Mitochondria Transcription Factor A: A Putative Target for the Effect of Melatonin on U87MG Malignant Glioma Cell Line - MDPI
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
molecules
Article
Mitochondria Transcription Factor A: A Putative
Target for the Effect of Melatonin on U87MG
Malignant Glioma Cell Line
Daiane G. Franco * ID
, Isabele F. Moretti and Suely K. N. Marie
Faculdade de Medicina FMUSP, Universidade de Sao Paulo, Sao Paulo, SP 01246903, Brazil;
isabelemoretti@gmail.com (I.F.M.); sknmarie@usp.br (S.K.N.M.)
* Correspondence: daianegfranco@yahoo.com.br; Tel.: +55-11-30618559
Academic Editor: Dun-Xian Tan
Received: 20 February 2018; Accepted: 7 May 2018; Published: 9 May 2018
Abstract: The disruption of mitochondrial activity has been associated with cancer development
because it contributes to regulating apoptosis and is the main source of reactive oxygen species (ROS)
production. Mitochondrial transcription factor A (TFAM) is a protein that maintains mitochondrial
DNA (mtDNA) integrity, and alterations in its expression are associated with mitochondrial damage
and cancer development. In addition, studies have shown that mitochondria are a known target
of melatonin, the pineal gland hormone that plays an important anti-tumorigenic role. Thus,
we hypothesized that melatonin decreases the expression of TFAM (RNA and protein) in the human
glioblastoma cell line U87MG, which disrupts mtDNA expression and results in cell death due to
increased ROS production and mitochondrial damage. Our results confirm the hypothesis, and also
show that melatonin reduced the expression of other mitochondrial transcription factors mRNA
(TFB1M and TFB2M) and interfered with mtDNA transcription. Moreover, melatonin delayed
cell cycle progression and potentiated the reduction of cell survival due to treatment with the
chemotherapeutic agent temozolomide. In conclusion, elucidating the effect of melatonin on TFAM
expression should help to understand the signaling pathways involved in glioblastoma progression,
and melatonin could be potentially applied in the treatment of this type of brain tumor.
Keywords: melatonin; mitochondria; TFAM; cancer; glioblastoma
1. Introduction
Mitochondria are double-membrane organelles that contain their own genetic material,
mitochondrial DNA (mtDNA), which is circular, high-copy-number DNA [1,2]. In addition to
being an energy source for the cell that produces adenosine triphosphate (ATP) via oxidative
phosphorylation, mitochondria are important in other processes, such as calcium homeostasis,
fatty acid oxidation, the synthesis of reactive oxygen species (ROS), apoptosis, the cell cycle, and
proliferation. Accordingly, the disruption of mitochondrial activity is associated with several diseases,
including cancer [3–5]. Moreover, mtDNA associates with the internal membrane of mitochondria
via a nucleoprotein complex called nucleoid [6,7], and the most abundant protein component of this
structure is mitochondrial transcription factor A (TFAM), which belongs to the high mobility group
(HMG) protein family [7–9]. Although TFAM can bind to mtDNA in a nonspecific manner to promote
the packaging and maintenance of genetic material, TFAM also binds to the promoter region to regulate
transcription, together with two other transcription factors (TFAB1M and TFB2M) that form a complex
with mitochondrial RNA polymerase (POLRMT) [10]. In addition, TFAM also regulates the replication
of mtDNA by controlling its copy number [6,11].
Molecules 2018, 23, 1129; doi:10.3390/molecules23051129 www.mdpi.com/journal/moleculesMolecules 2018, 23, 1129 2 of 16
TFAM has been considered a potential target for cancer therapy since changes in its expression
have been detected in several types of cancer [12–15]. In glioma, the TFAM RNA and protein levels
are upregulated, compared to non-neoplastic brain tissue [12,13]. Nevertheless, the protein levels of
TFAM positively correlated with the malignancy of glioma [15], higher RNA levels of TFAM correlated
with a better prognosis among patients with grade IV glioma (glioblastoma, GBM) [13].
Glioblastoma, or grade IV astrocytoma, as classified by the World Health Organization (WHO),
is a tumor of glial origin restricted to the central nervous system (CNS) that is highly invasive
to the surrounding cerebral parenchyma [16,17]. Despite advances in knowledge regarding the
molecular biology of astrocytomas that have improved diagnosis and treatment, the prognosis of this
condition remains poor, with a maximum survival of 24 months [16–18]. The treatment of this cancer
currently involves a combination of surgery, radiotherapy, and chemotherapy with temozolomide
(TMZ) [16,19–22].
Melatonin (N-acetyl-5-methoxytryptamine), a hormone synthesized from serotonin, acts on
several cellular processes, including proliferation, differentiation, invasion, and apoptosis, to result in
different effects on non-tumor cells and tumor cells [23,24]. In normal cells, melatonin increases viability
and acts as an antioxidant, whereas it activates apoptosis and increases the cellular content of reactive
oxygen species (ROS) in tumor cells, effects that depend on mitochondrial activity [25,26]. The use
of melatonin as an adjuvant cancer chemotherapy has shown promising results in relation to both
improving the efficacy of treatment and reducing the incidence of side effects. The effects of melatonin
on mitochondria have been widely explored, however little is known about the effects of melatonin
on mtDNA and TFAM expression. We hypothesized that melatonin can reduce the expression of
mitochondrial transcription factors (TFAM, TFMB1M, and TFB2M) to decrease the viability of cancer
cells due to an imbalance in mitochondrial activity. Using a GBM cell line (U87MG), we showed that
melatonin decreased the level of mitochondrial transcription factors, induced mitochondrial membrane
depolarization to cause apoptosis, increased intracellular oxidative stress, and delayed the cell cycle.
When used as an adjuvant therapy with TMZ, melatonin enhanced the efficacy of chemotherapy by
further decreasing cell viability/proliferation.
2. Results
2.1. Melatonin Decreased the Expression of TFAM, TFB1M, and TFB2M
We first investigated the ability of melatonin to change the gene expression of transcription factors
that act on mitochondria, and the effect of this change on the proper functioning of the organelle and,
consequently, the cell. Incubation with melatonin (1 mM or 3 mM) for 72 h reduced the expression of the
transcriptions factors TFAM (Vehicle: 1.01 ± 0.05%; Mel 1 mM: 0.73 ± 0.10%; Mel 3 mM: 0.66 ± 0.07%),
TFB1M (Vehicle: 1.04 ± 0.06%; Mel 1 mM: 0.46 ± 0.05%; Mel 3 mM: 0.41 ± 0.07%), and TFB2M (Vehicle:
1.02 ± 0.05%; Mel 1 mM: 0.50 ± 0.03%; Mel 3 mM: 0.47 ± 0.10%), compared to the vehicle control
group (Figure 1A–C).
2.2. Melatonin Decreased the Content of TFAM Protein
Western blotting analysis showed that expression of TFAM at the protein level was decreased
following melatonin (3 mM) treatment compared to the vehicle group (ethanol 0.9%), but for the
1 mM concentration, the melatonin effect was variable and the result was not statistically significant.
