PG-Priming Enhances Doxorubicin Influx to Trigger Necrotic and Autophagic Cell Death in Oral Squamous Cell Carcinoma - MDPI
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Journal of
Clinical Medicine
Article
PG-Priming Enhances Doxorubicin Influx to Trigger
Necrotic and Autophagic Cell Death in Oral
Squamous Cell Carcinoma
Shian-Ren Lin and Ching-Feng Weng *
Department of Life Science and Institute of Biotechnology, National Dong Hwa University,
Hualien 97401, Taiwan; d9813003@gms.ndhu.edu.tw
* Correspondence: cfweng@gms.ndhu.edu.tw; Tel.: +886-3-8903637
Received: 17 September 2018; Accepted: 18 October 2018; Published: 21 October 2018
Abstract: Synergistic effects between natural compounds and chemotherapy drugs are believed to
have fewer side effects with equivalent efficacy. However, the synergistic potential of prodigiosin
(PG) with doxorubicin (Dox) chemotherapy is still unknown. This study explores the synergistic
mechanism of PG and Dox against oral squamous cell carcinoma (OSCC) cells. Three OSCC cell lines
were treated with different PG/Dox combinatory schemes for cytotoxicity tests and were further
investigated for cell death characteristics by cell cycle flow cytometry and autophagy/apoptosis
marker labelling. When OSCC cells were pretreated with PG, the cytotoxicity of the subsequent
Dox-treatment was 30% higher than Dox alone. The cytotoxic efficacy of PG-pretreated was found
better than those of PG plus Dox co-treatment and Dox-pretreatment. Increase of Sub-G1 phase
and caspase-3/LC-3 levels without poly (ADP-ribose) polymeras (PARP) elevation indicated both
autophagy and necrosis occurred in OSCC cells. Dox flux after PG-priming was further evaluated
by rhodamine-123 accumulation and Dox transporters analysis to elucidate the PG-priming effect.
PG-priming autophagy enhanced Dox accumulation according to the increase of rhodamine-123
accumulation without the alterations of Dox transporters. Additionally, the cause of PG-triggered
autophagy was determined by co-treatment with endoplasmic reticulum (ER) stress or AMP-activated
protein kinase (AMPK) inhibitor. PG-induced autophagy was not related to nutrient deprivation and
ER stress was proved by co-treatment with specific inhibitor. Taken together, PG-priming autophagy
could sensitize OSCC cells by promoting Dox influx without regulation of Dox transporter. The
PG-priming might be a promising adjuvant approach for the chemotherapy of OSCC.
Keywords: prodigiosin; doxorubicin; priming; influx; autophagy
1. Introduction
Doxorubicin (Adriamycin, Dox), isolated from soil bacteria Streptomyces peucetius, is the first
member of anthracyclines (including daunorubicin, epirubicin, and idarubicin) [1,2]. The main
action of Dox is intercalating within DNA pairs, which leads to the inhibition of topoisomerase
IIβ and results in cell cycle blockage [3]. Rendering non-tissue specific characteristics, Dox gets
wide indications including leukemia, neuroblastoma, breast carcinoma, ovarian carcinoma, and most
recurrent or metastatic cancer [4]. Apart from these, the non-specific characteristics of Dox cause some
severe adverse effects in cancer patients including immunosuppression, bone marrow suppression,
hepatotoxicity, cardiotoxicity, and mucositis [5,6]. The cause of side effects is from the inhibition
of cell division as well as reactive oxygen species (ROS) by-products (doxorubicin-semiquinone,
doxorubicinol, dexrazoxane, and 7-deoxy-doxorubicinone) during metabolism of doxorubicin in
mitochondria [2,7–9]. As a result, these adverse effects might limit the applicable dosage and cancer
J. Clin. Med. 2018, 7, 375; doi:10.3390/jcm7100375 www.mdpi.com/journal/jcm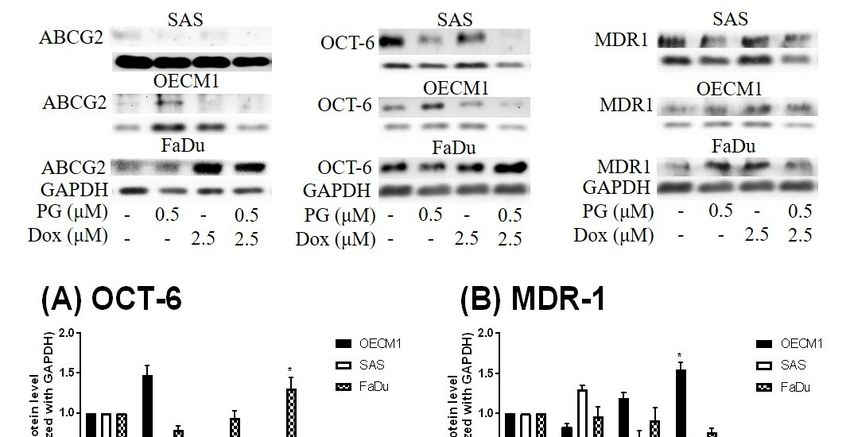
J. Clin. Med. 2018, 7, 375 2 of 17
treating efficacy of Dox. Accordingly, the alternative approach or new formulation to attenuate Dox
side effects and enhance Dox efficacy turns out to be a crucial issue for Dox use in the regimen of
chemotherapy. Recently, nanocarriers have exerted a favorable theme and some research has focused on
this topic to dissolve these obstacles, such as Dox encapsulated in pH-sensitive, ultrasonic-responsive,
or co-capsulated with MDR-1 inhibitor in PEGylated, liposome, or PLGA nano-carrier, which also
promote Dox uptake [10–14]. However, cytotoxicity of nanoparticle conjugated Dox was 10 times
lower than free-form Dox, which also restricted the use in cancer treatment [15].
A long treatment period with a low dose chemotherapeutic drug might induce chemoresistance
within cancer cells and subsequently toxicity could affect its use [16]. Notably, prevailing mechanisms
of chemoresistance could be classified into the following seven phases: drug flux, DNA damage
repair, cell death inhibition, epithelial-mesenchymal transition (EMT), drug target alteration, drug
inactivation, and epigenetics [17]. In Dox resistance, dug efflux would be the most concerning
phase [16]. Dox can import into cells through solute carrier family 22 member 16 (SLC22A16, also
known organic cation transporter 6, oct-6) and export by ATP-binding cassette transporter family
members, in which multidrug-related protein 1 (MDR-1 or p-glycoprotein) and breast cancer resistance
protein (BCRP or ABCG2) are involved [3]. These proteins will be regulated (upregulation of exporter
and downregulation of importer) during long-term exposure to a non-toxic dose of Dox [18]. Thereby,
numerous studies have put more attention to reducing MDR over-expression to reverse multidrug
resistance. CRISPER/Cas-9 gene editing, ursodeoxycholic acid, or Zingiber officinale Roscoe, have been
reported to down-regulate ABCB1 gene expressions in chemo-resistant cancer cells [19–21].
Prodigiosin (PG, PubChem CID: 5351169) is a red prodiginine pigment isolated from various
bacteria including Serratia marcescens, Pseudoalteromonas rubra, Hahella chejuensis, and actinomycete
bacteria [22–25]. Even though the original biological function in producer bacteria remains
unclear, PG has been identified with numerous biological activities including antimicrobial [26–29],
antimalarial [26,27,30], and antitumor [26,27,31–34] activities. Moreover, PG showed apoptotic
inducing property in many cancer types such as lung cancer [35–37], breast cancer [38,39],
colorectal cancer [40–42], leukemia [43,44], and hepatocellular carcinoma [45] without normal cell
cytotoxicity [41,46]. Recently, PG has also been identified as an autophagy inducer in OSCC cells [47,48].
However, the application of PG as an adjuvant in chemotherapy is still unknown.
2. Experimental Section
2.1. Research Aims
This study was conducted to explore the potential of PG combined with doxorubicin in anti-cancer
activity by using oral squamous cell carcinoma (OSCC) cells as a test platform. Next, experiments
tested the synergistic effects of PG and Dox against OSCC cells to evaluate the adjuvant potential of
PG for cancer therapy. Furthermore, the underlying molecular mechanisms of enhanced doxorubicin
cytotoxicity under PG-priming were also investigated.
2.2. Reagents
Cell-cultured medium and reagents were purchased from Thermo-Fisher (Waltham, MA, USA).
