouse strains from the Mouse Genomes Project (31) UCSC genome browsers. In the Ensembl browser, the hub
to create orthology relationships for the conserved annota- has been added to the Track Hub Registry (accessed via
tions and the identification of patterns of pseudogene gain the ‘Custom tracks’ section), and can be connected to
and loss between strains (32) and give a prototype for work by searching for ‘GENCODE update’. Alternatively, the
annotating human pseudogenes leveraging variation across data can be added as a custom track in both Ensembl
the human population. and UCSC browsers (http://ftp.ebi.ac.uk/pub/databases/
gencode/update trackhub/hub.txt). Additionally, a track-
hub of updates to genes associated with COVID-19
DATA ACCESS can be accessed in the same way (http://ftp.ebi.ac.uk/
GENCODE gene sets are currently updated up to four pub/databases/gencode/covid19 trackhub/hub.txt). In the
times each year for both human and mouse. Each release ‘COVID-19 genes’ track data view, transcript models
is versioned and made available immediately upon release that are unchanged with respect to release GENCODE
from Ensembl (6) and https//www.gencodegenes.org with 35/Ensembl 101 are coloured blue, whereas new models or
release on the UCSC Genome Browser (33) normally fol- pre-existing models that have been modified are shown in
lowing shortly thereafter. The current human release is orange (Figure 3). We also offer BED and gtf files for these
GENCODE 35 (August 2020) and the current mouse re- annotations.
lease is GENCODE M25 (April 2020). Additional infor- We have made available the public ‘Synonymous
mation and previous releases can be found at https//www. Constraint’ track hub in the UCSC Genome Browser
gencodegenes.org. that shows protein-coding regions under synonymous
GENCODE is the now the standardised default human constraint, indicating an overlapping function, and
and mouse annotation for both the Ensembl and UCSC synonymous accelerated regions, indicating a high mu-
genome browsers following a transition of UCSC’s mouse tation rate (https://data.broadinstitute.org/compbio1/
annotation in April 2019. Data is presented through all of SynonymousConstraintTracks/trackHub/).
the standard interfaces from both resources. Supported GENCODE annotation is available on the
To expedite public access to updated annotation be- GRCh38 human reference assembly and the GRCm38
tween releases, all annotation changes are made freely mouse reference assembly. Selected human releases are
available within 24 h via the ‘GENCODE update’ Track mapped back to the GRCh37 assembly and made available
Hub, which can be accessed at both the Ensembl and from UCSC and https://www.gencodegenes.org as a serviceD922 Nucleic Acids Research, 2021, Vol. 49, Database issue
to the community. The resulting mapping are not manu- noncoding RNAs with capture long-read sequencing. Nat Genet., 49,
ally checked and may have errors especially in complicated 1731–1740.
6. Harrow,J., Denoeud,F., Frankish,A., Reymond,A., Chen,C.K.,
regions of the human genome. We recommend use of the Chrast,J., Lagarde,J., Gilbert,J.G., Storey,R., Swarbreck,D. et al.
GRCh38 annotations if possible. (2006) GENCODE: producing a reference annotation for ENCODE.
Training about the GENCODE annotation and its use Genome Biol., 7, S4.
is available from the Ensembl and UCSC training team 7. Harrow,J., Frankish,A., Gonzalez,J.M., Tapanari,E., Diekhans,M.,
and user support is available from the Ensembl and UCSC Kokocinski,F., Aken,B.L., Barrell,D., Zadissa,A., Searle,S. et al.
(2012) GENCODE: the reference human genome annotation for The
helpdesks. ENCODE Project. Genome Res., 22, 1760–1774.
8. Howald,C., Tanzer,A., Chrast,J., Kokocinski,F., Derrien,T.,
Walters,N., Gonzalez,J.M., Frankish,A., Aken,B.L., Hourlier,T. et al.
CONCLUSION (2012) Combining RT-PCR-seq and RNA-seq to catalog all genic
elements encoded in the human genome. Genome Res., 22, 1698–1710.
The GENCODE consortium leverages the best available 9. Aken,B.L., Ayling,S., Barrell,D., Clarke,L., Curwen,V., Fairley,S.,
data, analysis and tools to continually improve the gene an- Fernandez Banet,J., Billis,K., Garcı́a Girón,C., Hourlier,T. et al.
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
notation of the human and mouse reference genomes. We (2016) The Ensembl gene annotation system. Database (Oxford),
have developed new methods and workflows to take ad- 2016, baw093.
10. Yates,A.D., Achuthan,P., Akanni,W., Allen,J., Allen,J.,
vantage of the increasing quality and volume of data, and Alvarez-Jarreta,J., Amode,M.R., Armean,I.M., Azov,A.G.,
in particular long transcriptomic data, while maintaining Bennett,R. et al. (2020) Ensembl 2020. Nucleic Acids Res., 48,
the specificity afforded by expert human oversight. We ex- D682–D688.
pect our ability to use new data to improve our coverage 11. Kokocinski,F., Harrow,J. and Hubbard,T. (2010) AnnoTrack–a
of novel genes and alternatively spliced transcripts will al- tracking system for genome annotation. BMC Genomics, 11, 538.