(Figure 2 and Supplementary material—Figure S1, Table S2).organelle and, consequently, the cell. Incubation with melatonin (1 mM or 3 mM) for 72 h reduced
the expression of the transcriptions factors TFAM (Vehicle: 1.01 ± 0.05%; Mel 1 mM: 0.73 ± 0.10%; Mel
3 mM: 0.66 ± 0.07%), TFB1M (Vehicle: 1.04 ± 0.06%; Mel 1 mM: 0.46 ± 0.05%; Mel 3 mM: 0.41 ± 0.07%),
and TFB2M (Vehicle: 1.02 ± 0.05%; Mel 1 mM: 0.50 ± 0.03%; Mel 3 mM: 0.47 ± 0.10%), compared to the
Molecules
vehicle 2018, 23,
control 1129 (Figure 1A–C).
group 3 of 16
Molecules 2018, 23, x FOR PEER REVIEW 3 of 16
Figure 1. Melatonin inhibits the expression of mitochondrial transcription factor A (TFAM), TFB1M,
and TFB2M—Cultured U87MG cells were incubated with melatonin (1 mM or 3 mM) for 72 h, and
the medium was exchanged every 24 h. The relative mRNA expression levels of each gene were
quantified by qRT-PCR using the geometric mean of the following normalizing genes: Hypoxanthime
phosphoribosyl transferase (HPRT), glucuronidase-beta (GUS-B), and TATA-Box binding protein
(TBP) [27]. The data are expressed as the relative quantification (2−ΔΔCt) compared to the vehicle-
treated groups (ethanol 0.3% or 0.9%). Gene expression did not differ in cells treated with vehicle or
Figure
0.3% and Melatonin
1. 0.9% inhibits
ethanol, the expression
and these groups wereof mitochondrial
represented astranscription
a single group.factor A (TFAM),
From TFB1M,
left to right are
and TFB2M—Cultured
presented the results for U87MG
TFAM (A),cells were incubated
TFB1M with melatonin
(B), and TFB2M (1 mM
(C). * p < 0.05, or 3with
tested mM)anfor 72 h, and
analysis of
the medium was exchanged every 24 h. The relative mRNA expression levels of
variance followed by the Bonferroni post-hoc correction using GraphPad Prism version 5, comparing
® each gene were
quantified
the effect ofby qRT-PCRtousing
melatonin the geometric
the vehicle group. mean of the following normalizing genes: Hypoxanthime
phosphoribosyl transferase (HPRT), glucuronidase-beta (GUS-B), and TATA-Box binding protein
(TBP) [27].Decreased
2.2. Melatonin The data are
theexpressed
Content ofas TFAM
the relative quantification (2−∆∆Ct ) compared to the vehicle-treated
Protein
groups (ethanol 0.3% or 0.9%). Gene expression did not differ in cells treated with vehicle or 0.3% and
Western blotting analysis showed that expression of TFAM at the protein level was decreased
0.9% ethanol, and these groups were represented as a single group. From left to right are presented
following melatonin
the results (3 mM)
for TFAM treatment
(A), TFB1M compared
(B), and to the
TFB2M (C). * p 0.05 compared to vehicle. The statistical analysis consisted of an ANOVA followed by Bonferroni’s
represented as a single bar. * p > 0.05 compared to vehicle. The statistical analysis consisted of an
post-hoc test.
ANOVA followed by Bonferroni’s post-hoc test.
2.3. Melatonin Decreased the Transcription of mtDNA but Did Not Affect Replication
2.3. Melatonin Decreased the Transcription of mtDNA but Did Not Affect Replication
Since the transcription factors TFAM, TFB1M, and TFB2M are directly related to the regulation of
Since the transcription factors TFAM, TFB1M, and TFB2M are directly related to the regulation
transcription and mtDNA replication, we evaluated the expression of the MT-ND1 gene to ascertain
of transcription and mtDNA replication, we evaluated the expression of the MT-ND1 gene to
if the effect of melatonin on transcription factors was reflected in mitochondrial gene expression
ascertain if the effect of melatonin on transcription factors was reflected in mitochondrial gene
and mtDNA copy number. To examine mitochondrial gene expression and mtDNA copy number,
expression and mtDNA copy number. To examine mitochondrial gene expression and mtDNA copy
we used the aforementioned primer for the NADH dehydrogenase 1 gene and mRNA and DNA
number, we used the aforementioned primer for the NADH dehydrogenase 1 gene and mRNA and
extracted from U87MG cells treated with 1 mM or 3 mM of melatonin for 72 h, respectively. Melatonin
DNA extracted from U87MG cells treated with 1 mM or 3 mM of melatonin for 72 h, respectively.
reduced the expression of the mtDNA gene MT-ND1 (Vehicle: 1.01 ± 0.05%; Mel 1 mM: 0.54 ± 0.06%;
Melatonin reduced the expression of the mtDNA gene MT-ND1 (Vehicle: 1.01 ± 0.05%; Mel 1 mM:
Mel 3 mM: 0.62 ± 0.12%) (Figure 3A), but despite the reduction in TFAM, TFB1M, and TFB2M
0.54 ± 0.06%; Mel 3 mM: 0.62 ± 0.12%) (Figure 3A), but despite the reduction in TFAM, TFB1M, and
TFB2M expression, mtDNA replication appeared unchanged, since the number of copies of
mitochondrial genetic material remained the same after treatment with melatonin (Figure 3B).Molecules 2018, 23, 1129 4 of 16
expression, mtDNA replication appeared unchanged, since the number of copies of mitochondrial
Molecules 2018, 23, x FOR PEER REVIEW 4 of 16
genetic material remained the same after treatment with melatonin (Figure 3B).
Figure 3. Melatonin inhibits mitochondrial NADH dehydrogenase 1 gene expression but does not affect
Figure 3. Melatonin inhibits mitochondrial NADH dehydrogenase 1 gene expression but does not
mitochondrial DNA (mtDNA) replication—Cultured U87MG cells were incubated with melatonin
affect mitochondrial DNA (mtDNA) replication—Cultured U87MG cells were incubated with
(1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h. The relative expression of
melatonin (1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h. The relative
the NADH dehydrogenase 1 gene (A) and mtDNA copy number (B) were determined by qRT-PCR,
expression
using of the NADH
mitochondrial RNA and dehydrogenase 1 gene respectively.
DNA as a template, (A) and mtDNA copyare
The data number (B) were
expressed determined
as the relative
by qRT-PCR,
quantification (2 using
− ∆∆Ct mitochondrial RNA and DNA as a template, respectively. The data are
) compared to the vehicle-treated groups (ethanol 0.3% or 0.9%). Gene expression expressed
didasnot
thediffer
relative quantification
treated with(2vehicle
−ΔΔCt) compared to the vehicle-treated groups (ethanol 0.3% or 0.9%).
in cells or 0.3% and 0.9% ethanol, and these groups were represented
as Gene expression
a single bar * p < did not tested
0.05, differ in cellsan
with treated with
analysis ofvehicle or 0.3%
variance and 0.9%
followed ethanol,
by the and these
Bonferroni groups
post-hoc
were represented
correction as a single
using GraphPad Prismbar * p < 5,
® version 0.05, tested with
comparing an analysis
the effect of variance
of melatonin followed
to the vehicle by the
group.
Bonferroni post-hoc correction using GraphPad Prism® version 5, comparing the effect of melatonin
to the vehicle group.
