MassComp, a lossless compressor for mass spectrometry data - bioRxiv
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
MassComp, a lossless compressor for mass spectrometry
data
Ruochen Yang1 , Xi Chen2 , and Idoia Ochoa2∗
ruocheny@usc.edu, xichen30@illinois.edu, idoia@illinois.edu
1
Electrical Engineering Department, University of Southern California, Los Angeles, CA, USA.
2
Electrical and Computer Engineering Department, University of Illinois at Urbana-Champaign,
Urbana, IL, USA.
∗
To whom correspondence should be addressed.
Abstract
Background: Mass Spectrometry (MS) is a widely used technique in biology research, and
has become key in proteomics and metabolomics analyses. As a result, the amount of MS data
has significantly increased in recent years. For example, the MS repository MassIVE contains
more than 123TB of data. Somehow surprisingly, these data are stored uncompressed, hence
incurring a significant storage cost. Efficient representation of these data is therefore paramount
to lessen the burden of storage and facilitate its dissemination.
Results: We present MassComp, a lossless compressor optimized for the numerical (m/z)-
intensity pairs that account for most of the MS data. We tested MassComp on several MS
data and show that it delivers on average a 46% reduction on the size of the numerical data,
and up to 89%. These results correspond to an average improvement of more than 27% when
compared to the general compressor gzip and of 40% when compared to the state-of-the-art
numerical compressor FPC. When tested on entire files retrieved from the MassIVE repository,
MassComp achieves on average a 59% size reduction. MassComp is written in C++ and freely
available at https://github.com/iochoa/MassComp.
Conclusions: The compression performance of MassComp demonstrates its potential to sig-
nificantly reduce the footprint of MS data, and shows the benefits of designing specialized
compression algorithms tailored to MS data. MassComp is an addition to the family of omics
compression algorithms designed to lessen the storage burden and facilitate the exchange and
dissemination of omics data.
Keywords: Mass Spectrometry, lossless compression, storage.
Background
High-resolution mass spectrometry (MS) is a powerful technique used to identify and quantify
molecules in simple and complex mixtures by separating molecular ions on the basis of their
mass and charge [1]. MS has become invaluable in the field of proteomics, which studies
dynamic protein products and their interactions [2]. Similarly, the field of metabolomics, which
aims at the comprehensive and quantitative analysis of wide arrays of metabolites in biological
samples, is developing thanks to the advancements in MS technology [3]. These fields are
rapidly growing, as they contribute towards a better understanding of the dynamic processes
involved in disease, with direct applications in prediction, diagnosis and prognosis [4, 5, 6].
1bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
As a result of this growth, the amount and size of MS data produced as part of proteomics
and metabolomics studies has increased by several orders of magnitude [7]. To facilitate the ex-
change and dissemination of these data, several centralized data repositories have been created
that make the data and results accessible to researchers and biologists alike. Examples of such
repositories include GPMDB (Global Proteome Machine Database) [8], PeptideAtlas/PASSEL
[9, 10], PRIDE [11, 12] and MassIVE (Mass Spectrometry Interactive Virtual Environment)
[13]. In particular, MassIVE contains more than 2 million files worth 123TB of storage, and
PRIDE contains around 7,000 projects and 74,000 assays.
MS data are mainly composed of the mass to charge ratios (m/z) and corresponding ion
counts, and are referred to as the (m/z)-intensity pairs. These data are generally stored in
the open XML (eXtensible Markup Language) formats mzXML [14] and mzML [15], after
conversion from the raw vendor formats (which may vary across technologies/instruments).
These formats facilitate exchange and vendor neutral analysis of mass spectrometry data, but
tend to be much larger than the raw data [16], as they include extensive additional metadata
(e.g., type of instrument employed). In addition, the mzXML and mzML files are structured
in a tab-delimited format containing human readable plain text, except for the (m/z)-intensity
pairs that are stored in binary format (base64). In particular, the mzXML format generally
contains data from several thousands of scans corresponding to a given experiment. Within
each scan, the (m/z)-intensity pairs are stored in the element “peaks”, and encoded in base64.