Prodigiosin was purified by Dr. Yu-Hsin Chen (Department of Life Science, National Dong-Hwa
University, Hualien, Taiwan). Liposome-coated doxorubicin (abbreviated as Dox) was obtained
from Dr. Ming-Fang Cheng (Division of Histology and Clinical Pathology, Hualian Army Forces
General Hospital, Hualien, Taiwan). Inhibitors used in this study were purchased from Santa Cruz
Biotechnology (Dallas, TX, USA). General chemicals were purchased from Sigma Aldrich (Merck
KGaA, Darmstadt, Germany). Polyvinylidene difluoride (PVDF) membrane used in Western blotting
was obtained from GE Healthcare (Chicago, IL, USA). The antibodies used in this study were obtained
from Santa Cruz Biotechnology, as shown in Table 1.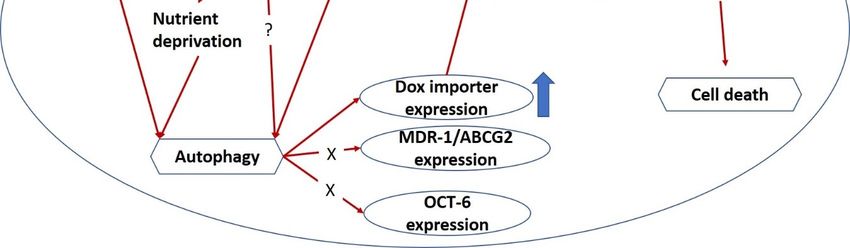
J. Clin. Med. 2018, 7, 375 3 of 17
Table 1. Antibodies used in this study.
Protein Host Source RRID MW (kDa) Dilution
MDR-1 Human Mouse AB_2565004 180 1:200
PARP1 Human Mouse AB_1127036 116 1:200
ABCG2 Human Mouse AB_629007 80 1:200
OCT-6 Human Mouse AB_10989254 46 1:200
GAPDH Human Mouse AB_1124759 37 1:1000
Caspase3 Human Mouse AB_1119997 32 1:200
LC3 I/II Human Mouse AB_2137722 15/18 1:200
HRP-conjugated 2nd Ab Mouse Goat AB_92635 1:5000
MW, molecular weight; RRID, Research Resource Identifiers.
2.3. Cell Culture
Cell lines used in this study were obtained from different sources: Human pharynx squamous
carcinoma FaDu from Dr. Chun-Shu Lin (Radiation Oncology Department, Tri-Service General
Hospital, Taipei, Taiwan), human oral squamous cell carcinoma cell line OECM1 and tongue carcinoma
cell line SAS from Professor Ta-Chun Yuan (Department of Life Science, National Dong Hwa University,
Hualien, Taiwan), and human bronchus epithelial cell BEAS-2b from American Type Culture Collection
(ATCC). OECM1 and SAS were cultured in Roswell Park Memorial Institute medium 1640 (RPMI 1640),
FaDu in minimum essential medium (MEM), and BEAS-2b in Dulbecco’s Modified Eagle Medium
(DMEM) and medium was changed every 2 days. All culture media were mixed with 10% fetal
bovine serum (FBS) and 1% antibiotic-antimycotic and cultured within 37 ◦ C, 5% CO2 incubator
(Thermo-Fisher). Cells were detached by 0.25% trypsin/ethylenediaminetetraacetic acid (EDTA)
for further experiments. All experiments were obtained within 20 passages concerning uniformity
and reproducibility.
2.4. Cytotoxicity Assay
Cytotoxicity was determined using a colorimetric assay by MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) previously described in the
literature [48]. The optical density (OD) alteration of mitochondrial enzymatic activity was converted
into the cell numbers according to the cell viability or cytotoxicity. Briefly, 1 × 104 cells per wells were
seeded in 96-well plate and incubated in culture conditions overnight. Then, cells were divided into
the six following treatment groups: (1) PG-Dox group: treated with PG followed by Dox, (2) Dox-PG
group treated with Dox and then PG, and (3) PG + Dox group: treated PG and Dox at the same time,
respectively. An additional three groups performed the same treatments with the above-described and
replaced Dox with cisplatin. All treatments were carried out in 12 h and 1 mg/mL of MTT solution
was added and further incubated for 4 h at 37 ◦ C as treatment finished. Finally, liquid in wells was
replaced by dimethyl sulfoxide (DMSO), and the absorbance at 570 nm was measured by Multiskan™
FC microplate photometer (Thermo-Fisher). Cytotoxicity of each treatment was represented by cell
viability which calculated from the absorbance ratio at 570 nm between treated and untreated groups.
To understand the cause of PG- and Dox-induced cell death, inhibitor recovering assays were also
performed following the above protocol. Autophagy inhibitors (bafilomycin A1 and 3-methyladenine),
endoplasmic reticulum (ER) stress inhibitors (tauroursodeoxycholic acid, TUDC), and AMPK inhibitors
(dorsomorphin, CC) were cotreated with various concentrations of PG or Dox, respectively.
2.5. Cell Cycle Analysis
Cell cycle analysis was carried out by flow cytometry. Firstly, 1 × 106 cells/well of OSCC were
seeded into 6-well plates and incubated in culture condition overnight. To understand drug-pretreated
effect, cells were treated with PG for 12 h, Dox for 12 h, or PG for 12 h followed by Dox for additional
12 h, respectively. After treatment, cells including culture medium were collected using trypsin/EDTA
J. Clin. Med. 2018, 7, 375 4 of 17
and washed by phosphate buffer saline (PBS) twice before being fixed with pre-cooled 70% ethanol/PBS
overnight. After fixation, cells were washed twice by PBS and stained with staining buffer (20 µg/mL
of propidium iodide, 0.1% Triton X-100, 0.2 mg/mL RNase A) at 37 ◦ C for 1 h. The fluorescent intensity
in the cells was measured by a flow cytometer (CytomicsTM FC500, Beckman, Fullerton, CA, USA).
Data from 104 cells were collected for each data file. Fluorescent intensities for each cell line were
acquiesced and plotted by flow cytometer software. The gating of each phase was based on the
acquisition histogram of untreated controls. Phases of each group were collected and the average of
each phase was calculated within the groups.
2.6. Doxorubicin Flux Analysis
Efflux and influx of Dox was determined by indirect method which used rhodamine 123, a
fluorescent Dox transporter substrate, detected as an indicator described previously [49,50]. Briefly,
1 × 104 cells/well of OSCC were seeded in 96-well plate and incubated in culture condition overnight.
After cell confluence, cells were divided into four groups and then treated with various regimens,
respectively. For Rhodamine-influx: short-term influx assay: 0.5 µM of PG in full-culture medium
was added and incubated for 1 h and replaced culture medium with 2 µM of rhodamine 123 in PBS
for additional 1 h; long-term influx assay: the same treatment as short-term influx assay instead the
incubation duration of PG from 1 h to 12 h. For Rhodamine-efflux: short-term efflux assay: 2 µM
of rhodamine 123 in PBS was firstly incubated for 1 h and followed by 0.5 µM of PG in full-culture
medium for additional 1 h; long-term efflux assay: the same as treatment as short-term influx assay
instead the incubation duration of PG from 1 h to 12 h. After incubation, cells were trebly washed by
PBS and lysed with 0.1% triton X-100 and the fluorescent intensity was determined at 485/538 nm by
α-screen multi-plate reader (Perkin Elmer, Waltham, MA, USA). These rhodamine flux studies were
used to estimate DOX flux.
2.7. Western Blotting
The detail protocol of Western blotting was described in our previous study [51]. In brief, 1 × 106
cells/well of OSCC cells were seeded into a 6-well plate and treated the same as with cell cycle
analysis. Cells were washed by PBS and lysed by radioimmunoprecipitation assay buffer (RIPA). Then,
30 µg of total proteins from cell lysates were electrophoretically separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) and transferred into polyvinylidene difluoride (PVDF)
membrane. Proteins of interest were identified via incubation with appropriate primary followed by
horseradish peroxidase (HRP)-conjugated secondary antibodies and exposed to the iBright imaging
system (Thermo-Fisher) for monitoring intensity of signals after soaking in enhance chemiluminescent
(ECL) reagents. Data acquisition was also performed by the iBright imaging system, and signal
intensity was normalized with GAPDH as an internal control.
2.8. Statistical Analysis
All quantified results were shown as mean ± standard deviation (SD) of three independent
experiments. Significant analysis used a one-way ANOVA, followed by Dunnett’s test. A data
histogram was built by GraphPad Prism 7.04 (La Jolla, CA, USA).