2.4. Melatonin Induced ROS Production
2.4.ToMelatonin
verify that melatonin
Induced ROSincreases oxidative stress in U87MG cells, we evaluated the production of
Production
superoxides as an indicator of ROS production using cytometry, based on the reaction of total cellular
To verify that melatonin increases oxidative stress in U87MG cells, we evaluated the production
superoxide with dihydroethidium (DHE). The result showed that melatonin increased ROS production
of superoxides as an indicator of ROS production using cytometry, based on the reaction of total
to 20.73 ± 1.03% at a concentration of 1 mM, and 23.62 ± 4.56% at a concentration of 3 mM, compared
cellular superoxide with dihydroethidium (DHE). The result showed that melatonin increased ROS
to the vehicle group (14.97 ± 1.89%, Figure 4A). To verify if the increase of ROS induced by melatonin
production to 20.73 ± 1.03% at a concentration of 1 mM, and 23.62 ± 4.56% at a concentration of 3 mM,
has an effect on cell viability, a known ROS scavenger, N-acetyl-L-cysteine (NAC, 10 mM), was used.
compared to the vehicle group (14.97 ± 1.89%, Figure 4A). To verify if the increase of ROS induced by
Figure 4B shows that the antioxidant agent reverts the 1 mM melatonin-induced viability reduction
melatonin has an effect on cell viability, a known ROS scavenger, N-acetyl-L-cysteine (NAC, 10 mM),
and about 40% the effect of melatonin 3 mM.
was used. Figure 4B shows that the antioxidant agent reverts the 1 mM melatonin-induced viability
reduction
2.5. Melatoninand aboutMitochondria
Induced 40% the effect of melatoninand
Depolarization 3 mM.
Apoptosis
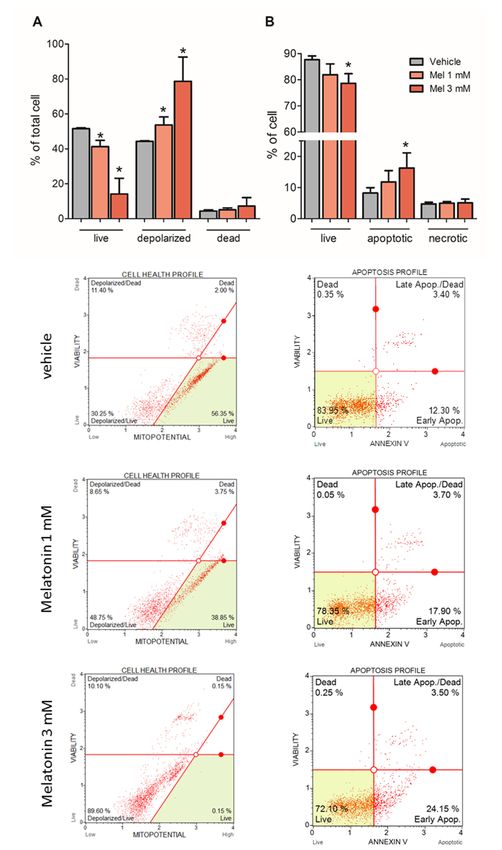
Two different cytometry experiments were performed to assess the ability of melatonin to induce
cell death. First, the depolarization of mitochondria was measured to verify if the loss of mitochondrial
inner membrane potential is associated with the early stage of apoptosis. The incubation of U87MG
with 1 mM and 3 mM melatonin for 72 h decreased the percentage of live cells in the vehicle group
from 51.55 ± 047% to 41.25 ± 3.61% and 14.14 ± 9.00%, respectively. Moreover, the percentage of cells
with depolarized mitochondria increased from 44.25 ± 0.4% in the vehicle group to 53.65 ± 4.60% and
78.66 ± 13.87% in the melatonin-treated groups (Figure 5A).
Second, apoptotic and necrotic cells were measured by staining phosphatidyl serine with Annexin
V and 7-AAD, respectively. The results showed that the percentage of live cells in the vehicle group
86.99 ± 1.42% decreased to 83.31 ± 3.80% and 78.63 ± 3.71% in the groups treated with 1 mM and 3 mM
melatonin, respectively. Specifically, the number of apoptotic cells, but not necrotic cells, increased.
Apoptotic cells increased to 11.79 ± 3.61% and 16.27 ± 4.86% in the melatonin group, whereas the
proportion of apoptotic cells was 8.26 ± 1.71% in the vehicle group (Figure 5B). The rates of apoptotic
Figure 4. Melatonin increases reactive oxygen species (ROS) production—(A) U87MG cells were
incubated with melatonin (1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h. ROSTo verify that melatonin increases oxidative stress in U87MG cells, we evaluated the production
of superoxides as an indicator of ROS production using cytometry, based on the reaction of total
cellular superoxide with dihydroethidium (DHE). The result showed that melatonin increased ROS
production to 20.73 ± 1.03% at a concentration of 1 mM, and 23.62 ± 4.56% at a concentration of 3 mM,
Molecules 2018, 23, 1129 5 of 16
compared to the vehicle group (14.97 ± 1.89%, Figure 4A). To verify if the increase of ROS induced by
melatonin has an effect on cell viability, a known ROS scavenger, N-acetyl-L-cysteine (NAC, 10 mM),
was
cellsused. Figure
did not 4Bbetween
differ shows that
the the antioxidant
vehicle agent reverts
groups (ethanol 0.3% the
and10.9%);
mM melatonin-induced
therefore, the meanviability
of these
reduction
rates was and about 40%
considered as athe effect
single of melatonin 3 mM.
group.
Figure 4. Melatonin increases reactive oxygen species (ROS) production—(A) U87MG cells were
Figure 4. Melatonin increases reactive oxygen species (ROS) production—(A) U87MG cells were
incubated with melatonin (1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h.
incubated with melatonin (1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h. ROS
ROS production was assessed by cytometry using the Muse® Cell Oxidative Stress kit. The results are
presented as the percentage of cells positively labeled for superoxide radicals. ROS production and
cell viability did not differ in cells treated with vehicle or 0.3% and 0.9% ethanol, and these groups are
consequently represented as a single bar. (B) U87MG cell were incubated with vehicle melatonin (1 mM
and 3 mM) and N-acetyl-L-cysteine (NAC, 10 mM) for 72 h and the medium was exchanged every 24 h.
Proliferation was assessed based on the reaction with PrestoBlue (Thermo Fisher Scientific), and the
fluorescence was read on a GloMax® 96 Microplate Luminometer (Promega Corporation). The results
were presented as a percentage of the control group. As the proliferation did not differ between the
vehicle-treated groups, they were represented as a single bar. * p < 0.05 compared to vehicle/control,
# p < 0.05 compared to the group treated with 1 mM melatonin, and + p < 0.05 compared to the group
treated with 3 mM melatonin. The statistical analysis consisted of an ANOVA followed by Bonferroni’s
Molecules 2018, 23, x FOR PEER REVIEW 6 of 16
post-hoc test.
Figure 5. Cont.Molecules 2018, 23, 1129 6 of 16
Figure 5.Figure
Melatonin induces
5. Melatonin mitochondrial
induces mitochondrial membrane depolarization
membrane depolarization andand apoptosis—U87MG
apoptosis—U87MG cells cells
were incubated with melatonin (1 mM or 3 mM) for 72 h, and the medium was
were incubated with melatonin (1 mM or 3 mM) for 72 h, and the medium was exchanged every 24 h. exchanged every 24 h.
(A) Mitochondrial polarization was evaluated by cytometry using a Muse® Mitopotential Assay Kit.
(A) Mitochondrial polarization was evaluated by cytometry using a Muse® Mitopotential Assay Kit.