The pairs can represent either single or double precision values, and this is specified in the
“peaks precision”. The number of pairs available for each scan varies, and it ranges from just
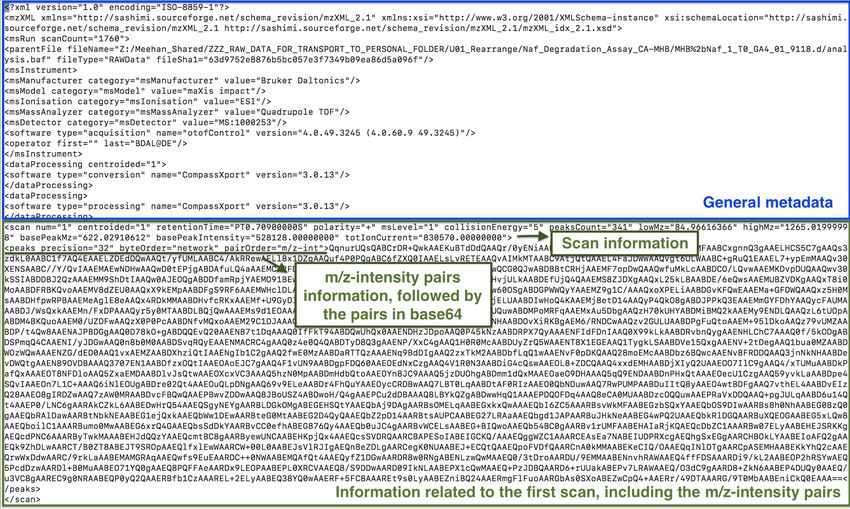
a few to thousands of them. See Fig. 1 for a snapshot of an mzXML file.
Figure 1: Example of the first lines of an mzXML file. After initial general information of the
format and instrument, the first scan is presented, in this case in single-precision (32 bits).
In addition of not being optimized for space saving due to the inherit characteristics of the
XML format, MS files are submitted and stored in the public repositories uncompressed, which
incurs a significant storage cost. Note that even though individual files may be small (e.g., less
than 100MBs in some cases), the combined total size of all submitted files can reach the order
of TBs. Still, little effort has been made to compress MS data. This is in contrast to other
omics disciplines, such as genomics, that have experienced an increasing effort in designing
specialized compression schemes to lessen the storage burden [17, 18, 19].
2bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
A family of numerical compression algorithms called MS-Numpress was presented in [16].
These algorithms are optimized for the compression of the floating point values corresponding to
the (m/z)-intensity pairs that characterize MS data. However, the proposed algorithms are lossy
and exhibit precision loss in the case where the m/z-intensity pairs represent double-precision
values. For example, the proposed compression algorithm numSlof for ion count data takes
the natural logarithm of values, multiplies by a scaling factor and then rounds to the nearest
integer. Similarly, the proposed compression algorithm numLin for (m/z) data multiplies the
values by a scaling factor and then rounds to the nearest integer. Further compression is then
achieved by the use of a linear predictor. Although not specifically designed to compress MS
data, general numerical compressors such as the state-of-the-art algorithm FPC [20] could be
used for this purpose, given that MS data are mainly composed of numerical data. In particular,
FPC is a fast lossless compressor optimized for linear streams of floating-point data. It uses
predictors in the form of hash tables to predict the next values in the sequence. The predicted
values are then XORed with the true values, and the resulting number of leading zero bytes
and the residual bytes are written as the output. However, general numerical compressors are
not tailored to MS data and thus better results (in compression ratio) are to be expected from
MS specialized compressors.
Here we introduce MassComp, a new specialized lossless compressor for MS data. Mass-
Comp is optimized for the compression of the mass to charge ratio (m/z)-intensity pairs that
characterize mass spectrometry (MS) data. However, for ease of use, MassComp works on
mzXML files. Briefly, MassComp extracts the (m/z)-intensity pairs from the mzXML file, and
compresses them effectively. Due to the different nature of the mass to charge (m/z) ratios
and the ion count (intensity) values, MassComp uses different compression strategies for each
of them. The remaining data from the mzXML file is extracted and compressed with the gen-
eral purpose compression algorithm gzip. MassComp is then able to reconstruct the original
mzXML file from the compressed data. gzip has been chosen for being the most common
general compressor available, and because several current MS computational tools can directly
work with gzip compressed files (e.g., peptide identification [21]).
We tested MassComp on several mzXML datasets from the MassIVE repository, and showed
that it is able to reduce the file sizes by almost 60% on average. Comparisons with gzip and
FPC on the numerical data show the benefit of designing specialized compressors tailored to MS
data. In particular, MassComp achieves an improvement of up to 51% and 85% in compression
ratio when compared to gzip and FPC, respectively. Comparison with MS-Numpress on double-
precision MS data shows that this family of algorithms perform better than MassComp in this
case. However, it is important to emphasize that the data can not be recovered exactly due to
the lossy nature of MS-Numpress when applied to double-precision data.
Results
MS files are stored uncompressed (i.e., there is no default compressor for MS data), and hence
we compare the performance of MassComp to that of the general lossless compressor gzip, the
state-of-the-art numerical compressor FPC [20], and the family of numerical compressors MS-
Numpress [16]. gzip was chosen for baseline performance over other general lossless compressors
as it is used in practice as the de-facto compressor for other omics data, such as genomics (e.g.,
for compression of FASTQ files [22] in public repositories and as the building block in the
widely used BAM format [23]). Results for FPC are shown for default “level” parameter 20
(simulation with other values produced similar results). MS-Numpress was run with the built-
in MS-Numpress compression option of the MSConvert GUI [24]. We selected the algorithms
numLin and numPic for the m/z and intensities, respectively, as well as the zlib option, as they
were found to offer the best compression performance.