12. Barnes,I.H.A., Ibarra-Soria,X., Fitzgerald,S., Gonzalez,J.M.,
low us to move towards a more complete representation of Davidson,C., Hardy,M.P., Manthravadi,D., Van Gerven,L.,
all gene features of known functional classes as we monitor Jorissen,M., Zeng,Z. et al. (2020) Expert curation of the human and
the emergence of new functional features that may require mouse olfactory receptor gene repertoires identifies conserved coding
annotation such as alternative translations of known coding regions split across two exons. BMC Genomics, 21, 196.
13. Steward,C.A., Roovers,J., Suner,M.M., Gonzalez,J.M.,
genes, non-canonical translations in, for example, lncRNAs Uszczynska-Ratajczak,B., Pervouchine,D., Fitzgerald,S., Viola,M.,
and mRNA with multiple functions. Stamberger,H., Hamdan,F.F. et al. (2019) Re-annotation of 191
developmental and epileptic encephalopathy-associated genes
unmasks de novo variants in SCN1A. NPJ Genom. Med., 4, 31.
FUNDING 14. Zhou,Y., Hou,Y., Shen,J., Huang,Y., Martin,W. and Cheng,F. (2020)
Network-based drug repurposing for novel coronavirus
National Human Genome Research Institute of the Na- 2019-nCoV/SARS-CoV-2. Cell Discov., 6, 14.
tional Institutes of Health [U41HG007234]; the content 15. Gordon,D.E., Jang,G.M., Bouhaddou,M., Xu,J., Obernier,K.,
is solely the responsibility of the authors and does not White,K.M., O’Meara,M.J., Rezelj,V.V., Guo,J.Z., Swaney,D.L. et al.
necessarily represent the official views of the National In- (2020) A SARS-CoV-2 protein interaction map reveals targets for
drug repurposing. Nature, 583, 459–468.
stitutes of Health; Wellcome Trust [WT108749/Z/15/Z, 16. Rozenblatt-Rosen,O., Stubbington,M.J.T., Regev,A. and
WT200990/Z/16/Z]; European Molecular Biology Labo- Teichmann,S.A. (2017) The Human Cell Atlas: from vision to reality.
ratory; Swiss National Science Foundation through the Na- Nature, 550, 451–453
tional Center of Competence in Research ‘RNA & Disease’ 17. Schoggins,J.W. and Rice,C.M. (2011) Interferon-stimulated genes and
their antiviral effector functions. Curr. Opin. Virol., 1, 519–525.
(to R.J.); Medical Faculty of the University of Bern (to 18. Pertea,M., Shumate,A., Pertea,G., Varabyou,A., Breitwieser,F.P.,
R.J). Funding for open access charge: National Institutes Chang,Y.C., Madugundu,A.K., Pandey,A. and Salzberg,S.L. (2018)
of Health. CHESS: a new human gene catalog curated from thousands of
Conflict of interest statement. Paul Flicek is a member of the large-scale RNA sequencing experiments reveals extensive
Scientific Advisory Boards of Fabric Genomics, Inc., and transcriptional noise. Genome Biol., 28, 208.
19. an Heesch,S., Witte,F., Schneider-Lunitz,V., Schulz,J.F., Adami,E.,
Eagle Genomics, Ltd. Faber,A.B., Kirchner,M., Maatz,H., Blachut,S., Sandmann,C.L.
et al. (2019) The translational landscape of the human heart. Cell,
178, 242–260.
REFERENCES 20. Armstrong,J., Hickey,G., Diekhans,M., Fiddes,I.T., Novak,A.M.,
1. Lin,M.F., Jungreis,I. and Kellis,M. (2011) PhyloCSF: a comparative Deran,A., Fang,Q., Xie,D., Feng,S., Stiller,J. et al. (2020) Progressive
genomics method to distinguish protein coding and non-coding Cactus is a multiple-genome aligner for the thousand-genome era.
regions. Bioinformatics, 27, i275–82. Nature, 587, 246–251.
2. Raney,B.J., Dreszer,T.R., Barber,G.P., Clawson,H., Fujita,P.A., 21. Mudge,J.M., Jungreis,I., Hunt,T., Gonzalez,J.M., Wright,J.C.,
Wang,T., Nguyen,N., Paten,B., Zweig,A.S., Karolchik,D. et al. (2014) Kay,M., Davidson,C., Fitzgerald,S., Seal,R., Tweedie,S. et al. (2019)
Track data hubs enable visualization of user-defined genome-wide Discovery of high-confidence human protein-coding genes and exons
annotations on the UCSC Genome Browser. Bioinformatics, 30, by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome
1003–1005. Res., 29, 2073–2087.