3. Results
3.1. Cytotoxicity Change of PG/Dox Treated Strategies
In this study, three combined manners (pretreatment, cotreatment, and posttreatment) of PG/Dox
were tested in OSCC. In pretreatment and post-treatment approaches, chemicals were previously
treated for 12 h and subsequently washed out, followed by new chemical treatment for additional
12 h. Therefore, the term “PG-pretreatment” would be defined as a “PG-priming” procedure in the
subsequent section. The cell viability of all tested OSCC cells were declined in all combined strategiesJ. Clin. Med. 2018, 7, x FOR PEER REVIEW 5 of 16
J. Clin. Med. 2018, 7, 375 5 of 17
procedure in the subsequent section. The cell viability of all tested OSCC cells were declined in all
combined strategies except cotreatment in SAS (p < 0.05). In three combined strategies,
PG-pretreatment
except cotreatment gotinthe
SAShighest reducing
(p < 0.05). levels
In three (as compared
combined strategies,with Dox alone) thangot
PG-pretreatment those of the
the highest
other
reducingtwo strategies, as shown with
levels (as compared in Figure
Dox 1A.
alone)This result
than posed
those of thetheother
potential of PG-pretreatment
two strategies, as shownas in
PG-priming in OSCC. When doubling the concentrations of PG, cell viability
Figure 1A. This result posed the potential of PG-pretreatment as PG-priming in OSCC. When doubling was the same as that of
single concentration,
the concentrations revealed
of PG, 0.5 μM
cell viability wasofthePG, samewhich
as thatwas the concentration,
of single maximum concentration
revealed 0.5 for µM
PG-priming.
of PG, whichAlso, was extending
the maximum the PG-priming
concentration period up to 24 h, the
for PG-priming. cytotoxicity
Also, extendingofthe Dox failed to
PG-priming
exhibit
period an up additive potentiation.
to 24 h, the cytotoxicity These results
of Dox indicated
failed thatan12additive
to exhibit h of PG-priming
potentiation.might reach
These the
results
maximum effect (data not shown). Moreover, with PG-priming in normal
indicated that 12 h of PG-priming might reach the maximum effect (data not shown). Moreover, with cell lines BEAS-2b, the cell
viability
PG-priming of Dox treatment
in normal didBEAS-2b,
cell lines not show the viability
the cell decreaseof asDoxmuch as OSCC,
treatment did noteven
showthough the
the decrease
concentrations of PG and Dox were twice higher than that of OSCC,
as much as OSCC, even though the concentrations of PG and Dox were twice higher than that of as shown in Figure 1B. This
result
OSCC,indicated
as shownthat PG-priming
in Figure 1B. Thiswas more
result effective
indicated that and less toxic
PG-priming wasthan
more that of Dox
effective andalone. An
less toxic
additional experiment was to investigate whether the PG-priming effect
than that of Dox alone. An additional experiment was to investigate whether the PG-priming effect could also be observed in
golden chemotherapy drug cisplatin, however, the cytotoxic enhancement
could also be observed in golden chemotherapy drug cisplatin, however, the cytotoxic enhancement in in PG/Dox combination
could
PG/Dox not combination
be found in PG/cisplatin combination,
could not be found as showncombination,
in PG/cisplatin in Figure 1C. asTaking
shown all results1C.
in Figure together,
Taking
PG-priming could enhance Dox cytotoxicity in OSCC cells through a Dox-related
all results together, PG-priming could enhance Dox cytotoxicity in OSCC cells through a Dox-related mechanism. In
subsequent experiments, the type of cell death triggered by PG-priming
mechanism. In subsequent experiments, the type of cell death triggered by PG-priming and the and the underlying
mechanism
underlyingwere furtherwere
mechanism investigated.
further investigated.
(A) PG/Dox-OSCC (B) PG/Dox-BEAS-2b
0.7 0.7
OECM1
0.6 0.6
SAS
0.5 0.5
FaDu
0.4 * 0.4
OD
OD
0.3 * 0.3
* *
0.2 0.2
0.1 0.1
0.0 0.0
PG (M ) - 0.5 - 0.5 0.5 - 0.5 0.5 - 0.5 PG (M ) - 0.5 - 0.5 1 - 1
DOX (M ) - - 2.5 2.5 - 2.5 2.5 - 2.5 2.5 DOX (M ) - - 2.5 2.5 - 5 5
PG - DOX DOX - PG PG+DOX
(C) PG/Cisplatin-OSCC
0.65
OECM1
0.52 * SAS
FaDu
0.39
OD
0.26
0.13
0.00
PG (M ) - 1 - 1
Cisplatin (M ) - - 5 5
Figure 1. Alteration of cytotoxicity in sequential PG (prodigiosin)/Dox (doxorubicin) and PG/cisplatin
combination
Figure in oral squamous
1. Alteration cell carcinoma
of cytotoxicity (OSCC) and
in sequential PG BEAS-2b cells. (A) OSCC
(prodigiosin)/Dox and (B) normal
(doxorubicin) and
bronchus cells-BEAS-2b were treated with various schemes of PG and Dox, and (C) Cisplatin
PG/cisplatin combination in oral squamous cell carcinoma (OSCC) and BEAS-2b cells. (A) OSCC substituted
and
Dox
(B) for 12 bronchus
normal h and analyzed cell viability
cells-BEAS-2b were by 3-(4, with
treated 5-dimethylthiazol-2-yl)-2,
various schemes of PG 5-diphenyltetrazolium
and Dox, and (C)
bromide (MTT)
Cisplatin assay.Dox
substituted The results
for 12 were
h andrepresented as mean
analyzed cell ± SDby
viability from three
3-(4, individual experiments.
5-dimethylthiazol-2-yl)-2,
* p < 0.05 as compared bromide
5-diphenyltetrazolium with Dox(MTT)
alone.assay. The results were represented as mean ± SD from three
individual experiments. * p < 0.05 as compared with Dox alone.
3.2. Identification of Cell Death Characteristics
3.2. Identification of Cellof
Numerous types Death Characteristics
cell death were found in cells, but the most common types were apoptosis and
autophagy, respectively. These two cell-death types could be distinguished by analyzing apoptosis and
autophagy-related protein and cell markers, and the most obvious marker would be cell cycle analysis.J. Clin. Med. 2018, 7, x FOR PEER REVIEW 6 of 16
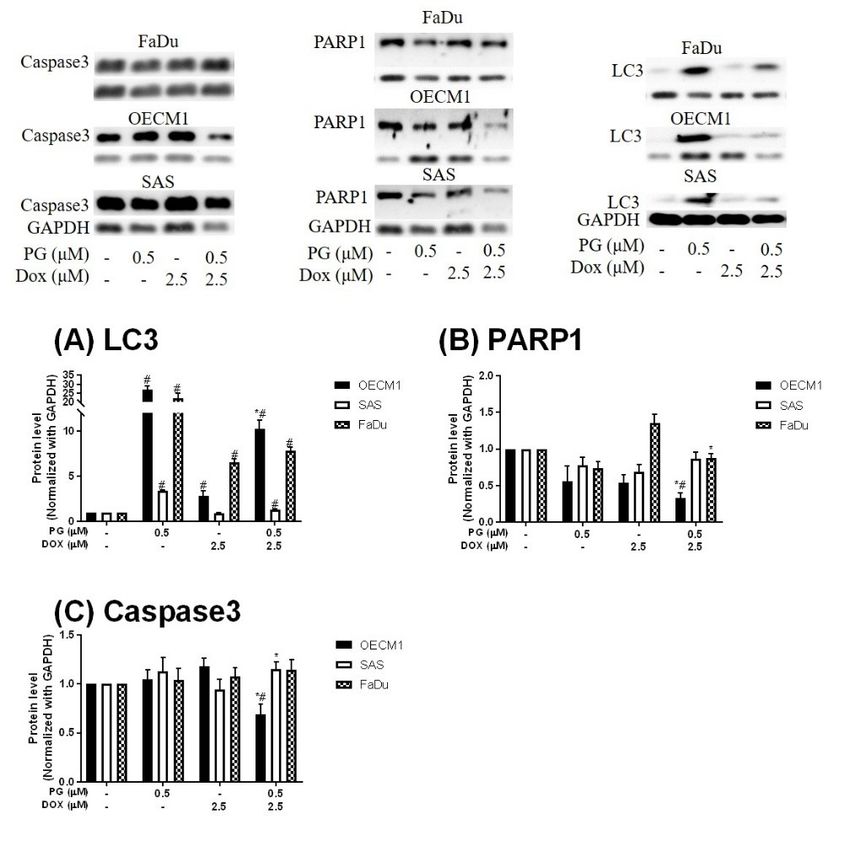
Numerous types of cell death were found in cells, but the most common types were apoptosis
and autophagy, respectively. These two cell-death types could be distinguished by analyzing
J. Clin. Med. 2018, 7, 375 6 of 17
apoptosis and autophagy-related protein and cell markers, and the most obvious marker would be
cell cycle analysis. In cell cycle analysis of SAS, Sub-G1 was significantly increased, but non-cleaved
InPARP1 andanalysis
cell cycle caspase-3 protein
of SAS, Sub-Glevels were not decreased after PG-priming, as shown in Figures 2B
1 was significantly increased, but non-cleaved PARP1 and caspase-3
and 3B,C.