All experiments were run in a machine running CentOS Linux version 7, with an Intel(R)
Xeon(R) CPU E5-2698 v4 @ 2.20GHz and 512GB of RAM, except for FPC and MS-Numpress,
3bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
which were run in a ThinkPad T460s laptop running Windows with 64 bit operating system,
Intel Core i7-6600U CPU @ 2.60GHz, and 8GB memory.
For the analysis, we randomly selected three experiments from the MassIVE repository, and
considered all mzXML files within them. The corresponding MassIVE IDs are MSV000080896,
MSV000080905 and MSV000081123, and they can be retrieved from ftp://massive.ucsd.
edu/ followed by the ID. These experiments contain, respectively, 600MB, 4GB, and 400MB
worth of mzXML files. All selected files contain single-precision (m/z)-intensity pairs, and
hence we also selected the raw files 110620 fract scxB05,
121213 PhosphoMRM TiO2 discovery, and ADH 100126 mix used in [16], in which the m/z-
intensity pairs represent double-precision values. Hereafter we refer to them as 110, 121 and
ADH. Their corresponding raw size is 16.63MB, 508.63MB, and 538MB, respectively. Conver-
sion from mzXML to mzML format, as well as conversion of the raw files to either mzXML or
mzML format, was done with MSConvert [24].
mzXML Pairs FPC C.R. gzip C.R. MassComp C.R. Gain gzip Gain FPC
MSV000080896 (single-precision)
9122 25 18 17.71 1.61 12 33.33 9.4 47.78 21.67 46.92
9123 25 18 17.68 1.78 12 33.33 9.4 47.78 21.67 46.83
9124 25 18 17.97 0.17 13 27.78 9.5 47.22 26.92 47.13
MSV000080905 (single-precision)
54 7.1 4 3.84 4.00 3.7 7.50 2.8 30.00 24.32 27.08
05 7.7 4.4 3.71 15.68 4.1 6.82 3.1 29.55 24.39 16.44
07 9 4.7 4.48 4.68 4.3 8.51 3.2 31.91 25.58 28.57
MSV000081123 (single-precision)
neg42 11 5.1 4.91 3.73 4.5 11.76 3.5 31.37 22.22 28.72
pos75 6.7 3.6 3.43 4.72 3.2 11.11 2.5 30.56 21.88 27.11
tag71 3.9 1.7 1.51 11.18 1.5 11.76 1.1 35.29 26.67 27.15
Raw Data (double-precision)
110 69 50 41.56 16.88 21 58 11 78 47.62 73.53
121 2,500 1,900 1,257 33.84 986 48.11 856 54.95 13.18 31.90
ADH 1,900 1,500 1,082 27.85 320 78.67 154 89.73 51.88 85.77
Average single-precision
5.28 16.88 36.83 23.92 32.89
Average double-precision
26.19 61.59 74.23 37.56 63.73
Average all
10.51 28.06 46.18 27.33 40.60
Table 1: Results for FPC, gzip and MassComp when tested on the m/z-intensity pairs of some
of the considered files, both in single and double precision. All sizes are expressed in MBs.
Column mzXML denotes the size of the mzXML file, whereas Pairs contains the size of only
the numerical data. Best results are highlighted in bold. C.R. denotes compression ratio,
computed as 100 − compressed size ∗ 100/size of pairs. The gain of MassComp is computed as
1 − MassComp size/other size.
Since FPC only works on numerical data, we first compared the performance of MassComp
to that of gzip and FPC when applied only to the (m/z)-intensity pairs. MS-Numpress is
omitted in this experiment as we were unable to run it solely on the numerical data. Table 1
shows the results for 3 randomly selected files from each of the MassIVE experiments and the
raw data after conversion to the mzXML format (all presented sizes are expressed in MBs). FPC
only works on plain little-endian numerical files, and hence the (m/z)-intensity pairs extracted
from the mzXML files were converted into the little-endian format prior to compression with
4bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
FPC. No conversion is made prior to compression with gzip and MassComp. The results show
that MassComp achieves the smallest compressed size on the numerical data, offering space
savings ranging from 29% to 89%. This corresponds to an average improvement in compression
ratio of 27% and 40% when compared to gzip and FPC, respectively. Also, note that FPC is
outweighted by gzip in all tested data. This may be due to the small number of pair elements
found in some of the scans, which may worsen the prediction performed by FPC. For example,
note that FPC obtains the best compression ratio on double precision files 121 and ADH, which
both contain scans with tens of thousands of pair elements, whereas the selected single precision
files contain generally scans with less than a thousand elements, leading to worse compression
ratios.
