The Glucocorticoid Triamcinolone Acetonide Inhibits Osmotic Swelling of Retinal Glial Cells via Stimulation of Endogenous Adenosine Signaling
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
0022-3565/05/3153-1036 –1045$20.00
THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 315, No. 3
Copyright © 2005 by The American Society for Pharmacology and Experimental Therapeutics 92353/3063312
JPET 315:1036–1045, 2005 Printed in U.S.A.
The Glucocorticoid Triamcinolone Acetonide Inhibits Osmotic
Swelling of Retinal Glial Cells via Stimulation of Endogenous
Adenosine Signaling
Ortrud Uckermann, Franziska Kutzera, Antje Wolf, Thomas Pannicke,
Andreas Reichenbach, Peter Wiedemann, Sebastian Wolf, and Andreas Bringmann
Paul Flechsig Institute of Brain Research (O.U., T.P., A.R.), Interdisziplinäres Zentrum für Klinische Forschung (O.U., A.W.), and
Department of Ophthalmology and Eye Clinic (F.K., P.W., A.B.), University of Leipzig Medical Faculty, Leipzig, Germany; and
Department for Ophthalmology, University Bern, Inselspital, Bern, Switzerland (S.W.)
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Received July 13, 2005; accepted August 31, 2005
ABSTRACT
The glucocorticoid triamcinolone acetonide is clinically used for lated at 3 days after transient retinal ischemia, in retinas of eyes
the treatment of macular edema. However, the edema-resolv- with lipopolysaccharide-induced ocular inflammation, and in
ing mechanisms of triamcinolone are incompletely understood. control retinas in the presence of Ba2⫹ (1 mM), H2O2 (200 M),
Since cell swelling is a central cause of cytotoxic edema in the arachidonic acid (10 M), or prostaglandin E2 (30 nM). The
brain and retina, we determined the effects of triamcinolone inhibiting effect of triamcinolone on osmotic glial cell swelling
acetonide on the swelling of retinal ganglion and Müller glial was mediated by stimulation of transporter-mediated release of
cells in acutely isolated retinas from rats and guinea pigs in situ. endogenous adenosine and subsequent A1 receptor activation,
Triamcinolone acetonide (100 M) had no effect on the swelling resulting in an elevation of the intracellular cAMP level and
of ganglion cells that was evoked in isolated whole mounts of activation of the protein kinase A, and, finally, in an opening of
the guinea pig retina by acute application of glutamate (1 mM) extrusion pathways for K⫹ and Cl⫺ ions. The inhibitory effect on
or high K⫹ (50 mM). However, triamcinolone reversed the os- the cytotoxic swelling of glial cells may contribute to the fast
motic swelling of Müller glial cells in retinas of the rat that was edema-resolving effect of vitreal triamcinolone observed in hu-
observed under various experimental conditions: in retinas iso- man patients.
Cystoid macular edema is an important complication of swelling and cyst formation, occurs after breakdown of the
various ocular diseases such as diabetic retinopathy and blood-retina barriers (Gass et al., 1985) induced by vascular
retinal vein occlusion. In patients with uveitis or diabetic endothelial growth factor (VEGF) (Antcliff and Marshall,
retinopathy, macular edema is the major cause of severe 1999; Marmor, 1999) and/or inflammatory mediators (Derev-
visual deterioration. Although the precise cause of cystoid janik et al., 2002). Vascular leakage results in serum protein
macular edema is still uncertain, inflammatory and isch- extravasation and osmotically driven water inflow into the
emic-hypoxic conditions have been causally implicated in the retinal tissue. In addition to this vasogenic mechanism, wa-
development of retinal edema (Tso, 1982; Yanoff et al., 1984; ter accumulation within retinal neurons and glial cells (re-
Guex-Crosier, 1999; Marmor, 1999). Extracellular (vaso- sulting in cell swelling, i.e., cytotoxic or intracellular edema)
genic) fluid accumulation, as a main causative factor of tissue may contribute to the edema in the ischemic and postisch-
emic retina (Pannicke et al., 2004; Uckermann et al., 2004b).