3. O’Leary,N.A., Wright,M.W., Brister,J.R., Ciufo,S., Haddad,D., 22. Jungreis,I., Chan,C.S., Waterhouse,R.M., Fields,G., Lin,M.F. and
McVeigh,R., Rajput,B., Robbertse,B., Smith-White,B., Ako-Adjei,D. Kellis,M. (2016) Evolutionary dynamics of abundant stop codon
et al. (2016) Reference sequence (RefSeq) database at NCBI: current readthrough. Mol. Biol. Evol., 33, 3108–3132.
status, taxonomic expansion, and functional annotation. Nucleic 23. Loughran,G., Jungreis,I., Tzani,I., Power,M., Dmitriev,R.I.,
Acids Res., 44, D733–D745 Ivanov,I.P., Kellis,M. and Atkins,J.F. (2018) Stop codon readthrough
4. The UniProt Consortium (2019) UniProt: a worldwide hub of protein generates a C-terminally extended variant of the human vitamin D
knowledge. Nucleic Acids Res., 47, D506–D515 receptor with reduced calcitriol response. J. Biol. Chem., 293,
5. Lagarde,J., Uszczynska-Ratajczak,B., Carbonell,S., Pérez-Lluch,S., 4434–4444.
Abad,A., Davis,C., Gingeras,T.R., Frankish,A., Harrow,J., Guigo,R. 24. Khan,Y.A., Jungreis,I., Wright,J.C., Mudge,J.M., Choudhary,J.S.,
et al. (2017) High-throughput annotation of full-length long Firth,A.E. and Kellis,M. (2020) Evidence for a novel overlappingNucleic Acids Research, 2021, Vol. 49, Database issue D923
coding sequence in POLG initiated at a CUG start codon. BMC annotation database for long non-coding RNAs. Nucleic Acids Res.,
Genet., 21, 25. 46, D308–D314.
25. Perez-Riverol,Y., Bai,J., Bernal-Llinares,M., Hewapathirana,S., 30. Hon,C.C., Ramilowski,J.A., Harshbarger,J., Bertin,N.,
Kundu,D.J., Inuganti,A., Griss,J., Mayer,G., Eisenacher,M., Pérez,E. Rackham,O.J., Gough,J., Denisenko,E., Schmeier,S., Poulsen,T.M.,
et al. (2019) The PRIDE database and related tools and resources in Severin,J. et al. (2017) An atlas of human long non-coding RNAs
2019: improving support for quantification data. Nucleic Acids Res., with accurate 5 ends. Nature, 543, 199–204.
47, D442–D450. 31. Lilue,J., Doran,A.G., Fiddes,I.T., Abrudan,M., Armstrong,J.,
26. Rodriguez,J.M., Rodriguez-Rivas,J., Di Domenico,T., Vázquez,J., Bennett,R., Chow,W., Collins,J., Collins,S., Czechanski,A. et al.
Valencia,A. and Tress,M.L. (2018) APPRIS 2017: principal isoforms (2018) Sixteen diverse laboratory mouse reference genomes define
for multiple gene sets. Nucleic Acids Res., 46, D213–D217. strain-specific haplotypes and novel functional loci. Nat. Genet., 50,
27. Carninci,P., Kvam,C., Kitamura,A., Ohsumi,T., Okazaki,Y., Itoh,M., 1574–1583.
Kamiya,M., Shibata,K., Sasaki,N., Izawa,M. et al. (1996) 32. Sisu,C., Muir,P., Frankish,A., Fiddes,I., Diekhans,M., Thybert,D.,
High-efficiency full-length cDNA cloning by biotinylated CAP Odom,D.T., Flicek,P., Keane,T.M., Hubbard,T. et al. (2020)
trapper. Genomics, 37, 327–336. Transcriptional activity and strain-specific history of mouse
28. Iyer,M.K., Niknafs,Y.S., Malik,R., Singhal,U., Sahu,A., Hosono,Y., pseudogenes. Nat. Commun., 11, 3695.
Barrette,T.R., Prensner,J.R., Evans,J.R., Zhao,S. et al. (2015) The 33. Kent,W.J., Sugnet,C.W., Furey,T.S., Roskin,K.M., Pringle,T.H.,
Downloaded from https://academic.oup.com/nar/article/49/D1/D916/6018430 by guest on 25 March 2021
landscape of long noncoding RNAs in the human transcriptome. Zahler,A.M. and Haussler,D. (2002) The human genome browser at
Nat. Genet., 47, 199–208. UCSC. Genome Res., 12, 996–1006.
29. Fang,S., Zhang,L., Guo,J., Niu,Y., Wu,Y., Li,H., Zhao,L., Li,X.,
Teng,X., Sun,X. et al. (2018) NONCODEV5: a comprehensiveYou can also read

















