Num. Files Average mzXML gzip C.R. [std] MassComp C.R. [std] Gain gzip
MSV000080896 9 34 299 168 43.81 [0.39] 113 62.21 [0.94] 32.74
MSV000080905 514 8 4,100 2,500 39.02 [1.62] 1,800 56.10 [0.81] 28.00
Plate1 123 7.96 979 591 39.63 [1.33] 455 53.52 [0.74] 23.01
Plate2 132 8.33 1,100 633 42.45 [1.59] 486 55.82 [0.73] 23.22
Plate3 135 8.15 1,100 690 37.27 [1.61] 528 52.00 [0.84] 23.48
Plate4 124 8.06 999 608 39.14 [1.79] 467 53.25 [0.88] 23.19
MSV000081123 54 8 406 218 46.31 [3.01] 165 59.36 [1.90] 24.31
Average 43.05 [2.95] 59.22 [2.22] 28.35
Table 2: Results for gzip and MassComp when tested on whole mzXML. Results show the total
size of the compressed files when considering all mzXML files within each MassIVE experiment,
as well as the compression ratio (denoted by C.R.). We also included the standard deviation,
denoted by std. The compression ratio and the gain of MassComp with respect to gzip is
computed as in Table 1. Results for experiment MSV000080905 are split into different Plates,
as provided in the MassIVE repository, in addition to the overall performance. All sizes are
expressed in MBs.
To further assess the benefits of MassComp, Table 2 shows the results of MassComp and gzip
when applied to the entire collection of files from the selected MassIVE experiments, and when
considering the entire files (i.e., not only the numerical data). MS-Numpress is not included
in this analysis as it only works on mzML files. The mzXML files of MassIVE experiment
MSV000080905 are organized in 4 different folders, namely Plate1, Plate2, Plate3 and Plate4,
and hence we also show the results for each of them individually. For each experiment we also
specify the number of files and the average size of each of them (columns Num. Files and
Average, respectively). All sizes are expressed in MBs, and the best results are highlighted in
bold. We also specify the compression ratio of each algorithm, as well as the gain of MassComp
with respect to gzip (these metrics are computed in the same way as in Table 1). We observe that
MassComp consistently outperforms gzip on the tested files, with compression gains ranging
from 24% to 32%. The performance of MassComp is also more consistent across files of a given
experiment, as indicated by the standard deviation (only for experiment MSV000080896 this
is not the case, which corresponds to the one with the least amount of files). In addition,
MassComp offers on average space savings of almost 60%. For example, the space needed to
store MS data for experiment MSV000080905 is decreased from 4.1GB to 1.8GB, showing the
potential of MassComp to significantly reduce the footprint of MS data.
Table 3 shows the compression and decompression times of both gzip and MassComp when
applied to all mzXML files of the selected MassIVE experiments. As it can be observed, the
compression time of gzip and MassComp is comparable. However, gzip is faster at decompres-
sion, since MassComp needs to reassemble the mzXML files from the compressed data. For
example, for experiment 80896, MassComp and gzip employ 33 and 23 seconds for compression,
5bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
respectively, whereas they employ 6 minutes and 4 seconds for decompression. In addition to
the time needed to reassemble the file, a significant amount of time (up to 50%) is spent in the
arithmetic decoder (see Methods section), and hence further optimization of this step1 could
greatly improve the decompression speed.
Finally, in Table 4 we show the comparison of MassComp to MS-Numpress when applied
to the same randomly selected files of the MassIVE experiments shown in Table 1, as well as to
the double-precision data. Note that a fair comparison is difficult to make, as MassComp works
on mzXML files, whereas MS-Numpress can only be applied to mzML files. For this reason,
we refrain from highlighting the smallest sizes in bold. Nevertheless, for the double-precision
data both the mzXML and mzML files occupy a similar space, and hence we can conclude that
MS-Numpress provides better compression performance in this case. For example, file ADH
occupies 1.90GB in mzXML format and 1.96GB in mzML format, and the compressed size
of MassComp and MS-Numpress is 469MB versus 145MB, respectively. However, note that
MS-Numpress is lossy in this case, and hence the exact numerical values can not be recovered.
C.T. gzip D.T. gzip C.T. MassComp D.T. MassComp
MSV000080896 23 4 33 360
MSV000080905 241 47 359 3,980
Plate1 59 11 85 930
Plate2 59 12 90 990
Plate3 65 13 97 1,100
Plate4 58 11 87 960
MSV000081123 24 4 34 320
Table 3: Compression and decompression times of gzip and MassComp when applied to all
mzXML files of the selected MassIVE experiments. All times are expressed in seconds. Best
performance is highlighted in bold.
Discussion
The above results demonstrate the benefits of designing specialized compressor schemes tailored
to the specific data, in this case Mass Spectrometry data. In particular, we have presented
MassComp, a lossless compressor optimized for the m/z-intensity values that characterize MS
data. MassComp is able to reduce the sizes of the m/z-intensity pairs by 37% and 74% on
average, on single and double precision data, respectively. In contrast, the general compressor
gzip achieves on average 28% reduction in size, whereas the numerical compressor FPC attains
on average only 10% reduction. Note that even though single MS files may be small, a single
experiment generally produces several files, which can account for several tens of GBs. Efficient
compressed representations can therefore alleviate storage requirements for MS data.