This work was supported by Interdisziplinäres Zentrum für Klinische For-
During diabetic retinopathy, the swelling of ganglion cell
schung at the Faculty of Medicine of the University of Leipzig Project C21 and bodies and their processes precedes the loss of these cells and
by DFG Grants BR 1249/2-1 and GRK 1097/1. the gliosis of the inner retinal layers (Duke-Elder and Do-
Article, publication date, and citation information can be found at
http://jpet.aspetjournals.org. bree, 1967). There are good arguments for a contribution of
doi:10.1124/jpet.105.092353. glial (Müller) cell swelling to the development of cystoid
ABBREVIATIONS: VEGF, vascular endothelial growth factor; LPS, lipopolysaccharide; PGE2, prostaglandin E2; LY341495, (2S)-2-amino-2-
[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; CSC, 8-(3-chlorostyryl) caffeine;
CPA, N(6)-cyclopentyladenosine; MPEP, 2-methyl-6-(phenylethynyl)-piperidine; BAPTA/AM, bis-(o-aminophenoxy)ethane-N,N,N⬘,N⬘-tetra-acetic
acid acetoxymethyl ester; pCPT, 8-(4-chlorophenylthio); IBMX, 3-isobutyl-1-methylxanthine; H-89, N-[2-((p-bromocinnamyl)amino)ethyl]-5-iso-
quinolinesulfonamide; NPPB, 5-nitro-2-(3-phenylpropylamino)benzoic acid; NBTI, N-nitrobenzylthioinosine; ARL-67156, 6-N,N-diethyl-D-,␥-
dibromomethylene ATP; AOPCP, adenosine-5⬘-O-(␣,-methylene)-diphosphonate; DMSO, dimethyl sulfoxide.
1036Triamcinolone Inhibits Glial Cell Swelling 1037
macular edema, e.g., in cases without significant angio- Materials and Methods
graphic vascular leakage (Fine and Brucker, 1981; Yanoff et
Materials. Mitotracker Orange (chloromethyltetramethylro-
al., 1984). A similar situation has been described for the
samine) was purchased from Molecular Probes (Eugene, OR). Ket-
brain where the postischemic or traumatic edema is charac- amine was obtained from Ratiopharm (Ulm, Germany) and xylazine
terized by swelling of astroglial cells (Kimelberg, 1995). In from BayerVital (Leverkusen, Germany). PGE2 was delivered from
the retina, dysregulated fluid-absorbing mechanisms nor- Calbiochem (San Diego, CA), and LY341495 was from Tocris Cook-
mally carried out by pigment epithelial and glial cells (Bring- son Inc. (Bristol, UK). Triamcinolone acetonide, acetazolamide, ara-
mann et al., 2004) may contribute to edema formation, re- chidonic acid, 4-bromophenacyl bromide, indomethacin, 8-cyclopen-
sulting in an ineffective redistribution of fluid from the retina tyl-1,3-dipropylxanthine (DPCPX), 8-(3-chlorostyryl) caffeine (CSC),
into the blood. The observation that clinically significant N(6)-cyclopentyladenosine (CPA), 2-methyl-6-(phenylethynyl)-piper-
diabetic macular edema occurs only when (in addition to idine (MPEP), bis-(o-aminophenoxy)ethane-N,N,N⬘,N⬘-tetra-acetic
acid acetoxymethyl ester (BAPTA/AM), 8-(4-chlorophenylthio)
vascular leakage) also the active transport mechanisms of
(pCPT)-cAMP, forskolin, 3-isobutyl-1-methylxanthine (IBMX), 2,3-
the blood-retinal barriers are dysfunctional (Mori et al., dideoxyadenosine, H-89, flufenamic acid, 5-nitro-2-(3-phenylpro-
2002) underlines the importance of disturbed fluid clearance pylamino)benzoic acid (NPPB), N-nitrobenzylthioinosine (NBTI),
in the development of retinal edema. ARL-67156, adenosine-5⬘-O-(␣,-methylene)-diphosphonate (AOPCP),
Anti-inflammatory corticosteroids are commonly used to and all other substances used were purchased from Sigma Chemie
treat inflammatory eye diseases, and among them, triamcin- (Deisenhofen, Germany).
olone acetonide (9␣-fluoro-16␣-hydroxyprednisolone) has the
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Animal Models. All experiments were carried out in accordance
advantage to form crystals that represent local depots for the with applicable German laws and with the Association for Research
in Vision and Ophthalmology Statement for the Use of Animals in
sustained release over relatively long periods of time. Intra-
Ophthalmic and Vision Research. Adult Long-Evans rats (250 –350
vitreal triamcinolone is used clinically for the rapid resolu-
g) were used to induce transient retinal ischemia in one eye of the
tion of macular edema (Ip et al., 2003; Massin et al., 2004). animals; the other eye remained untreated and served as control.