protein levelsThese
were results revealed
not decreased that
after PG-priming
PG-priming, prior toinDox
as shown Figurestreatment
2B and would lead results
3B,C. These to SAS
undergoing necrosis. Moreover, while PG combined with autophagic
revealed that PG-priming prior to Dox treatment would lead to SAS undergoing necrosis. Moreover, inhibitor (bafilomycin A1
while PG combined with autophagic inhibitor (bafilomycin A1 (BA1) and 3-methyladenine (3MA), cellin
(BA1) and 3-methyladenine (3MA), cell viability of PG-priming could be recovered, as shown
Figure 4.
viability of This phenomenon
PG-priming could might indicateasthat
be recovered, bothinnecrosis
shown Figure 4.and Thisautophagy
phenomenon weremight
activated after
indicate
that both necrosis and autophagy were activated after PG-priming and the autophagy would be ain
PG-priming and the autophagy would be a major clue, which was further confirmed by increases
LC3 clue,
major proteinwhichlevels,
wasas shown
further in Figureby3A.
confirmed In FaDu
increases cells,
in LC3 Sub-Glevels,
protein 1 phase was not significantly
as shown in Figure 3A.
increased after PG/Dox treatment, as shown in Figure 2C. Also,
In FaDu cells, Sub-G1 phase was not significantly increased after PG/Dox treatment, non-cleaved PARP1 and caspase-3
as shown in
protein levels while PG-priming followed by Dox treatment were decreased
Figure 2C. Also, non-cleaved PARP1 and caspase-3 protein levels while PG-priming followed by Dox when compared with
Dox alone,
treatment as shown
were decreasedin Figure
when 3B,C.
comparedThe results
with Dox alsoalone,
showed the necrosis
as shown in Figureactivation
3B,C. within FaDu
The results
cells similar to SAS cells. Unlike SAS cells, this cell viability could not be
also showed the necrosis activation within FaDu cells similar to SAS cells. Unlike SAS cells, this cell recovered by autophagy
inhibitor,
viability as shown
could not beinrecovered
Figure 4. InbyFaDu cells, LC3
autophagy protein
inhibitor, aslevels
shown were also significantly
in Figure 4. In FaDuincreased
cells, LC3in
the PG/Dox
protein levelsgroup,
were alsoas shown in Figure
significantly 3A. These
increased results
in the revealed
PG/Dox group, thatasboth
shown necrosis and 3A.
in Figure autophagy
These
were activated in FaDu cells and necrosis would be the main cause of
results revealed that both necrosis and autophagy were activated in FaDu cells and necrosis would cytotoxicity, whereas OECM1
becells
theshowed
main cause different patterns from
of cytotoxicity, the above
whereas OECM1 two cells
cells lines.
showedThedifferent
sub-G1 phasepatternsdidfrom
not significantly
the above
increase either in the non-cleaved PARP1 or caspase-3 protein levels,
two cells lines. The sub-G1 phase did not significantly increase either in the non-cleaved PARP1 as shown in Figures 2A and
or
3B,C. Taken together, PG-priming cell death of OECM1 was not related
caspase-3 protein levels, as shown in Figures 2A and 3B,C. Taken together, PG-priming cell death of to apoptosis or necrosis. On
the other
OECM1 washand, the cell
not related viability of
to apoptosis orOECM1
necrosis.after
On thePG-priming
other hand, could be viability
the cell recovered by autophagy
of OECM1 after
inhibitors, as shown in Figure 4. Also, LC3 protein levels were increased
PG-priming could be recovered by autophagy inhibitors, as shown in Figure 4. Also, LC3 protein 10-fold, as shown in Figure
3A. These
levels two results
were increased gave as
10-fold, a clear
shownclue for autophagy
in Figure 3A. Theseintwo OECM1
resultsby PG-priming.
gave a clear clueConsidering
for autophagy all
inabove
OECM1 results, induced cell Considering
by PG-priming. death characteristics
all aboveinresults,
three different
inducedcell celllines
death bycharacteristics
PG-priming illustrated
in three
different cell lines by PG-priming illustrated in different configurations. OECM1posed
in different configurations. OECM1 showed autophagy, and SAS and FaDu showedboth cell death
autophagy,
andandSASautophagy.
and FaDu In the both
posed subsequent
cell deathexperiments,
and autophagy. theInpotential
the subsequentpathways of PG-enhanced
experiments, the potentialDox
cytotoxicity
pathways were under investigation.
of PG-enhanced Dox cytotoxicity were under investigation.
Figure 2. Alteration of cell cycle in (A) OECM1, (B) SAS, and (C) FaDu cells. OSCC cells were treated
Figure
with 2. Alteration
PG/Dox of cell
for 12/12 cycle in
h prior to (A) OECM1,
staining with(B) SAS, and (C)
propidium FaDu
iodide cells.
(PI) andOSCC cells were
fluorescent treated
intensity
with
was PG/Doxby
analyzed forflow
12/12 h prior toThe
cytometry. staining
resultswith
werepropidium iodide
represented ± SD
(PI) and
as mean fluorescent
from threeintensity was
individual
analyzed by* pflow
experiments. cytometry.
< 0.05 The with
as compared results
DOX were represented as mean ± SD from three individual
alone.
experiments. * p < 0.05 as compared with DOX alone.J. Clin. Med. 2018, 7, x FOR PEER REVIEW 7 of 16
J. Clin. Med. 2018, 7, 375 7 of 17
J. Clin. Med. 2018, 7, x FOR PEER REVIEW 7 of 16
Figure 3. Expression of (A) LC3, (B) PARP1, and (C) Caspase3 in OSCCs after PG-priming. OSCC
cells were
Figure treated withofPG/Dox
3. Expression for(B)
(A) LC3, 12/12 h andand
PARP1, then desired
(C) protein
Caspase3 levels were
in OSCCs afteranalyzed by Western
PG-priming. OSCC
Figure 3. Expression of (A) LC3, (B) PARP1, and (C) Caspase3 in OSCCs after PG-priming. OSCC
blotting.
cells wereThe results
treated withwere
PG/Doxnormalized
for 12/12with
h andGAPDH and protein
then desired represented
levelsas mean
were ± SD by
analyzed from three
Western
cells were treated with PG/Dox for 12/12 h and then desired protein levels were analyzed by Western
individualThe
blotting. experiments.
results wereMolecular weight:
normalized withPARP1,
GAPDH 116and
KDa; GAPDH, 37
represented as KDa; ± SD from
meanCaspase3, 34 three
KDa;
blotting. The results were normalized with GAPDH and represented as mean ± SD from three
individual
LC3, 18 KDa. experiments. Molecular with
# p < 0.05 compared weight: PARP1,control;
untreated 116 KDa;* pGAPDH,
< 0.05 as37 KDa; Caspase3,
compared 34 alone.
with Dox KDa; LC3,
individual experiments. Molecular weight: PARP1, 116 KDa; GAPDH, 37 KDa; Caspase3, 34 KDa;
18 KDa. # p < 0.05 compared with untreated control; * p < 0.05 as compared with Dox alone.
LC3, 18 KDa. # p < 0.05 compared with untreated control; * p < 0.05 as compared with Dox alone.
0.7
OECM1
0.60.7
SAS
OECM1
0.50.6
* * FaDu
SAS
0.40.5 *
**
OD
* FaDu
*
0.30.4 **
OD
0.20.3 *
0.10.2
0.1
0.0
0.0 ) -
PG (M 0.5 0.5 0.5 0.5 0.5
DOXPG (M ) -)
(M - 2.5
0.5 2.5
0.5 2.5
0.5 2.5
0.5 2.5
0.5
3MA (mM)
DOX (M -) - -2.5 10
2.5 -
2.5 10
2.5 -
2.5
BA1
3MA(nM)
(mM)- - -- -10 1- 10
- 1-
BA1 (nM) - - - 1 - 1
PG + inhibitor Dox + inhibitor
PG + inhibitor Dox + inhibitor
1st 12 h X PG PG + 3MA PG + BA1 PG PG
1st 12 h X PG PG + 3MA PG + BA1 PG PG
2nd 12 h X Dox Dox Dox Dox + 3MA Dox + BA1
2nd 12 h X Dox Dox Dox Dox + 3MA Dox + BA1
Figure
Figure 4. Alteration
4. Alteration of cell
of of
cell viability
viability combined with autophagy inhibitor. OSCC
OSCC cells
cells were
were treated
treated
Figure 4. Alteration cell viabilitycombined
combinedwith withautophagy
autophagy inhibitor.
inhibitor. OSCC cells were treated
with
with PG
PG PG + inhibitor/DOX
+ inhibitor/DOX or PG/Dox + inhibitor and cell viability was analyzed. Table under figure
with + inhibitor/DOXororPG/Dox
PG/Dox++ inhibitor
inhibitor and cell viability
and cell viability was
wasanalyzed.
analyzed.Table
Tableunder
underfigure
figure
was the
was the scheme
scheme of treatment.
ofof
treatment. “X”
“X”meant
meant incubated
incubatedwith
withcomplete
complete medium
mediumwithout
withoutPG or Dox. The
was the scheme treatment. “X” meant incubated with complete medium without PG orPG orThe
Dox. Dox.
results
The were
results represented
were represented as mean
as mean± SD
± SDfrom
fromthree individual
three individual experiments.
experiments. **pp <
<
results were represented as mean ± SD from three individual experiments. * p < 0.05 as compared 0.05
0.05 as
as compared
compared
with
with PG/Dox.