For ease of use, MassComp accepts mzXML files, and compresses the remaining data (i.e.,
everything but the pairs) using the general compressor gzip. One of the drawbacks of MassComp
in its current form is the decompression times, which are higher than those of gzip. The
reasons are mainly the need of reassembling the mzXML file and the use of a multi-symbol
arithmetic encoder. However, the running time could be greatly improved by compressing and
decompressing the m/z-intensity pairs of each scan in parallel, as well as the metadata. Other
improvements in decompression times could come from using a binary arithmetic encoder rather
1
This is out of the scope of this paper but will be considered in future versions of the algorithm. See the
Discussion section for details.
6bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
File ID mzXML MassComp C.R. mzML MS-Numpress C.R.
MSV000080896
9122 25 11.64 53.44 53.85 21.84 59.44
9123 25 11.58 53.68 53.77 21.81 59.44
9124 25 11.72 53.12 54.53 22.07 59.53
MSV000080905
54 7.1 4.74 33.24 20.91 14.75 29.46
05 7.7 5.05 34.42 22.17 15.05 32.12
07 9 5.23 41.89 22.92 15.25 33.46
MSV000081123
neg42 11 7.045 35.95 34.46 25.73 25.33
pos75 6.7 4.568 31.82 20.34 14.21 30.14
TAG71 3.9 2.782 28.67 13.36 10.52 21.26
110 69 27.08 60.75 80.92 25.42∗ 68.59
121 2,500 1,104.93 55.80 2,587.45 307.07∗ 88.13
ADH 1,900 469.51 75.29 1,965.63 145.25∗ 92.61
∗
Lossy compression
Table 4: Compression performance of MassComp and MS-Numpress, the latter run with num-
Lin, numPic and zlib as it was found to offer the best performance. Since MassComp works on
mzXML files and MS-Numpress on mzML files, an exact comparison is not possible, and hence
we refrain from highlighting the smallest compressed sizes in bold. All sizes are expressed in
MBs.
than a multi-symbol one. This can be achieved by first binarizing the symbols to be compressed,
as done in video coding standards by means of CABAC [25], for example (note however that
some loss in compression ratio may be expected in this case). Another improvement could come
from incorporating the compression method of MassComp for the m/z-intensity pairs directly
into the current formats for storing MS data, that is, mzXML and mzML files. Note that this
would reduce the files by 46% on average (see Table 1). Then, downstream applications that
use the MS data could decompress each scan as needed.
Finally, note that MassComp is completely lossless, in contrast to MS-Numpress that is
lossy for double-precision data. Future extensions of MassComp could consider lossy options
for these data. However, such an extension should be accompanied by an exhaustive analysis on
how the loss in precision may affect the downstream applications that use MS data in practice
(see [16] for some preliminary results on this regard). This analysis should include several data
sets and applications, as done for the case of lossy compression of quality scores present in
genomic data [26]. Further work could also include support for random access of the pairs
corresponding to the different scans.
Conclusions
As a key technique for proteomics and metabolomics analyses, mass spectrometry (MS) is
widely used in biology research. As a result, the amount of MS data has significantly increased
in recent years. For example, the MS repository MassIVE contains more than 123TB of data.
Somehow surprisingly, these data are stored uncompressed, hence incurring a significant storage
cost. Efficient representation of these data is therefore paramount to lessen the burden of
storage and facilitate its dissemination. This has been the case in other omics datasets, such
as genomics, where there has been a growing interest in designing specialized compressors for
raw and aligned genomic data (see [17] and references therein).
7bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
We have presented MassComp, a specialized lossless compressor for MS data. MS data is
mainly composed of mass to charge ratio (m/z)-intensity pairs stored in base64 format. These
pairs correspond to floating-point numerical data, which are generally difficult to compress. Due
to the different nature of m/z and intensity values, MassComp employs different compression
strategies for each of them. We tested the performance of the proposed algorithm on several
datasets retrieved from the MassIVE repository, as well as on some of the datasets used in
[16]. When tested only on the numerical pairs, we show that MassComp outperforms both
the general lossless compressor gzip and the numerical compressor FPC in compression ratio.
In particular, MassComp exhibits up to 51% and 85% improvement when compared to gzip
and FPC, respectively. In addition, MassComp is able to reduce the size of the pairs by 46%
on average, in contrast to gzip and FPC, which on average reduce the sizes by 28% and 10%,
respectively. When tested on the whole mzXML files, MassComp showed a 28% improvement
with respect to gzip, and an average compression ratio of 59%. Finally, MS-Numpress offers
better compression results for double-precision data, however the algorithm is lossy, whereas
MassComp is lossless, in that the data can be recovered exactly.