However, the mechanisms by which triamcinolone exerts its Anesthesia was induced with intramuscular ketamine (100 mg/kg)
fast edema-resolving effect are incompletely understood. and xylazine (5 mg/kg). The anterior chamber of the treated eye was
There is evidence that triamcinolone inhibits vasogenic cannulated from the pars plana with a 27-gauge infusion needle,
edema and inflammation. Triamcinolone decreases vascular connected to a bag containing normal saline. The intraocular pres-
leakage (Ando et al., 1994), reduces the secretion of VEGF by sure was increased to 160 mm Hg for 60 min by elevating the saline
pigment epithelial cells during oxidative stress (Matsuda et bag. Sham treatment (i.e., using the same procedures without ele-
vation of the saline bag) does not induce alterations of the osmotic
al., 2005), down-regulates the expression of the VEGF gene
swelling characteristics of Müller glial cells when compared with
in vascular smooth muscle cells (Nauck et al., 1998), and
control (Pannicke et al., 2004). Ocular inflammation alters the swell-
reduces the vitreal level of VEGF in patients with diabetic ing characteristics of Müller glial cells in a similar fashion as isch-
retinopathy (Brooks et al., 2004). Furthermore, triamcino- emia reperfusion (Pannicke et al., 2005). We used an animal model of
lone decreases the paracellular permeability of cultured ep- uveoretinitis induced by intravitreal injection of LPS (Becker et al.,
ithelial cells, down-regulates the inflammatory expression of 2000). During ketamine/xylazine anesthesia, LPS from Escherichia
endothelial adhesion molecules (Penfold et al., 2000), and coli 055:B5 (0.5%; Sigma-Aldrich) dissolved in 2 l of saline was
inhibits leukocyte-endothelial interactions in the diabetic injected into one eye of the animals. At 3 days after transient retinal
retina of the rat (Tamura et al., 2005). However, it is not ischemia or at 7 days after intravitreal injection of LPS, the animals
were killed by CO2, and the retinas were removed.
known whether triamcinolone, in addition to its inhibitory
Glial Cell Swelling. To determine volume changes of Müller glial
action on vasogenic edema, also has an effect on the devel- cells evoked by hypotonic stress, the cross-sectional area of Müller
opment of cytotoxic (intracellular) edema. Inhibition of cyto- cell somata in the inner nuclear layer of retinal slices was measured.
toxic edema may contribute to the rapid edema-resolving Acutely isolated retinal slices (thickness, 1 mm) were placed in a
effect of triamcinolone observed in human patients. There- perfusion chamber and loaded with the vital dye Mitotracker Orange
fore, we investigated whether triamcinolone inhibits the (10 M). It has been shown that Mitotracker Orange is taken up
swelling of retinal neurons or glial cells. Neuronal cell swell- selectively by Müller glial cells, whereas neurons remain unstained
ing was evoked by application of glutamate or high K⫹ onto (Uckermann et al., 2004a). The stock solution of the dye was pre-
acutely isolated whole mounts of the guinea pig retina, which pared in dimethyl sulfoxide (DMSO) and resolved in extracellular
solution that contained 136 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM
is known to rapidly induce swelling of retinal neurons by
MgCl2, 10 mM HEPES, and 11 mM glucose, adjusted to pH 7.4 with
activation of ionotropic glutamate receptors, resulting in in- Tris. The recording chamber was continuously perfused with extra-
tracellular Na⫹ and Cl⫺ overload and water influx (Ucker- cellular solution; test substances were added by rapid changing of
mann et al., 2004b). Müller cell swelling was investigated in the perfusate. The hypotonic solution was made by adding distilled
slices of the rat retina during exposure of a hypotonic solu- water. Ba2⫹ (1 mM) was added to the extracellular solution, and was
tion (a situation that resembles hypoxia-induced cytotoxic preincubated for 10 min. H2O2, PGE2, and triamcinolone were ap-
edema in the brain). We investigated the osmotic swelling of plied simultaneously with the hypotonic solution. A stock solution of
retinal glial (Müller) cells during various experimental con- triamcinolone was made by using DMSO; vehicle alone had no effect
on cell volume. Blocking substances were preincubated for 10 to 45
ditions: in a rat model of transient retinal ischemia reperfu-
min. All experiments were performed at room temperature. The
sion, in a model of ocular inflammation induced by intravit-
slices were examined by using a confocal laser scanning microscope
real injection of lipopolysaccharide (LPS), during acute LSM 510 Meta (Carl Zeiss GmbH, Jena, Germany). Mitotracker
application of the K⫹ channel blocker Ba2⫹ or of arachidonic Orange was excited at 543 nm, and emission was recorded with a
acid and prostaglandin E2 (PGE2), respectively, and during 560-nm long-pass filter. During the experiments, the Mitotracker
oxidative stress induced by acute application of H2O2. Orange-stained somata in the inner nuclear layer were recorded at1038 Uckermann et al.