PG/Dox.
with PG/Dox.J. Clin. Med. 2018, 7, 375 8 of 17
J. Clin. Med. 2018, 7, x FOR PEER REVIEW 8 of 16
3.3.3.3. DoxorubicinFlux
Doxorubicin FluxAffected
Affectedby
byPG-Induced
PG-Induced Autophagy
Autophagy
ToTo measure
measure the the possible
possible action
action of PG-induced
of PG-induced autophagy,
autophagy, we examined
we examined the Dox the
fluxDox flux in
in PG-priming
PG-priming OSCC cells. This Dox flux was determined by rhodamine-123 (R123) accumulation.
OSCC cells. This Dox flux was determined by rhodamine-123 (R123) accumulation. R123 is a green
R123 is a green fluorescent dye which acts as a Dox-transporter substrate over decades [52].
fluorescent dye which acts as a Dox-transporter substrate over decades [52]. Accordingly, PG and Dox
Accordingly, PG and Dox are all red fluorescence and PG is stronger fluorescence than that of Dox.
are all red fluorescence and PG is stronger fluorescence than that of Dox. After PG/Dox combination
After PG/Dox combination treatment, PG will interfere with the measurement of Dox
treatment, PG will interfere with the measurement of Dox fluorescent-intensity. Also, the less cytotoxic
fluorescent-intensity. Also, the less cytotoxic nature of R123 could eliminate the interference of cell
nature of R123 could eliminate the interference of cell death caused by Dox. Therefore, R123 was
death caused by Dox. Therefore, R123 was employed as an indicator to indirectly determine the
employed
Dox-fluxas inan indicator
this study. Intoshort-term
indirectly priming
determine (1 the Dox-flux in did
h), PG-priming this not
study. In short-term
enhance priming (1 h),
R123 accumulation,
PG-priming
which revealed PG did not allosterically regulate Dox transporter, as shown in Figureregulate
did not enhance R123 accumulation, which revealed PG did not allosterically 5A.
Dox transporter, as shown in Figure 5A. Subsequently, PG-priming showed additional
Subsequently, PG-priming showed additional 50–70% of R123 accumulation for long-term priming 50–70% of R123
accumulation
(12 h). Moreover, the enhancing R123 could be attenuated by autophagy inhibitor, as shown in by
for long-term priming (12 h). Moreover, the enhancing R123 could be attenuated
autophagy
Figure 5B.inhibitor, as shown
This result indicatedin Figure 5B. This result
that PG-priming indicated
either enhanced that
DoxPG-priming either enhanced
importer expressions or
Dox importer
reduced expressions
exporter or reduced
expressions. exporterDox
When checking expressions.
transporterWhen
levels,checking
however,Dox transporter
the importer levels,
(Oct-6)
however,
was notthe importer decreased,
significantly (Oct-6) wasand notexporters
significantly
(MDR-1decreased, and exporters
and ABCG2) (MDR-1 in
slightly increased and ABCG2)
OECM1
and decreased
slightly increasedininSAS and FaDu,
OECM1 as shownin
and decreased inSAS
Figure
and6.FaDu,
This result was not
as shown associated
in Figure with
6. This previous
result was not
results. Itwith
associated might be postulated
previous asmight
results. It an indication of an unknown
be postulated but important
as an indication mechanism
of an unknown of Dox
but important
transport. of Dox transport.
mechanism
Figure 5. Rhodamine 123 accumulation after PG pretreatment. OSCC cells were treated with
Figure 5.forRhodamine
PG/R123 123 accumulation
(A) short term after
(1/1 h) and (B) PGterm
long pretreatment. OSCC cells
(12/1 h) followed were treated
by analyzed with
fluorescent
PG/R123 for (A) short term (1/1 h) and (B) long term (12/1 h) followed by analyzed fluorescent
intensity within cells. The results were represented as mean ± SD from three individual experiments.
# p < 0.05 as compared with R123 alone; * p < 0.05 compared with PG/R123 combination.J. Clin. Med. 2018, 7, x FOR PEER REVIEW 9 of 16
J. Clin.intensity
Med. 2018,within
7, 375 cells. The results were represented as mean ± SD from three individual experiments.9 of 17
# p < 0.05 as compared with R123 alone; * p < 0.05 compared with PG/R123 combination.
Figure 6. Protein levels of Doxorubicin-related importer and exporter after PG-priming. OSCC cells
Figure
were 6. Protein
treated levels offor
with PG/Dox Doxorubicin-related
12/12 h and then were importer andforexporter
analyzed Importer after PG-priming.
OCT-6 OSCC
(A) and exporter
MDR-1 (B) treated
cells were and ABCG2 (C) protein
with PG/Dox for levels
12/12 hbyand
Western blotting.
then were The results
analyzed were normalized
for Importer OCT-6 (A)with
and
GAPDH
exporter and represented
MDR-1 as mean(C)
(B) and ABCG2 ± SD from levels
protein three individual
by Western experiments. Molecular
blotting. The weight:
results were MDR-1,
normalized
170
withKDa;
GAPDHABCG2,and72 KDa; OCT-6,
represented as 58 KDa;
mean GAPDH,
± SD 37 KDa.
from three * p < 0.05
individual compared with
experiments. Dox alone.
Molecular weight:
MDR-1, 170 KDa; ABCG2, 72 KDa; OCT-6, 58 KDa; GAPDH, 37 KDa. * p < 0.05 compared with Dox
3.4. ER Stress and Energy Deprivation Analysis in PG-Priming OSCC Cells
alone.
PG could activate autophagy of OSCC cells as proven by the previous section and literature [48].
3.4. ER Stresspart,
In this final and Energy
the aimDeprivation Analysis
was to find in PG-Priming
the trigger OSCC autophagy
of PG-induced Cells in OSCC cells. As we
noted,PG could activate autophagy of OSCC cells as proven by the previous response)
the two known triggers of autophagy are ER stress (unfolded protein section and and energy
literature
deprivation, which may be involved in the PG-priming reaction. ER stress was determined
[48]. In this final part, the aim was to find the trigger of PG-induced autophagy in OSCC cells. As we by adding
ER stress
noted, theinhibitor TUDC
two known while energy
triggers deprivation
of autophagy wasstress
are ER blocked by addition
(unfolded of AMPK
protein specific
response) andinhibitor
energy
CC. When we combined TUDC or CC with PG and Dox treatment, cell viability
deprivation, which may be involved in the PG-priming reaction. ER stress was determined of three OSCC cells by
lines
adding were
ERnot significantly
stress inhibitor changed,
TUDC whileas shown
energyindeprivation
Figure 7, (data
wasofblocked
FaDu notby shown).
additionThis result
of AMPK
further
specificpostulated that
inhibitor CC. PG-induced
When autophagy
we combined TUDCand Doxwith
or CC flux PG
increase
and Doxwere not caused
treatment, cellbyviability
ER stress
of
and energy deprivation.
three OSCC cells lines were not significantly changed, as shown in Figure 7, (data of FaDu not
shown). This result further postulated that PG-induced autophagy and Dox flux increase were not
caused by ER stress and energy deprivation.J. Clin. Med. 2018, 7, 375
x FOR PEER REVIEW 10 of
10 of 17
16
0.7
OECM1
0.6
SAS
0.5
0.4
OD
0.3
* *
0.2
0.1
0.0
PG (M ) - 0.5 0.5 0.5 0.5 0.5 0.5 0.5
Dox (M ) - 2.5 2.5 2.5 2.5 2.5 2.5 2.5
CC (M ) - - 0.5 - - 0.5 - -
TUDC (M ) - - - 25 100 - 25 100
PG + inhibitor Dox + inhibitor
Figure 7. Cell viability change when PG/Dox combined with ER stress and energy deprivation
Figure 7. Cell viability change when PG/Dox combined with ER stress and energy deprivation
inhibitors. OSCC cells were treated with PG + inhibitor/Dox or PG/Dox + inhibitor and were analyzed
inhibitors. OSCC cells were treated with PG + inhibitor/Dox or PG/Dox + inhibitor and were
for cell viability. The inhibitors contained tauroursodeoxycholic acid (TUDC) and dorsomorphin
analyzed for cell viability. The inhibitors contained tauroursodeoxycholic acid (TUDC) and
(compound C, CC). The results were represented as mean ± SD from three individual experiments.
dorsomorphin (compound C, CC). The results were represented as mean ± SD from three individual
* p < 0.05 compared with PG/Dox.
experiments. * p < 0.05 compared with PG/Dox.