These results demonstrate the potential of MassComp to significantly reduce the footprint
of MS data, and show the benefits of designing specialized compression algorithms tailored to
MS data. MassComp is an addition to the family of omics compression algorithms designed to
lessen the storage burden and facilitate the exchange and dissemination of omics data.
Methods
MassComp is a specialized lossless compressor for MS data. In particular, MassComp is opti-
mized to compress the (m/z)-intensity pairs, and applies the general lossless compressor gzip to
the remaining data. In its current format, MassComp accepts as input mzXML files. However,
note that various conversion software are available to convert between the mzML and mzXML
formats [24, 27], and hence the proposed algorithm MassComp can be potentially applied to
mzML files as well. Furthermore, the (m/z)-intensity pairs data is equivalent in both formats,
and hence the proposed compression method could be applied seamlessly after extraction of
these pairs.
Due to the different nature of the mass to charge (m/z) ratios and the ion count (intensity)
values, MassComp uses different compression strategies for each of them. Thus MassComp
first extracts the (m/z)-intensity pairs from the available scans, and after decoding the base64
data, separates the pairs into m/z and intensity, and encodes each category individually. In the
following we describe each of the strategies employed by the proposed algorithm MassComp in
more detail.
Base64 decoding
MassComp decodes the base64 symbols of the (m/z)-intensity pairs and expresses each value
in the IEEE 754 standard for single- or double-precision floating-point format. For each file,
MassComp automatically detects the adequate precision.
For single-precision, each symbol in the IEEE 754 standard occupies 4 bytes (32 bits) in
computer memory, and it is able to represent a wide dynamic range of values by using a floating
point, with 6 to 9 significant decimal digits precision. Specifically, the first bit is a sign bit,
followed by an exponent width of 8 bits and a significand precision (fraction) of 23 bits. See
Fig. 2 for an example. For double-precision, the format occupies 8 bytes (64 bits) instead, with
1 bit for the sign, 11 bits for the exponent, and 52 for the fraction.
The compression of the mass-to-charge ratios and ion counts (intensities) differ slightly for
single and double precision. In the following we first focus on single precision, and then show
how the methods are extended for double precision.
8bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
Figure 2: Example for representing value 0.15625 in the IEEE 754 standard. The value can
be computed from the binary representation as: (−1)b31 (1 + 23 −i (e−127)
P
P7 i=1 b23−i 2 )2 , with e =
i
b
i=0 23+i 2 .
Mass-to-charge ratio (m/z) compression
Single-precision: In most cases, ion scan is sequential, leading to m/z values that are smooth
and confined. MassComp takes advantage of this and implements a variation of delta encod-
ing for them. In particular, MassComp first converts each IEEE 754 standard single-precision
floating-point value into its equivalent hexadecimal representation (corresponding to 8 hexadec-
imal symbols). Differences of adjacent values are then calculated for each digit. Derived from
the smoothness of mass-to-charge ratio values, the computed differences contain many zeros
at front. The length of the front zeros is encoded by means of an arithmetic encoder. The
output of the arithmetic encoder together with the remaining non-zero parts of the difference
are written to the output. Due to the uniformity of the non-zero parts no further compression
is applied to them. This process is depicted in Fig. 3.
The number of (m/z)-intensity pairs varies greatly from scan to scan, ranging from a few
pairs to several thousands of them. To account for those scans with fewer pairs, the employed
arithmetic encoder is not adaptive and hence the symbol frequencies are first computed and
stored in the output. Note that an adaptive scheme needs to compress several values before it
can learn the statistics of the data, and thus it may perform poorly in these cases.
Figure 3: Schematic of the encoding performed by the proposed algorithm MassComp for the
compression of the mass-to-charge (m/z) ratio values.
The decompressor interprets encoded mass-to-charge ratios by zero-padding the front zeros
of its differences and adding the previous decompressed hexadecimal values.
Double-precision: The method used to compress the m/z values in double-precision for-
mat is very similar to the single precision case. However, some modifications are needed, as in
9bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
the double-precision format the corresponding hexadecimal representation occupies 16 symbols
instead. We observe that in this case, when taking the difference between adjancent values,
several zeros appear in the last positions. Hence in addition to the front-zeros, we also encode
the number of back-zeros with an arithmetic encoder. The remaining of the method remains
the same.
Intensity (ion count) compression
Single-precision: Like most floating-point data, the intensity values exhibit high precision.
In addition, unlike the mass to charge ratio values, they are not smooth and increasing, mak-
ing them more difficult to compress efficiently. However, these data are generated by mass
spectrometers, which have a limitation of range. As discussed above, the first 9 bits in the
single-precision IEEE 754 floating-point format correspond to the sign and exponent, and thus
they are very likely to be the same across different intensity values. Furthermore, due to the
finite precision of the mass spectrometer, bits from last positions (i.e., bits corresponding to
the fraction) sometimes also share similarity with previous values.