The depolarized group was represented by the sum of depolarized live and dead cells. Membrane
The depolarized group
polarization did notwas represented
differ by with
in cells treated the sum ofordepolarized
vehicle 0.3% and 0.9% live and and
ethanol, dead cells.
these Membrane
groups
polarization
weredid not differ
represented in cells
as a single bar.treated with was
(B) Apoptosis vehicle or 0.3%
evaluated and 0.9%
by cytometry ethanol,
using the Museand
® these groups
Annexin
V & Dead Cell Assay Kit. The necrotic group represents the sum of dead cells and
were represented as a single bar. (B) Apoptosis was evaluated by cytometry using the Muse® Annexin late apoptotic/dead
cells presented in the representative plots. * p > 0.05 compared to vehicle. The statistical analysis
consisted of an ANOVA followed by Bonferroni’s post-hoc test.Molecules 2018, 23, x FOR PEER REVIEW 7 of 16
V & Dead Cell Assay Kit. The necrotic group represents the sum of dead cells and late apoptotic/dead
cells presented in the representative plots. * p > 0.05 compared to vehicle. The statistical analysis
Molecules 2018, 23,
consisted of 1129
an ANOVA followed by Bonferroni’s post-hoc test. 7 of 16
2.6. Melatonin Arrested U87MG Cells at the G0/G1 Phase of the Cell Cycle
2.6. Melatonin Arrested U87MG Cells at the G0/G1 Phase of the Cell Cycle
In addition to the effect of melatonin on apoptosis, we investigated its ability to alter the cell
cycle.InAccordingly,
addition to the effectcells
U87MG of melatonin on apoptosis,
were incubated we investigated
with either 1 mM or 3 its
mMability to alter
melatonin forthe
72cell
h.
cycle. Accordingly, U87MG cells were incubated with either 1 mM or 3 mM melatonin
Melatonin increased G0/G1 cell cycle arrest in U87MG cells at both 1 mM (71.92 ± 2.47%) and 3 mM for 72 h.
Melatonin
(77.77 increased
± 2.73%), G0/G1
compared cell vehicle
to the cycle arrest
groupin (64.85
U87MG cells at(Figure
± 1.20%) 6). (71.92 ± 2.47%) and 3 mM
both 1 mM
(77.77 ± 2.73%), compared to the vehicle group (64.85 ± 1.20%) (Figure 6).
Melatoninslows
Figure6.6. Melatonin
Figure slowsthethetransition
transitionfrom
from thethe G0/G1
G0/G1totoSSphase
phaseof ofthe
thecell
cell cycle—U87MG
cycle—U87MGcells cells
were incubated
were incubated with
with melatonin
melatonin (1 (1 mM
mM or or 33 mM)
mM) forfor 72
72 h,
h,and
andthe
themedium
mediumwas wasexchanged
exchangedeveryevery2424h.
The cell ®
h. The cellcycle
cyclephases
phaseswere
wereevaluated
evaluatedby bycytometry
cytometryusing
usingthe
theMuse
Muse® Cell
Cell Cycle Assay Kit.
Cycle Assay Kit. The
Thecell
cell
cycle distribution did not differ in cells treated with vehicle or 0.3% and 0.9% ethanol,
cycle distribution did not differ in cells treated with vehicle or 0.3% and 0.9% ethanol, and these and these groups
were represented
groups as a single
were represented asbar. * p > 0.05
a single bar.compared
* p > 0.05tocompared
vehicle. The
to statistical analysis
vehicle. The consisted
statistical of an
analysis
ANOVA followed
consisted by Bonferroni’s
of an ANOVA followed by post-hoc test. post-hoc test.
Bonferroni’s
2.7.Melatonin
2.7. MelatoninPotentiated
Potentiatedthe
theEffect
EffectofofTemozolomide
Temozolomide (TMZ)
(TMZ) to
to Reduce
Reduce Cell
Cell Viability
Viability
To determine
To determinethe thepotential
potentialsynergistic
synergisticeffect
effectof
ofmelatonin
melatoninand andthe
thechemotherapeutic
chemotherapeuticdrug drugTMZ,
TMZ,
we incubated
we incubated U87MG
U87MG cellscellsfor
for 72
72 hh with
with 11 mMmM oror 33 mM
mM of of melatonin
melatonin in in combination
combination withwith TMZ
TMZ
(0.8 mM).
(0.8 mM). Tumor
Tumor cell viability
viability was
was measured
measuredby bydetecting
detectingthethefluorescence
fluorescenceemitted
emittedbybythe
thereduction
reductionof
the PrestoBlue ® reagent by living cells. The results obtained showed that 1 mM and 3 mM melatonin
of the PrestoBlue reagent by living cells. The results obtained showed that 1 mM and 3 mM
®
reduced cell
melatonin viability/proliferation
reduced by 10% and by
cell viability/proliferation 34%,10%respectively.
and 34%,TMZ reduced cell
respectively. TMZ viability
reducedby 45%.
cell
Therefore,
viability bythe addition
45%. of 1 mM
Therefore, the and 3 mMofmelatonin
addition 1 mM and increased the effect by
3 mM melatonin 49% andthe
increased 87%, respectively
effect by 49%
(Figure
and 87%,7). Cells treated
respectively with7).
(Figure vehicles (ethanol
Cells treated 0.3%
with or 0.9%,
vehicles dimethylsulfoxide
(ethanol (DMSO) 0.1%, or a
0.3% or 0.9%, dimethylsulfoxide
combination
(DMSO) 0.1%,thereof) are represented
or a combination as aare
thereof) single bar in Figure
represented as a 6single
becausebarthese vehicles
in Figure did not these
6 because affect
cell viability and the viability did not significantly differ between these groups.
vehicles did not affect cell viability and the viability did not significantly differ between these groups.Molecules
Molecules2018,
2018,23,
23,x1129
FOR PEER REVIEW 8 8ofof16
16
Melatoninpotentiates
Figure7.7. Melatonin
Figure potentiatesthe
theeffect
effectof
oftemozolomide
temozolomide(TMZ) (TMZ)on on cell
cell proliferation/survival—
proliferation/survival—
U87MG cells were incubated with melatonin (1 mM or 3 mM) in combination not
U87MG cells were incubated with melatonin (1 mM or 3 mM) in combination or withwith
or not TMZ (0.8 mM)
TMZ (0.8
for 72 h, and the medium was exchanged every 24 h. Proliferation was assessed based
mM) for 72 h, and the medium was exchanged every 24 h. Proliferation was assessed based on the on the reaction
with PrestoBlue (Thermo(Thermo
Fisher Scientific), and the fluorescence was read on a GloMax ®
reaction with PrestoBlue Fisher Scientific), and the fluorescence was read on a 96 Microplate
GloMax ® 96
Microplate Luminometer (Promega Corporation). The results are presented as a percentage of theof
Luminometer (Promega Corporation). The results are presented as a percentage of the vehicle
each group. Proliferation did not differ between the vehicle-treated groups, and these groups were
vehicle of each group. Proliferation did not differ between the vehicle-treated groups, and these
represented as a single bar. * p < 0.05 compared to vehicle, # p < 0.05 compared to the group treated with
groups were represented as a single bar. * p < 0.05 compared to vehicle, # p < 0.05 compared to the
1 mM melatonin, and + p < 0.05 compared to the group treated with 3 mM melatonin. The statistical
group treated with 1 mM melatonin, and + p < 0.05 compared to the group treated with 3 mM
analysis consisted of an ANOVA followed by Bonferroni’s post-hoc test.
melatonin. The statistical analysis consisted of an ANOVA followed by Bonferroni’s post-hoc test.