4. Discussion
4. Discussion
The present study is the first demonstration of PG-enhanced Dox influx by activating autophagy
in OSCCThe cells.
present study
Based on isthethe Doxfirst
fluxdemonstration
experiments, the of enhancing
PG-enhanced Dox influx
mechanism of Doxby influx
activating
was
autophagy
neither in OSCC
related to known cells. Based on
importers northe Dox flux
exporters. PGexperiments, the enhancing
could be a potential adjuvantmechanism of Dox
for Dox treatment.
influxthis
Also, wasstudy
neither relatedthe
excluded to characteristics
known importers nor exporters.
of known PG could be
autophagy-triggers inaPG-induced
potential adjuvant
autophagy, for
Dox treatment. Also, this study excluded the characteristics of
which posed a new site for autophagy triggering mechanisms. In this work, we first report about known autophagy-triggers in
PG-induced
the autophagy, which
autophagy-activating property posed a newassite
of Dox, thisfor autophagy
activity triggering
was known to bemechanisms.
inhibited byIn this
the work,
intrinsic
we first report about
activation of autophagy. the autophagy-activating property of Dox, as this activity was known to be
inhibited by the intrinsic activation of autophagy.
Due to the non-tissue specific nature and high-cardiotoxicity, several studies have tried to discover
naturalDue to the non-tissue
compounds that could specific nature and
synergistically high-cardiotoxicity,
potentiate the efficacy of Dox several studies
without have tried
elevating normal to
discover natural compounds that could synergistically potentiate
cell toxicity. In a previous report, gambogic acid, a xanthonoid from Garcinia hanburyi, sensitized the efficacy of Dox without
elevatingcancer
ovarian normal cell
cells toxicity.
toward DoxInthrough
a previous report, gambogic
accumulation of ROS [53]. acid,Nitidine
a xanthonoid
chloride,from Garcinia
an alkaloid,
hanburyi, sensitized
synergized ovarianincancer
Dox cytotoxicity breastcells
cancertoward Dox through
cell through PI3K/Akt accumulation of ROS [54].
signaling pathway [53]. Gingerol
Nitidine
chloride, anDox
synergized alkaloid,
againstsynergized
liver cancer Doxcells,
cytotoxicity
leading to in G2/M
breast cancer cell through
arrest [55]. Not onlyPI3K/Akt signaling
pure compounds;
pathwayextract
phenolic [54]. Gingerol
of flaxseedsynergized Dox against
oil also promoted Dox liver cancer
efficacy cells,
against leading
breast to cells
cancer G2/M[56].
arrest [55]. Not
Evodiamine,
only pure compounds; phenolic extract of flaxseed oil also promoted
a major element of Evodiae fructus, reversed chemoresistance in multi-drug resistant breast cancer Dox efficacy against breast
cancer cells [56]. Evodiamine, a major element of Evodiae fructus,
cells through the Ras/MEK/ERK signaling pathway [57]. Additionally, neferine could combat Dox reversed chemoresistance in
multi-drugthrough
resistance resistant ROSbreast cancer cells
accumulation and through
Fas signaling the Ras/MEK/ERK
pathway in lung signaling pathway
cancer [58]. [57].
In gastric
Additionally, neferine could combat Dox resistance through ROS accumulation
cancer, curcumin and formononetin posed different mechanisms to enhance Dox cytotoxicity [59,60]. and Fas signaling
pathway
These in lung
natural cancerexhibit
compounds [58]. In gastric cancer,
potentiation for main curcumin
applications andinformononetin
cancer treatment, posed different
and also play
amechanisms
supporting to enhance
role in Dox Doxregimen cytotoxicity
to overcome[59,60].the These natural
limitation compounds
of Dox exhibit potentiation for
usage [61–66].
mainTheapplications
first aiminofcancer treatment,
this study was and also play
to explore thea supporting
synergistic role effect in within
Dox regimen
PG/Dox to overcome
regimen.
the combined
PG limitation with
of Dox usagechemotherapy
current [61–66]. agents was studied in breast cancer and found that PG could
The paclitaxel
facilitate first aim of this study
sensitivity in was to explore human
triple-negative the synergistic effect within
breast carcinoma cellsPG/Dox regimen. PG
via down-regulating
combined with current chemotherapy agents was studied in breast
survivin expression, an anti-apoptotic protein that acts as a caspase inhibitor [67,68]. Our cancer and found that PGresults
could
facilitate paclitaxel
demonstrated sensitivitythat
new evidence in triple-negative
PG acts as an adjuvanthuman breast carcinoma cells
with conventional via down-regulating
chemotherapeutic drugs,
survivin expression, an anti-apoptotic
such as paclitaxel and Dox as well. protein that acts as a caspase inhibitor [67,68]. Our resultsJ. Clin. Med. 2018, 7, 375 11 of 17
The synergism of natural compounds with Dox could be found in priming fashion, nevertheless
the co-treatment is addressed in main efforts [53–60]. While priming with CDK inhibitor in
triple-negative breast cancer cell MDA-MB-231, Dox-induced DNA double-strand break would
be activated and resulted in cytotoxic enhancement [69]. Cyclophosphamide, a conventional
chemotherapeutic drug that acts as an intercalator of DNA, could increase HER2-targeted liposomal
Dox accumulation in breast cancer cells [70]. An in vivo study focused on Dox efficacy after mitomycin
C and carboplatin (two conventional chemotherapeutic drugs) pretreatment in human metastatic
breast cancer-bearing mice. The results showed inhibition growth of xenografted tumors and reducing
expressions of p-glycoprotein [71]. In our study, we showed that PG potentiated Dox cytotoxicity only
in a pretreatment fashion (as a PG-priming effect), which was the first report of natural compound that
primed cancer cells to be sensitized with Dox, and posed the potential of PG as an adjuvant using Dox
as a chemotherapeutic agent. The clinical application of PG-priming might provide a great clue for
reducing the dosage of Dox and dampening the side effects of Dox.
Due to red fluorescent nature [72], we preliminarily examined PG influx into OSCC cells. The data
showed that PG could enter OSCC cells within 1 h and reached saturation after 1.5 h exposure (data
not shown). When OSCC cells were primed with PG for 1 h, R123 fluorescent intensity did not
significantly accumulate. This gave clear insight into PG action that did not allosterically modulate
Dox importer or exporter activity. A previous study has also indicated that PG was not the substrate of
multidrug resistance-related protein including MDR-1, BCRP (ABCG2), and MRP2 [73]. Again, our
study confirmed that Dox efflux protein was not allosterically activated by PG-priming and further
exposed that PG did not allosterically mediate Oct-6 activity.
By R123 accumulation assay, PG did not allosterically control Dox transporter activity but affected
transporter expression in long-term priming, as shown in Figure 5. To our best knowledge, the Dox
uptake of cells via Oct-6 and excretion of Dox by MDR-1 and ABCG2 have been reported [3,74]. In
our study, Dox influx significantly increased in PG-priming for 12 h, which hypothesized that Oct-6
might be up-regulated or MDR-1/ABCG2 down-regulated. However, Oct-6 was down-regulated after
PG/Dox treatments in Western blotting. Furthermore, expression levels of MDR-1 and ABCG2 did
not show significant reductions. These results proposed a new Dox flux mechanism that needs to be
further investigated.
PG was known as apoptotic and autophagic inducer in previous studies [47,75]. According to
a recent study, PG induced apoptosis via inhibiting Bcl-2, activating Bak/Bax, intercalating DNA
leading to suppress the cell cycle [75]. However, the action of PG-induced autophagy has not been fully
explored yet. Remarkably, autophagy was triggered by stresses, such as ER stress (unfolded protein
response), nutrient deprivation, and oxidative stress [76–79]. Hence, these cellular stresses would be
the trigger clue of PG-induced autophagy. However, our test exposed that using ER stress inhibitor
(TUDC) and nutrient deprivation inhibitor (CC) could not restore the cell viability of PG/Dox. Also,
our data showed that PG did not elevate ROS level within OSCC cells (data not shown). All-known
causes of autophagy, including ER stress, nutrient deprivation, and oxidative stress, were excluded
from the trigger clue of PG-induced autophagy in this study, which the new potential mechanism of
autophagy activation necessitates to be further elucidated.