Hence, we developed a compression method based on “match” compression for the single-
precision floating-point intensity values. Briefly, MassComp first looks for a perfect match
of several predefined bits of the current value with previously compressed ones. If a match
is found, a pointer indicating the position to the previous value is stored, together with the
residual (i.e., the non-matching bits). If a match is not found, the pointer is set to zero and
the unmatched data is stored.
Figure 4: Schematic of the method employed by MassComp to compress the intensity values.
Initially, MassComp inspects the first 50 intensity values and decides searching a perfect
match for the first 8 bits only, or the first 8 bits together with the last 16. This decision is
done adaptively for each scan, and it is based on the number of matches found for each case
(i.e., the one with more matches is selected). Though the first 9 bits are the sign bit and the
exponent, MassComp only searches for a match in the first 8 bits to achieve a balance between
compression ratio and speed. The base64 intensity values are first converted to hexadecimal,
and thus it is more efficient to implement the searching algorithm in an integer number of
hexadecimal symbols (and hence multiple of 4 bits). In addition, working and operating with
bytes is more efficient. The match is sought within the last 15 compressed intensity values,
and thus the pointer can take values in {0, 1, . . . , 15}. Recall that 0 is reserved to indicate no
10bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
match. To summarize, intensity values are encoded in three blocks: pointers, residuals, and
unmatched data. The pointers are further compressed with an arithmetic encoder. Fig. 4
shows an example of the described method to compress the intensity values. All experimental
designed choices, such as inspecting the first 50 intensity values, deciding on a match for the
first 8 bits or also the last 16, as well as looking for a match to only the previous 15 values,
were decided based on simulations, as these were found to offer the best trade-off between
compression performance and speed.
Decompression works as follows. MassComp first reads the pointer, which indicates whether
a matched to a previous value was found or not. If the pointer is zero, the decompressor ex-
tracts 32 bits from the unmatched binary block. If the pointer is non-zero, the corresponding
previously decoded value is found and the matched bits extracted. These bits are then com-
bined with the residual bits (i.e., the non matching bits extracted from the residual block) to
reconstruct the intensity value.
Double-precision: Recall that each intensity values in double-precision is expressed with
16 hexadecimal symbols. The method employed to compress these values is similar to that of
the single-precision format. However, in this case we look for a match of either the first 8 bits
or the first 8 bits together with the last 32 bits. The pointers, residuals, and unmatched data
are then encoded in the same way as in the single precision version.
Implementation details:
MassComp is implemented in C++, and works in Windows and Linux. The code is freely avail-
able for download at https://github.com/iochoa/MassComp. The input file to MassComp is
an mzXML file, but we also provide scripts to facilitate the compression and decompression of
several mzXML files within a directory.
To parse the original mzXML file and reconstruct it from the compressed data, MassComp
uses the C++ XML parser TinyXML-2 [28]. After parsing the file, the m/z-intensity pairs are
effectively compressed and the output stored in a binary file. The remaining metadata is stored
in another file, and the two files are then further compressed with the general lossless compressor
algorithm gzip. At the time of decompression these two files are extracted, and after decoding
of the m/z-intensity pairs from the binary file, the original mzXML file is reconstructed. Both
the encoder and decoder detect the precision of the m/z-intensity pairs automatically, and use
the single or double precision method described above accordingly.
Availability of data and material
All data used in the manuscript is available online. MassComp is written in C++ and freely
available for download at https://github.com/iochoa/MassComp, together with instructions
to install and run the software.
Funding
Publication of this article was sponsored by the UORTSP of Tsinghua University, grant number
2018-182799 from the Chan Zuckerberg Initiative DAF, an advised fund SVCF, and an SRI
grant from UIUC.
References
[1] Alan G Marshall et al., “Fourier transform ion cyclotron resonance mass spectrometry: a
primer,” Mass spectrometry reviews, vol. 17, no. 1, pp. 1–35, 1998.
11bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
[2] Ruedi Aebersold and Matthias Mann, “Mass spectrometry-based proteomics,” Nature,
vol. 422, no. 6928, pp. 198–207, 2003.
[3] Katja Dettmer, Pavel A Aronov, and Bruce D Hammock, “Mass spectrometry-based
metabolomics,” Mass spectrometry reviews, vol. 26, no. 1, pp. 51–78, 2007.
[4] Felix S Oppermann, Florian Gnad, et al., “Large-scale proteomics analysis of the human
kinome,” Molecular & Cellular Proteomics, vol. 8, no. 7, pp. 1751–1764, 2009.
[5] Mohamad Bakar et al., “Metabolomics–the complementary field in systems biology: a
review on obesity and type 2 diabetes,” Molecular BioSystems, vol. 11, no. 7, pp. 1742–
1774, 2015.
[6] Trevor T Duarte and Charles T Spencer, “Personalized proteomics: the future of precision
medicine,” Proteomes, vol. 4, no. 4, pp. 29, 2016.