3.3.Discussion
Discussion
Mitochondriaare
Mitochondria arecentral
centralorganelles
organellesininthe
thedevelopment
developmentofofcancer
cancerbecause
becausethey
theyare
areresponsible
responsible
forthe
for thebalance
balanceof ofbioenergetic
bioenergeticand andbiosynthetic
biosyntheticprocesses,
processes,and
andbecause
becausethey theyare
arethe
themain
mainsource
sourceof of
superoxides and are implicated in the intrinsic apoptosis pathway. Besides that,
superoxides and are implicated in the intrinsic apoptosis pathway. Besides that, the transcription the transcription
factorTFAM
factor TFAMisisessential
essentialforforthe
thereplication,
replication,transcription,
transcription,andandmaintenance
maintenanceofofmtDNA
mtDNAand, and,therefore,
therefore,
formitochondrial
for mitochondrial homeostasis
homeostasis [8–10].
[8–10]. Recent
Recent discoveries
discoverieshave
haveshown
shownthatthatmitochondria
mitochondria arearea target for
a target
melatonin [28], and melatonin has been shown to accumulate in mitochondria
for melatonin [28], and melatonin has been shown to accumulate in mitochondria against the against the concentration
gradient via active
concentration gradienttransport [29].transport
via active This phenomenon remains poorly
[29]. This phenomenon understood,
remains but melatonin
poorly understood, butis
known to prevent the inhibition of complexes I and IV induced by red ruthenium
melatonin is known to prevent the inhibition of complexes I and IV induced by red ruthenium [30]. [30]. In the present
study,
In we showed
the present study,that
weTFAM
showed andthat
other mitochondrial
TFAM and othertranscription
mitochondrial factors (TFB1M and
transcription TFB2M)
factors (TFB1Mmay
be targets
and TFB2M)ofmay melatonin in glioblastoma
be targets of melatonin in cells. The melatonin-induced
glioblastoma reduction in the
cells. The melatonin-induced expression
reduction in
the expression of these transcription factors was associated with reduced mitochondrial NADH1
of these transcription factors was associated with reduced mitochondrial NADH dehydrogenase
(MT-ND1) gene1 expression.
dehydrogenase (MT-ND1) gene In accordance
expression.with our results,with
In accordance Prunet-Marcassus and colleaguesand
our results, Prunet-Marcassus [31]
also showed that melatonin reduces the transcriptional content of mitochondria
colleagues [31] also showed that melatonin reduces the transcriptional content of mitochondria by by 44% in brown
adipocytes
44% in brown in adipocytes
the Siberianinhamster.
the Siberian hamster.
Evidenceindicates
Evidence indicatesthat
thatTFAM
TFAMhas hasbeen
beenshown
shownto toregulate
regulatemtDNA
mtDNAcopy copynumber
number[10,11],
[10,11],and andanan
unbalance in the number of mtDNA copies is associated with several neurodegenerative
unbalance in the number of mtDNA copies is associated with several neurodegenerative diseases and diseases and
cancer,including
cancer, includingglioblastoma
glioblastoma[13,32–34].
[13,32–34].Melatonin
Melatonindid didnot
notaffect
affectthe
themtDNA
mtDNAcopy copynumber
numberininthis this
study, despite
study, despite reducing
reducing mitochondrial
mitochondrialgene geneexpression,
expression,which
whichindicates
indicates that different
that differentmechanisms
mechanisms are
responsible for the control of mitochondrial gene expression and mtDNA replication
are responsible for the control of mitochondrial gene expression and mtDNA replication by melatonin. by melatonin.
In cultures of mouse C6 gliomas and Neuro2a mice, melatonin reverses the morphine- and nickelMolecules 2018, 23, 1129 9 of 16
In cultures of mouse C6 gliomas and Neuro2a mice, melatonin reverses the morphine- and nickel
chloride-induced reductions in mtDNA copy number, respectively. But, in accordance with our results,
melatonin alone does not interfere with mtDNA content [35,36].
Changes in mitochondrial gene expression or nuclear genes may result in the collapse of the
mitochondrial respiratory chain to increase ROS production, which, consequently, may trigger the
activation of apoptosis [3–5,37–40]. Our results showed that melatonin increased ROS production,
which may be a consequence of alterations in the expression of respiratory chain genes that depend
on the activity of the transcription factors TFAM, TFB1M, and TFB2M. This hypothesis might be
supported by the fact that the downregulation of TFAM expression has promoted ROS-dependent
activation of JNK/p38 MAPK and apoptosis, as reported in non-small cell lung cancer [41]. In addition,
in the cardiac muscle cell line, the downregulation of TFAM caused mitochondrial oxidative
phosphorylation dysfunction, resulting in increased ROS production [42]. Still, the overexpression
of TFAM inhibited mitochondrial ROS generation in HeLa cells [43] and prevented oxidative stress,
facilitating cardioprotection [44].
High concentrations of ROS can damage mtDNA and nuclear DNA and alter the expression of
oncogenes and tumor suppressor genes, which modifies the onset and progression of tumors [37].
Several studies demonstrated that melatonin influences the intracellular content of ROS by different
mechanisms. The protective role of melatonin by reducing oxidative stress in experimental models
of tissue damage is well known [24]. For example, in contrast to our results, in a non-cancerous
model of myocardial ischemia/reperfusion injury in type 1 diabetic rats, melatonin has increased
TFAM expression, reducing mitochondrial oxidative stress and enhancing its biogenesis [45]. However,
in cancer cells, two scenarios have been shown: A pro-oxidant activity in which melatonin induces the
increase of intracellular levels of ROS, leading to cell death, as was shown in the present study and
by others [26,46–54]; and melatonin reducing intracellular levels of ROS and inducing cell death by
different mechanisms, such as, for example, inhibiting the nuclear transcription factor kappa B (NF-κB)
nuclear activity, as reported in human glioma cells (T98 and U251) by Wang and colleagues [55],
and in rat glioma cells (C6) by Martín and colleagues [56]. Otherwise, the melatonin activation of
NF-κB has also been associated with an increase in intracellular oxidative stress in a model of human
monocyte (U937) culture [47,48], and in a primary cerebellar granule cell culture [57]. In the present
study, a known antioxidant, NAC, completely inhibited the decreased viability induced by 1 mM
melatonin in a U87MG cell culture. At a melatonin concentration of 3 mM, NAC inhibited this effect
by about 40%, indicating that melatonin-induced ROS increase is at least in part responsible for the
observed cell death. It is important to consider the concentration and the time of exposure to melatonin.
The pharmacological concentrations of melatonin used (1 mM and 3 mM) indicate that the observed
effects are independent of melatonin receptors (MT1 and MT2). Minor concentrations of melatonin
(1 µM and 100 µM) had no effect on cell proliferation or on the expression of TFAM (data not shown).
The concentration of 3 mM of melatonin presented a more consistent result regarding the reduction of
TFAM at the protein level. Although not statistically significant, a trend of TFAM protein decrease
was also observed at the concentration of 1 mM (supplemental material). Interestingly, 1 mM of
melatonin was enough to alter the mitochondrial gene (MT-ND1) expression. The exposition time to
melatonin also proved to be crucial for the observed results. Incubation times with melatonin of less
than 72 h were not sufficient to alter cell viability, as well as intracellular ROS content (data not shown).
Wang and colleagues [55] have also shown no change in cell proliferation after 24 h of incubation with
melatonin, although they have detected a decrease in intracellular ROS levels.