During the screening of an autophagic marker, we also found that Dox-induced autophagy in both
OECM1 and FaDu cells. It is well known that Dox was an apoptotic inducer via inhibiting cell cycle and
producing ROS [80]. The potentiated role of autophagy in Dox treatment is focused on the activation
of autophagy to ameliorate cardiotoxicity, and consequently inhibiting autophagy could promote Dox
sensitivity in cancer cells [81–84]. The autophagy-activating features of Dox suggested the unclear
field of Dox action. In the result of autophagy inhibitor recovery assay, autophagic inhibitors could
not recover Dox-induced cell death, which implies that Dox-induced autophagy might not be solely
involved in Dox-induced cell death, as shown in Figure 3.
Collectively, a model for the mechanical action of PG-priming autophagy potentiated Dox influx
is proposed, as shown in Figure 8. When PG entered OSCC cells, autophagy which was irrelevant toJ. Clin. Med. 2018, 7, 375 12 of 17
J. Clin. Med. 2018, 7, x FOR PEER REVIEW 12 of 16
nutrient deprivation,
irrelevant to nutrientERdeprivation,
stress, and ROS, was activated.
ER stress, and ROS,Subsequently,
was activated.PG-induced autophagy
Subsequently, could
PG-induced
up-regulate Dox importer expression and in terms translocated to cell membrane, and consequently
autophagy could up-regulate Dox importer expression and in terms translocated to cell membrane,
led
andtoconsequently
the enhancement
led toofthe
Dox influx resulting
enhancement in cell
of Dox death.
influx resulting in cell death.
Figure8.8.Potential
Figure Potentialmechanism
mechanismof of PG-priming
PG-priming doxorubicin
doxorubicin cytotoxicity
cytotoxicity enhancement.
enhancement. “X” “X” indicates
indicates not
not according
according to thistomechanism.
this mechanism. “?” illustrates
“?” illustrates still unknown.
still unknown. ER, endoplasmic
ER, endoplasmic reticulum;
reticulum; ROS,
ROS, reactive
reactivespecies.
oxygen oxygen species.
5.5.Conclusions
Conclusions
The
Thepresent
present study
study firstly
firstly demonstrated thethe potential
potential ofofPG
PGas
asan
anadjuvant
adjuvantfor
forDox
Doxtreatment
treatmentin
in OSCC
OSCC cells.
cells. PG-induced
PG-induced autophagy
autophagy waswas
not not associated
associated withwith
ER ER stress,
stress, nutrient
nutrient deprivation,
deprivation, and
and oxidative
oxidative stress.
stress. Also,
Also, the the enhancement
enhancement of Dox
of Dox influx
influx triggered
triggered by PG-primed
by PG-primed autophagy
autophagy diddid
not
not induce
induce viavia Dox
Dox transporter,such
transporter, suchasasMDR-1,
MDR-1,ABCG2,
ABCG2, and and OCT-6. The potential
potential mechanism
mechanism of of
PG-priming
PG-primingremains
remainsunclear
unclearandandwould
wouldbe beaafurther
furtherchallenge
challengefor
forPG
PGand
andDox
Doxinvestigation.
investigation.
Author
AuthorContributions:
Contributions:Conceptualization,
Conceptualization,S.-R.L. and C.-F.W.;
S.-R.L. methodology,
and C.-F.W.; S.-R.L.; software,
methodology, S.-R.L.; C.-F.W.;
software,validation,
C.-F.W.;
C.-F.W.; formal analysis, S.-R.L.; investigation, S.-R.L.; writing—original draft preparation, S.-R.L.; writing—review
validation, C.-F.W.; formal analysis, S.-R.L.; investigation, S.-R.L.; writing—original draft preparation,
and editing, C.-F.W.; supervision, C.-F.W.; project administration, C.-F.W.; funding acquisition, C.-F.W.
S.-R.L.;
writing—review and editing, C.-F.W.; supervision, C.-F.W.; project administration, C.-F.W.; funding
Funding: This
acquisition, research was funded by Ministry of Science and Technology, grant number 107-2320-B-259-003
C.-F.W.
(C.F. Weng).
Funding: This research was funded by Ministry of Science and Technology, grant number 107-2320-B-259-003
Acknowledgments: We sincerely thank Yu-Tong Chen from Kaoshiung Medical University who gave valuable
(C.F.inWeng).
help imaging of chemiluminescent Western blotting.
Acknowledgments:
Conflicts of Interest: We
The sincerely thank Yu-Tong
authors declare Chen from
no any conflict Kaoshiung Medical University who gave valuable
of interest.
help in imaging of chemiluminescent Western blotting.
References
Conflicts of Interest: The authors declare no any conflict of interest.
1. McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy
References
and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [CrossRef] [PubMed]
2.1. Damiani,
McGowan,R.M.; Moura,
J.V.; D.J.; R.;
Chung, Viau, C.M.; Caceres,
Maulik, R.A.; Henriques,
A.; Piotrowska, J.A.; Saffi,
I.; Walker, J.M.;J. Yellon,
Pathways of cardiac
D.M. toxicity:
Anthracycline
Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone.
Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. Arch. Toxicol. 2016, 90,
2. 2063–2076. [CrossRef] [PubMed]
Damiani, R.M.; Moura, D.J.; Viau, C.M.; Caceres, R.A.; Henriques, J.A.; Saffi, J. Pathways of cardiac toxicity:
Comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch. Toxicol. 2016, 90,
2063–2076.J. Clin. Med. 2018, 7, 375 13 of 17
3. Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin
pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [CrossRef]
[PubMed]
4. Johnson-Arbor, K.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publisher: Treasure Island, FL, USA,
2018.
5. Renu, K.; Abilash, V.G.; Pirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of
doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [CrossRef]
[PubMed]
6. Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; El Habak, K.; Mostafa, R.;
Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted
therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [CrossRef] [PubMed]
7. Asensio-Lopez, M.C.; Soler, F.; Pascual-Figal, D.; Fernandez-Belda, F.; Lax, A. Doxorubicin-induced oxidative
stress: The protective effect of nicorandil on HL-1 cardiomyocytes. PLoS ONE 2017, 12, e0172803. [CrossRef]
[PubMed]
8. Kwatra, M.; Kumar, V.; Jangra, A.; Mishra, M.; Ahmed, S.; Ghosh, P.; Vohora, D.; Khanam, R. Ameliorative
effect of naringin against doxorubicin-induced acute cardiac toxicity in rats. Pharm. Biol. 2016, 54, 637–647.
[CrossRef] [PubMed]
9. Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism.
J. Pharm. Pharmacol. 2016, 68, 729–741. [CrossRef] [PubMed]
10. Wang, Z.; He, Q.; Zhao, W.; Luo, J.; Gao, W. Tumor-homing, pH- and ultrasound-responsive
polypeptide-doxorubicin nanoconjugates overcome doxorubicin resistance in cancer therapy. J. Control.
Release 2017, 264, 66–75. [CrossRef] [PubMed]
11. Tang, J.; Zhang, L.; Gao, H.; Liu, Y.; Zhang, Q.; Ran, R.; Zhang, Z.; He, Q. Co-delivery of doxorubicin and
P-gp inhibitor by a reduction-sensitive liposome to overcome multidrug resistance, enhance anti-tumor
efficiency and reduce toxicity. Drug Deliv. 2016, 23, 1130–1143. [CrossRef] [PubMed]
12. Xue, H.; Yu, Z.; Liu, Y.; Yuan, W.; Yang, T.; You, J.; He, X.; Lee, R.J.; Li, L.; Xu, C. Delivery of miR-375 and
doxorubicin hydrochloride by lipid-coated hollow mesoporous silica nanoparticles to overcome multiple
drug resistance in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 5271–5287. [CrossRef] [PubMed]
13. Gupta, B.; Ramasamy, T.; Poudel, B.K.; Pathak, S.; Regmi, S.; Choi, J.Y.; Son, Y.; Thapa, R.K.; Jeong, J.H.;
Kim, J.R.; et al. Development of Bioactive PEGylated Nanostructured Platforms for Sequential Delivery of
Doxorubicin and Imatinib to Overcome Drug Resistance in Metastatic Tumors. ACS Appl. Mater. Interfaces
2017, 9, 9280–9290. [CrossRef] [PubMed]
14. Perillo, E.; Porto, S.; Falanga, A.; Zappavigna, S.; Stiuso, P.; Tirino, V.; Desiderio, V.; Papaccio, G.; Galdiero, M.;
Giordano, A.; et al. Liposome armed with herpes virus-derived gH625 peptide to overcome doxorubicin
resistance in lung adenocarcinoma cell lines. Oncotarget 2016, 7, 4077–4092. [CrossRef] [PubMed]
15. Kaminskas, L.M.; McLeod, V.M.; Kelly, B.D.; Sberna, G.; Boyd, B.J.; Williamson, M.; Owen, D.J.; Porter, C.J.
A comparison of changes to doxorubicin pharmacokinetics, antitumor activity, and toxicity mediated by
PEGylated dendrimer and PEGylated liposome drug delivery systems. Nanomedicine 2012, 8, 103–111.