[7] Attila Csordas, David Ovelleiro, et al., “Pride: quality control in a proteomics data
repository,” Database, vol. 2012, 2012.
[8] Robertson Craig et al., “Open source system for analyzing, validating, and storing protein
identification data,” Journal of proteome research, vol. 3, no. 6, pp. 1234–1242, 2004.
[9] Frank Desiere, Eric W Deutsch, et al., “The peptideatlas project,” Nucleic acids research,
vol. 34, no. suppl 1, pp. D655–D658, 2006.
[10] Terry Farrah, Eric W Deutsch, et al., “Passel: the peptideatlas srmexperiment library,”
Proteomics, vol. 12, no. 8, pp. 1170–1175, 2012.
[11] Lennart Martens, Henning Hermjakob, Philip Jones, et al., “Pride: the proteomics iden-
tifications database,” Proteomics, vol. 5, no. 13, pp. 3537–3545, 2005.
[12] Philip Jones, Richard G Côté, et al., “Pride: a public repository of protein and peptide
identifications for the proteomics community,” Nucleic acids research, vol. 34, no. suppl 1,
pp. D659–D663, 2006.
[13] massIVE, “Mass Spectrometry Interactive Virtual Environment,” https://massive.
ucsd.edu/ProteoSAFe/static/massive.jsp, [Accessed: August 2017].
[14] Patrick GA Pedrioli, Jimmy K Eng, et al., “A common open representation of mass
spectrometry data and its application to proteomics research,” Nature biotechnology, vol.
22, no. 11, pp. 1459–1466, 2004.
[15] Henning Hermjakob, “The hupo proteomics standards initiative–overcoming the fragmen-
tation of proteomics data,” Proteomics, vol. 6, no. S2, pp. 34–38, 2006.
[16] Johan Teleman et al., “Numerical compression schemes for proteomics mass spectrometry
data,” Molecular & Cellular Proteomics, vol. 13, no. 6, pp. 1537–1542, 2014.
[17] Ibrahim Numanagić et al., “Comparison of high-throughput sequencing data compression
tools,” nature methods, vol. 13, no. 12, pp. 1005, 2016.
[18] Lukasz Roguski, Idoia Ochoa, Mikel Hernaez, and Sebastian Deorowicz, “Fastore-a space-
saving solution for raw sequencing data,” bioRxiv, p. 168096, 2017.
[19] Greg Malysa, Mikel Hernaez, et al., “Qvz: lossy compression of quality values,” Bioinfor-
matics, vol. 31, no. 19, pp. 3122–3129, 2015.
12bioRxiv preprint first posted online Feb. 6, 2019; doi: http://dx.doi.org/10.1101/542894. The copyright holder for this preprint
(which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.
It is made available under a CC-BY-NC-ND 4.0 International license.
[20] Martin Burtscher and Paruj Ratanaworabhan, “Fpc: A high-speed compressor for double-
precision floating-point data,” IEEE Transactions on Computers, vol. 58, no. 1, pp. 18–31,
2009.
[21] Nathan J Edwards, “Peparml: A meta-search peptide identification platform for tandem
mass spectra,” Current protocols in bioinformatics, vol. 44, no. 1, pp. 13–23, 2013.
[22] Michael L Metzker, “Sequencing technologiesâĂŤthe next generation,” Nature reviews
genetics, vol. 11, no. 1, pp. 31, 2010.
[23] Heng Li, Bob Handsaker, et al., “The sequence alignment/map format and samtools,”
Bioinformatics, vol. 25, no. 16, pp. 2078–2079, 2009.
[24] MSconverter, “Data Conversion to GNPS Compatible Formats - .mzXML and
.mzML,” https://bix-lab.ucsd.edu/display/Public/Data+Conversion+to+GNPS+
Compatible+Formats+-+.mzXML+and+.mzML, [Accessed: August 2017].
[25] Detlev Marpe, Heiko Schwarz, and Thomas Wiegand, “Context-based adaptive binary
arithmetic coding in the h. 264/avc video compression standard,” IEEE Transactions on
circuits and systems for video technology, vol. 13, no. 7, pp. 620–636, 2003.
[26] Idoia Ochoa, Mikel Hernaez, Rachel Goldfeder, Tsachy Weissman, and Euan Ashley, “Ef-
fect of lossy compression of quality scores on variant calling,” Briefings in bioinformatics,
vol. 18, no. 2, pp. 183–194, 2016.
[27] FileConverter, “FileConverter – Converts between different MS file formats,”
http://ftp.mi.fu-berlin.de/pub/OpenMS/release1.9-documentation/html/TOPP_
FileConverter.html, [Accessed: August 2017].
[28] TinyXML-2, ,” http://www.grinninglizard.com/tinyxml2/, [Accessed: August 2017].
13You can also read

















