Our results showed that mitochondrial membrane depolarization was increased in cells incubated
with melatonin, which indicates a collapse of inner membrane polarization that triggers the opening
of mitochondrial transition pores (MTPs) and the release of cytochrome C and other pro-apoptotic
factors [28,38,58,59]. The increase in ROS production induced by melatonin might lead to mitochondrial
membrane depolarization and the activation of cell death, although confirmation of the activation of
intrinsic apoptosis needs to be proven. Melatonin is known to affect MTP in non-cancer cells, such asMolecules 2018, 23, 1129 10 of 16
in striatal neurons. In this model, melatonin prevents loss of mitochondrial membrane potential and
reduces the probability of MTP opening, which prevents cell death by apoptosis [60]. Whereas in
human promyelocytic leukaemia HL-60 cells, melatonin increases H2 O2 -induced ROS generation,
causing a decrease in mitochondrial membrane potential and cell death [51]. The opposing effects of
melatonin on cancer and non-cancer cells has been widely discussed and can be reviewed in [23,26].
In addition to the effects on oxidative stress and cell death, we showed that melatonin inhibits
the progression of the cell cycle in U87MG cells. A possible pathway to explain this cell cycle arrest
is through the physical interaction between the tumor suppressor protein p53 and TFAM [61–64].
Furthermore, p53 is a target of melatonin, which activates p53, and in its turn induces apoptosis and
arrests tumor cells in the G1/G0 to S transition of the cell cycle [26,65].
Glioblastoma is a very aggressive type of cancer with a very low survival rate. Therefore, new
therapeutic targets have been investigated, and TFAM is a strong candidate target since its expression
is altered in several types of cancer, including glioma [13,15], colorectal cancer [63], epithelial ovarian
carcinoma [66], bladder cancer [67], breast cancer [68], lung cancer [41,69], and colon cancer [70].
Moreover, the use of melatonin in the treatment of cancer has shown promising results. Specifically,
previous studies have demonstrated a possible antitumor role for melatonin in glioma models [47,48].
In addition, Kinker and colleagues [71] have recently demonstrated that human glioma cell lines (HOG,
T98G, and U87MG) produce melatonin, and the ability of cells to produce this hormone negatively
correlated with tumor malignancy.
Finally, our results showed that the combination of melatonin with temozolamide, TMZ,
potentiated its effects on cell survival, pointing at a promising combinatorial treatment for glioblastoma
patients. In summary, our results suggest that increased generation of melatonin-induced intracellular
ROS in U87MG glioblastoma cells may be an effect of melatonin on the expression of TFAM and
other mitochondrial transcription factors (TFB1M and TFB2M), leading to mitochondrial disruption.
Our study opens a new perspective to understand the mechanism of action of melatonin in tumor cells.
4. Materials and Methods
4.1. Cell Culture Conditions
The human malignant glioma cell line U87MG (American Type Culture Collection, ATCC) was
routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Carlsbad, CA,
USA), supplemented with 10% fetal bovine serum (FBS) (Life Technologies), 100 IU/mL penicillin, and
100 µg/mL streptomycin (Life Technologies), in a humidified atmosphere consisting of 5% CO2 in air
at 37 ◦ C. The cell line was authenticated by short tandem repeat DNA profiling using the GenePrint 10
System (Promega, Madison, WI, USA).
4.2. Extraction of RNA and DNA
Cells were plated 2 × 105 cells/mL in a 24-well plate and treated with 1 mM or 3 mM of melatonin
for 72 h, and the control groups were treated with 0.3% or 0.9% of ethanol vehicle, respectively.
The medium containing melatonin or vehicle was changed every 24 h. The cells were then digested with
RLT Plus buffer (QIAGEN, Hilden, Germany), syringe homogenized 10 times, and frozen at −80 ◦ C
before extracting the genetic material. Both RNA and DNA were extracted from the cell homogenate
using the AllPrep DNA/RNA Micro Kit (QIAGEN) following the protocol provided by the manufacturer.
The concentration (ng/µL) and purity of DNA and total RNA were determined by quantification on the
NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
4.3. Expression of TFAM, TFB1M, TFB2M, and NADH Dehydrogenase 1 (MT-ND1) by qRT-PCR
RNA was reverse transcribed using the Maxima First Strand cDNA Synthesis kit for qRT-PCR
(Thermo Scientific) according to the manufacturer’s specifications. Quantitative data were obtained
using SYBR green (Thermo Scientific) qRT-PCR on the ABI Prism 7500 sequence detector (AppliedMolecules 2018, 23, 1129 11 of 16
Biosystems, Foster City, CA, USA), and normalized in relation to the geometric mean of three
housekeeping genes: Hypoxanthime phosphoribosyltransferase (HPRT), glucuronidase beta (GUSB),
and TATA binding protein (TBP). The equation 2−∆∆Ct was applied to calculate the relative gene
expression levels [27]. The primers were designed to amplify 80–150 bp length amplicons, had a melting
temperature of 60 ◦ C, and were synthesized by Integrate DNA Technology (IDT, Coralville, IA, USA)
as follows (50 to 30 ); TFAM F: CTCCCCCTTCAGTTTTGTGT, TFAM R: GCATCGGG-TTCTGAGCTTT;
TFB1M F: ATGGCTCAGTACCTCTGCAATG, TFB1M R: TGGGCTGTATCAAGGGAGTGA; TFB2M F:
ATCCCGGAAATCCAGACTTGT, TFB2M R: GACCAAGGCTCCATGTGCA; NADH dehydrogenase
1 (MT-ND1) F: TGATGGCTAGGGTGACTTCAT, MT-ND1 R: CCTAGCCGTTTACTCAATCCT;
HPRT F: TGAGGATTTGGAAAGGGTGT, HPRT R: GAGCACACAGAGGGCTACAA; GUSB F:
GAAAATACGTGGTTGGAGAGCTCATT, GUSB R: CCGAGTGAAGATCCCCTTTTTA; TBP F:
AGGATAAGAGAGCCACGAACCA, TBP R: CTTGCTGCCAGTCTGGACTGT. PCR was carried out
as follows: 5 min at 50 ◦ C, 10 min at 95 ◦ C, 40 cycles at 95 ◦ C for 15 s, and 60 ◦ C for 1 min. The primer
concentrations used were 200–400 nM. All assays were carried out in duplicate and eventually repeated
when the standard deviation exceeded 0.4.
4.4. Mitochondrial DNA Copy Number Quantification
A single copy gene—hemoglobin beta (HBB)—was used as a reference to determine the number
of copies of mtDNA by SYBR Green qRT-PCR on an ABI Prism 7500 sequence detector (Applied
Biosystems). The primer sequence used to quantify the mtDNA copy number was the same as
that of NADH dehydrogenase 1, and the primer was used at a final concentration of 200 nM.
The sequences of HBB were as follows (50 –30 ): HBB F: GTGAAGGCTCATGGCAAGA and HBB
R: AGCTCACTCAGGTGTGGCAAAG (IDT). The cycle conditions were 10 min at 95 ◦ C, 40 cycles at
95 ◦ C for 15 s, and 60 ◦ C for 1 min. All assays were carried out in duplicate and eventually repeated
when the standard deviation exceeded 0.4. The relative mtDNA copy number was determined with
the equation 2−∆∆Ct [27].