[CrossRef] [PubMed]
16. Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer:
A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [CrossRef] [PubMed]
17. Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in
cancer: An overview. Cancers 2014, 6, 1769–1792. [CrossRef] [PubMed]
18. Bradley, G.; Juranka, P.F.; Ling, V. Mechanism of multidrug resistance. Biochim. Biophys. Acta 1988, 948,
87–128. [CrossRef]
19. Liu, T.; Li, Z.; Zhang, Q.; De Amorim Bernstein, K.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z.
Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to
reverse drug resistance. Oncotarget 2016, 7, 83502–83513. [CrossRef] [PubMed]
20. Komori, Y.; Arisawa, S.; Takai, M.; Yokoyama, K.; Honda, M.; Hayashi, K.; Ishigami, M.; Katano, Y.; Goto, H.;
Ueyama, J.; et al. Ursodeoxycholic acid inhibits overexpression of P-glycoprotein induced by doxorubicin in
HepG2 cells. Eur. J. Pharmacol. 2014, 724, 161–167. [CrossRef] [PubMed]
21. Pereira, M.M.; Haniadka, R.; Chacko, P.P.; Palatty, P.L.; Baliga, M.S. Zingiber officinale Roscoe (ginger) as an
adjuvant in cancer treatment: A review. J. BUON 2011, 16, 414–424. [PubMed]J. Clin. Med. 2018, 7, 375 14 of 17
22. Laatsch, H.; Kellner, M.; Weyland, H. Butyl-meta-cycloheptylprodiginine—A revision of the structure of the
former ortho-isomer. J. Antibiot. 1991, 44, 187–191. [CrossRef] [PubMed]
23. Soliev, A.B.; Hosokawa, K.; Enomoto, K. Bioactive pigments from marine bacteria: Applications and
physiological roles. Evid. Based Complement. Alternat. Med. 2011, 2011, 670349. [CrossRef] [PubMed]
24. Chang, C.C.; Chen, W.C.; Ho, T.F.; Wu, H.S.; Wei, Y.H. Development of natural anti-tumor drugs by
microorganisms. J. Biosci. Bioeng. 2011, 111, 501–511. [CrossRef] [PubMed]
25. Marchal, E.; Smithen, D.A.; Uddin, M.I.; Robertson, A.W.; Jakeman, D.L.; Mollard, V.; Goodman, C.D.;
MacDougall, K.S.; McFarland, S.A.; McFadden, G.I.; et al. Synthesis and antimalarial activity of
prodigiosenes. Org. Biomol. Chem. 2014, 12, 4132–4142. [CrossRef] [PubMed]
26. Lapenda, J.C.; Silva, P.A.; Vicalvi, M.C.; Sena, K.X.; Nascimento, S.C. Antimicrobial activity of prodigiosin
isolated from Serratia marcescens UFPEDA 398. World J. Microbiol. Biotechnol. 2015, 31, 399–406. [CrossRef]
[PubMed]
27. Wang, Y.; Nakajima, A.; Hosokawa, K.; Soliev, A.B.; Osaka, I.; Arakawa, R.; Enomoto, K. Cytotoxic prodigiosin
family pigments from Pseudoalteromonas sp. 1020R isolated from the Pacific coast of Japan. Biosci. Biotechnol.
Biochem. 2012, 76, 1229–1232. [CrossRef] [PubMed]
28. Kimyon, O.; Das, T.; Ibugo, A.I.; Kutty, S.K.; Ho, K.K.; Tebben, J.; Kumar, N.; Manefield, M. Serratia
Secondary Metabolite Prodigiosin Inhibits Pseudomonas aeruginosa Biofilm Development by Producing
Reactive Oxygen Species that Damage Biological Molecules. Front. Microbiol. 2016, 7, 972. [CrossRef]
[PubMed]
29. Song, Y.; Liu, G.; Li, J.; Huang, H.; Zhang, X.; Zhang, H.; Ju, J. Cytotoxic and antibacterial angucycline-
and prodigiosin-analogues from the deep-sea derived Streptomyces sp. SCSIO 11594. Mar. Drugs 2015, 13,
1304–1316. [CrossRef] [PubMed]
30. Kancharla, P.; Lu, W.; Salem, S.M.; Kelly, J.X.; Reynolds, K.A. Stereospecific synthesis of
23-hydroxyundecylprodiginines and analogues and conversion to antimalarial premarineosins via a Rieske
oxygenase catalyzed bicyclization. J. Org. Chem. 2014, 79, 11674–11689. [CrossRef] [PubMed]
31. Perez-Tomas, R.; Vinas, M. New insights on the antitumoral properties of prodiginines. Curr. Med. Chem.
2010, 17, 2222–2231. [CrossRef] [PubMed]
32. Sam, S.; Sam, M.R.; Esmaeillou, M.; Safaralizadeh, R. Effective Targeting Survivin, Caspase-3 and
MicroRNA-16-1 Expression by Methyl-3-pentyl-6-methoxyprodigiosene Triggers Apoptosis in Colorectal
Cancer Stem-Like Cells. Pathol. Oncol. Res. 2016, 22, 715–723. [CrossRef] [PubMed]
33. Yu, C.J.; Ou, J.H.; Wang, M.L.; Jialielihan, N.; Liu, Y.H. Elevated survivin mediated multidrug resistance and
reduced apoptosis in breast cancer stem cells. J. BUON 2015, 20, 1287–1294. [PubMed]
34. Chiu, W.-J.; Lin, S.-R.; Chen, Y.-H.; Tsai, M.-J.; Leong, M.; Weng, C.-F. Prodigiosin-Emerged
PI3K/Beclin-1-Independent Pathway Elicits Autophagic Cell Death in Doxorubicin-Sensitive and -Resistant
Lung Cancer. J. Clin. Med. 2018, 7, 321. [CrossRef] [PubMed]
35. Llagostera, E.; Soto-Cerrato, V.; Montaner, B.; Perez-Tomas, R. Prodigiosin induces apoptosis by acting on
mitochondria in human lung cancer cells. Ann. N.Y. Acad. Sci. 2003, 1010, 178–181. [CrossRef] [PubMed]
36. Llagostera, E.; Soto-Cerrato, V.; Joshi, R.; Montaner, B.; Gimenez-Bonafe, P.; Perez-Tomas, R. High cytotoxic
sensitivity of the human small cell lung doxorubicin-resistant carcinoma (GLC4/ADR) cell line to prodigiosin
through apoptosis activation. Anticancer Drugs 2005, 16, 393–399. [CrossRef] [PubMed]
37. Zhou, W.; Jin, Z.X.; Wan, Y.J. Apoptosis of human lung adenocarcinoma A549 cells induced by prodigiosin
analogue obtained from an entomopathogenic bacterium Serratia marcescens. Appl. Microbiol. Biotechnol.
2010, 88, 1269–1275. [CrossRef] [PubMed]
38. Soto-Cerrato, V.; Llagostera, E.; Montaner, B.; Scheffer, G.L.; Perez-Tomas, R. Mitochondria-mediated
apoptosis operating irrespective of multidrug resistance in breast cancer cells by the anticancer agent
prodigiosin. Biochem. Pharmacol. 2004, 68, 1345–1352. [CrossRef] [PubMed]
39. Monge, M.; Vilaseca, M.; Soto-Cerrato, V.; Montaner, B.; Giralt, E.; Perez-Tomas, R. Proteomic analysis of
prodigiosin-induced apoptosis in a breast cancer mitoxantrone-resistant (MCF-7 MR) cell line. Investig. New
Drugs 2007, 25, 21–29. [CrossRef] [PubMed]
40. Montaner, B.; Perez-Tomas, R. Prodigiosin-induced apoptosis in human colon cancer cells. Life Sci. 2001, 68,
2025–2036. [CrossRef]You can also read

















