4.5. Western Blot Analysis
Total protein lysates were prepared from U98MG cell cultures with RIPA lysis buffer and protease
inhibitor cocktail (Sigma-Aldrich) on ice. The protein concentration was determined using a NanoDrop
Microvolume Spectrophotometers (Thermo Scientific™). Total protein lysates (30 mg) were separated
by 4% to 12% polyacrylamide gel electrophoresis (Invitrogen, Carsbald, CA, USA) with 1× NuPAGE
MOPS SDS 20× (Invitrogen, Carsbald, CA, USA) running buffer. The proteins were electrophoretically
transferred to a Polyvinylidene Fluoride membrane (PVDF) through the semi-dry Trans-Blot® SD
system (Trans-Blot® Transfer Cell, Biorad, Hercules, CA, USA). The membrane was blocked with
5% skim milk and incubated with rabbit monoclonal primary anti-TFAM diluted 1:500, and with
mouse monoclonal primary anti-β-actina (clone AC-74, Sigma-Aldrich) diluted 1:5000, as a protein
loading control. The secondary antibodies used were anti-rabbit (1:1000) and anti-mouse IgG (1:5000)
conjugated to peroxidase (Sigma-Aldrich). The immune complexes were visualized using enhanced
chemiluminescence reagent (Western Lightning Chemiluminescence Reagent Plus, Perkin Elmer,
Waltham, MA, USA) and detected with UVITEC (Alliance 4.7) Cambridge, UK.
4.6. Evaluation of Oxidative Stress, Cell Cycle, Apoptosis, and Mitochondria Polarization
U87MG cells were treated with 1 mM or 3 mM melatonin for 72 h. Control groups were treated
with ethanol vehicle (0.3% or 0.9%, relative to the melatonin concentration). The medium was changed
every 24 h, and the melatonin or vehicle was replaced. The cells were then prepared for cytometric
assays to evaluate oxidative stress, the cell cycle, apoptosis, and mitochondrial membrane polarization
using the Muse® Cell Analyzer (Merck Millipore, Billerica, MA, USA) and appropriate reagent kits:
Muse® Cell Oxidative Stress Kit, Muse® Cell Cycle Assay Kit, Muse® Annexin V & Dead Cell Assay Kit
and Muse® Mitopotential Assay Kit, respectively, according to the manufacturer’s instructions.Molecules 2018, 23, 1129 12 of 16
4.7. Cell Viability/Proliferation
U87MG cells were treated with melatonin (1 mM or 3 mM) in the presence or absence of
temozolomide (TMZ 0.8 mM—Sigma-Aldrich, St. Louis, MO, USA) or N-Acetyl-L-cysteine (NAC 10 mM—
Sigma-Aldrich, St. Louis, MO, USA) for 72 h, and the medium and drug were replaced every
24 h. The control groups were treated with melatonin vehicle (0.3% or 0.9% ethanol), TMZ vehicle
(0.1% dimethylsulfoxide—DMSO), or a combination thereof (0.3% ethanol + DMSO 0.1% or 0.9% ethanol
+ 0.1% DMSO). NAC was diluted in DMEM. After the treatments, the cells were incubated with PrestoBlue
reagent (Invitrogen, Carlsbad, CA, USA) for 2 h, and the fluorescence was measured on a GloMax® 96
Microplate Luminometer (Promega Corporation, Madison, WI, USA).
4.8. Statistical Analysis
The results are reported as the mean ± s.e.m. of at least three independent experiments, and were
normalized to the groups treated with specific vehicles for each experiment. The differences between
experimental groups were tested with an analysis of variance followed by the Bonferroni post-hoc
correction using GraphPad Prism® version 5.
Supplementary Materials: The following are available online.
Author Contributions: D.G.F. and S.K.N.M. conceived, designed the experiments and contributed with the
analysis of the data; D.G.F. and I.F.M. performed the experiments; All authors critically revised the manuscript.
Funding: This research was funded by Fundação de Amparo a Pesquisa de São Paulo (FAPESP) grant
numbers [#2004/12133-6, #2013/02162-8, #2014/17220-6] and Conselho Nacional de Desenvolvimento Científico
e Tecnológico (CNPq) grant number [#305730/2015-0].
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Nass, N.M. The circularity of Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1966, 56, 1215–1222. [CrossRef]
[PubMed]
2. Van Bruggen, E.F.; Borst, P.; Ruttenberg, G.J.; Gruber, M.; Kroon, A.M. Circular mitochondrial DNA.
Biochim. Biophys. Acta 1966, 119, 437–439. [CrossRef]
3. Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update.
Cell Metab. 2016, 25, 57–71. [CrossRef] [PubMed]
4. Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities andopportunities. Biochim. Biophys. Acta 2017,
185, 655–664. [CrossRef] [PubMed]
5. Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74,
1999–2017. [CrossRef] [PubMed]
6. Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. DNA replication and transcription in mammalian
mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [CrossRef] [PubMed]
7. Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920.
[CrossRef] [PubMed]
8. Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human
mitochondrial DNA is packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [CrossRef] [PubMed]
9. Kanki, T.; Nakayama, H.; Sasaki, N.; Takio, K.; Alam, T.I.; Hamasaki, N.; Kang, D. Mitochondrial nucleoid
and transcription factor A. Ann. N. Y. Acad. Sci. 2004, 1011, 61–68. [CrossRef] [PubMed]
10. Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial
transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819,
921–929. [CrossRef] [PubMed]
11. Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.;
Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number
in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [CrossRef] [PubMed]Molecules 2018, 23, 1129 13 of 16
12. Lin, P.C.; Lin, J.K.; Yang, S.H.; Wang, H.S.; Li, A.F.; Chang, S.C. Expression of beta-F1-ATPase and
mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer:
Clinical data analysis and evidence from an in vitro study. Int. J. Colorectal. Dis. 2008, 23, 1223–1232.
[CrossRef] [PubMed]
13. Correia, R.L.; Oba-Shinjo, S.M.; Uno, M.; Huang, N.; Marie, S.K. Mitochondrial DNA depletion and
its correlation with TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating astrocytomas.
Mitochondrion 2011, 11, 48–53. [CrossRef] [PubMed]
14. Yoshida, Y.; Hasegawa, J.; Nezu, R.; Kim, Y.K.; Hirota, M.; Kawano, K.; Izumi, H.; Kohno, K. Clinical
usefulness of mitochondrial transcription factor A expression as a predictive marker in colorectal cancer
patients treated with FOLFOX. Cancer Sci. 2011, 102, 578–582. [CrossRef] [PubMed]
15. Jiang, J.; Yang, J.; Wang, Z.; Wu, G.; Liu, F. TFAM is directly regulated by miR-23b in glioma. Oncol. Rep.
2013, 30, 2105–2110. [CrossRef] [PubMed]
16. Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310,
1842–1850. [CrossRef] [PubMed]
17. Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.;
Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of
the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [CrossRef] [PubMed]
18. Mineo, J.F.; Bordron, A.; Baroncini, M.; Ramirez, C.; Maurage, C.A.; Blond, S.; Dam-Hieu, P. Prognosis
factors of survival time in patients with glioblastoma multiforme: A multivariate analysis of 340 patients.
Acta Neurochir. 2007, 149, 245–252. [CrossRef] [PubMed]
19. Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.;
Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment.
Genes Dev. 2007, 21, 2683–2710. [CrossRef] [PubMed]
20. Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment.
Annu. Rev. Pathol. 2014, 9, 1–25. [CrossRef] [PubMed]
21. Parker, N.R.; Correia, N.; Crossley, B.; Buckland, M.E.; Howell, V.M.; Wheeler, H.R. Correlation of MicroRNA
132 Up-regulation with an Unfavorable Clinical Outcome in Patients with Primary Glioblastoma Multiforme
Treated with Radiotherapy Plus Concomitant and Adjuvant Temozolomide Chemotherapy. Transl. Oncol.
2013, 6, 742–748. [CrossRef] [PubMed]
22. Ichimura, K.; Narita, Y.; Hawkins, C.E. Diffusely infiltrating astrocytomas: Pathology, molecular mechanisms
and markers. Acta Neuropathol. 2015, 129, 789–808. [CrossRef] [PubMed]
23. Sainz, R.M.; Mayo, J.C.; Rodriguez, C.; Tan, D.X.; Lopez-Burillo, S.; Reiter, R.J. Melatonin and cell death:
Differential actions on apoptosis in normal and cancer cells. Cell. Mol. Life Sci. 2003, 60, 1407–1426. [CrossRef]
[PubMed]
24. Luchetti, F.; Canonico, B.; Betti, M.; Arcangeletti, M.; Pilolli, F.; Piroddi, M.; Canesi, L.; Papa, S.; Galli, F.
Melatonin signaling and cell protection function. FASEB J. 2010, 24, 3603–3624. [CrossRef] [PubMed]
25. Radogna, F.; Nuccitelli, S.; Mengoni, F.; Ghibelli, L. Neuroprotection by melatonin on astrocytoma cell death.
Ann. N. Y. Acad. Sci. 2009, 1171, 509–513. [CrossRef] [PubMed]
26. Lanoix, D.; Lacasse, A.A.; Reiter, R.J.; Vaillancourt, C. Melatonin: The smart killer: The human trophoblast as
a model. Mol. Cell. Endocrinol. 2012, 348, 1–11. [CrossRef] [PubMed]
27. Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)). Method Methods 2001, 25, 402–408. [CrossRef] [PubMed]
28. Acuña Castroviejo, D.; Escames, G.; Carazo, A.; León, J.; Khaldy, H.; Reiter, R.J. Melatonin, mitochondrial
homeostasis and mitochondrial-related diseases. Curr. Top Med. Chem. 2002, 2, 133–151. [CrossRef] [PubMed]
29. Tan, D.X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving
Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [CrossRef] [PubMed]
30. Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D.
Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial
damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [CrossRef] [PubMed]
31. Prunet-Marcassus, B.; Ambid, L.; Viguerie-Bascands, N.; Pénicaud, L.; Casteilla, L. Evidence for a direct
effect of melatonin on mitochondrial genome expression of Siberian hamster brown adipocytes. J. Pineal Res.
2001, 30, 108–115. [CrossRef] [PubMed]Molecules 2018, 23, 1129 14 of 16
32. Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N.;
et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance
in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987. [CrossRef] [PubMed]
33. Dickinson, A.; Yeung, K.Y.; Donoghue, J.; Baker, M.J.; Kelly, R.D.; McKenzie, M.; Johns, T.G.; St. John, J.C.
The regulation of mitochondrial DNA copy number in glioblastoma cells. Cell Death Differ. 2013, 20,
1644–1653. [CrossRef] [PubMed]
34. Meng, S.; Han, J. Mitochondrial DNA copy number alteration in human cancers. N. Am. J. Med. Sci. 2013, 6,
22–25. [CrossRef]
35. Feng, Y.M.; Jia, Y.F.; Su, L.Y.; Wang, D.; Lv, L.; Xu, L.; Yao, Y.G. Decreased mitochondrial DNA copy number
in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be
salvaged by melatonin. Autophagy 2013, 9, 1395–1406. [CrossRef] [PubMed]
36. Xu, S.C.; He, M.D.; Lu, Y.H.; Li, L.; Zhong, M.; Zhang, Y.W.; Wang, Y.; Yu, Z.P.; Zhou, Z. Nickel exposure
induces oxidative damage to mitochondrial DNA in Neuro2a cells: The neuroprotective roles of melatonin.
J. Pineal Res. 2011, 51, 426–433. [CrossRef] [PubMed]
37. Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol.
2000, 279, L1005–L1028. [CrossRef] [PubMed]
38. De Miguel, M.; Cordero, M.D. Oxidative Therapy against Cancer. In Oxidative Stress and Diseases;
Lushchak, V.I., Gospodaryov, D.V., Eds.; InTech: Rijeka, Croatia, 2012; Available online: http://
www.intechopen.com/books/oxidative-stress-and-diseases/oxidative-therapy-against-cancer (accessed
on 4 April 2018).
39. Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species.
Antioxid. Redox Signal. 2013, 19, 546–558. [CrossRef] [PubMed]
40. Rinaldi, M.; Caffo, M.; Minutoli, L.; Marini, H.; Abbritti, R.V.; Squadrito, F.; Trichilo, V.; Valenti, A.; Barresi, V.;
Altavilla, D.; et al. ROS and Brain Gliomas: An Overview of Potential and Innovative Therapeutic Strategies.
Int. J. Mol. Sci. 2016, 17, 984. [CrossRef] [PubMed]
41. Xie, D.; Wu, X.; Lan, L.; Shangguan, F.; Lin, X.; Chen, F.; Xu, S.; Zhang, Y.; Chen, Z.; Huang, K.; et al.
Downregulation of TFAM inhibits the tumorigenesis of non-small cell lung cancer by activating ROS-
mediated JNK/p38MAPK signaling and reducing cellular bioenergetics. Oncotarget 2016, 7, 11609–11624.
[CrossRef] [PubMed]
42. Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail. Rev.
2016, 21, 499–517. [CrossRef] [PubMed]
43. Hayashi, Y.; Yoshida, M.; Yamato, M.; Ide, T.; Wu, Z.; Ochi-Shindou, M.; Kanki, T.; Kang, D.; Sunagawa, K.;
Tsutsui, H.; et al. Reverse of age-dependent memory impairment and mitochondrial DNA damage in
microglia by an overexpression of human mitochondrial transcription factor a in mice. J. Neurosci. 2008, 28,
8624–8634. [CrossRef] [PubMed]
44. Ikeda, M.; Ide, T.; Fujino, T.; Arai, S.; Saku, K.; Kakino, T.; Tyynismaa, H.; Yamasaki, T.; Yamada, K.; Kang, D.;
et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection
associated with limited mitochondrial oxidative stress. PLoS ONE 2015, 10, e0119687. [CrossRef] [PubMed]
45. Yu, L.; Gong, B.; Duan, W.; Fan, C.; Zhang, J.; Li, Z.; Xue, X.; Xu, Y.; Meng, D.; Li, B.; et al. Melatonin
ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial
function: Role of AMPK-PGC-1α-SIRT3 signaling. Sci. Rep. 2017, 7, 41337. [CrossRef] [PubMed]
46. Wölfler, A.; Caluba, H.C.; Abuja, P.M.; Dohr, G.; Schauenstein, K.; Liebmann, P.M. Prooxidant activity of
melatonin promotes fas-induced cell death in human leukemic Jurkat cells. FEBS Lett. 2001, 502, 127–131.
[CrossRef]
47. Albertini, M.C.; Radogna, F.; Accorsi, A.; Uguccioni, F.; Paternoster, L.; Cerella, C.; De Nicola, M.;
D’Alessio, M.; Bergamaschi, A.; Magrini, A.; et al. Intracellular pro-oxidant activity of melatonin deprives
U937 cells of reduced glutathione without affecting glutathione peroxidase activity. Ann. N. Y. Acad. Sci.
2006, 1091, 10–16. [CrossRef] [PubMed]
48. Cristofanon, S.; Uguccioni, F.; Cerella, C.; Radogna, F.; Dicato, M.; Ghibelli, L.; Diederich, M. Intracellular
prooxidant activity of melatonin induces a survival pathway involving NF-kappaB activation. Ann. N. Y.
Acad. Sci. 2009, 1171, 472–478. [CrossRef] [PubMed]You can also read

















