the focal plane of their largest extension. To assure that this focal Results
plane was precisely recorded, the cell soma investigated was contin-
uously refocused in the course of the experiments. Neuronal Cell Swelling. The swelling of neuronal cells
Neuronal Cell Swelling. To investigate neuronal cell swelling, was investigated in whole mounts of the guinea pig retina. It
the nonvascularized guinea pig retina was used, which (in compar- is known that acute application of glutamate (1 mM) to the
ison with the retina of the rat) has the advantage that the cell bodies whole mounts causes rapid swelling of neuronal cell bodies in
in the ganglion cell layer can be recorded without interference with the ganglion cell (by ⬃30%; P ⬍ 0.001) (Fig. 1B) and inner
blood vessels and astrocytes. The animals (550 – 650 g) were killed by
nuclear layers (not shown) (Uckermann et al., 2004b). Simul-
CO2, and the retinas were removed. Pieces of retinal whole mounts (5
taneously, the glial (Müller) cell fibers in the inner plexiform
mm2) were explanted and mechanically fixed in a perfusion chamber,
with their vitreal surface up. The whole mounts were loaded with layer decrease their thickness (by ⬃30%; P ⬍ 0.001) (Fig.
Mitotracker Orange resolved in extracellular solution. Elevation of 1C), caused by swelling of synaptic structures. A similar
the K⫹ concentration was generated by equimolar reduction of Na⫹. neuronal cell swelling can be observed during application of
Images were taken at two focal planes: the ganglion cell and inner a high-K⫹ (50 mM) solution to the acutely isolated retinal
plexiform layers (Fig. 1A). Reflected light was acquired by using a whole mounts, as well as in the ischemic guinea pig retina
650-nm HeNe laser and a 340-nm long-pass filter. just after reperfusion (Uckermann et al., 2004b). The high-
Data Analysis. To determine the extent of glial cell swelling, the K⫹-evoked neuronal cell swelling is mediated by stimulation
cross-sectional area of Mitotracker Orange-stained cell bodies in the
of endogenous glutamate release and subsequent activation
inner nuclear layer of retinal slices was measured manually using
of ionotropic glutamate receptors (Uckermann et al., 2004b).
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
the image analysis software of the laser scanning microscope. The
values are expressed in percentage of the control data measured To investigate a possible effect of triamcinolone on neuro-
before iso- or hypotonic challenge (100%). To determine the extent of nal cell swelling, the cross-sectional areas of neuronal cell
neuronal cell swelling, two parameters were estimated as described bodies in the ganglion cell layer and of Müller cell fibers in
previously (Uckermann et al., 2004b): the cross-sectional area of the inner plexiform layer were recorded. Extracellular appli-
neuronal cell bodies in the ganglion cell layer, as well as the cross- cation of triamcinolone acetonide (100 M) did not decrease
sectional area of Müller cell fibers that pass through the inner the magnitude of neuronal cell swelling in the ganglion cell
plexiform layer; a decrease of the fiber area is caused by swelling of
layer evoked either by glutamate (1 mM) or by high K⫹ (Fig.
synapses structures located in this layer. Data are expressed as
1D). Likewise, the glutamate- or high-K⫹-evoked shrinkage
means ⫾ S.E.M. Statistical analysis was made by using the Prism
program (GraphPad Software Inc., San Diego, CA); significance was of the glial cell profiles in the inner plexiform layer was not
determined by Mann-Whitney U test or by analysis of variance altered by triamcinolone (Fig. 1E). The data suggest that
followed by comparisons for multiple groups. Curve fits were made acute application of triamcinolone does not inhibit neuronal
by using the Boltzmann equation. cell swelling.
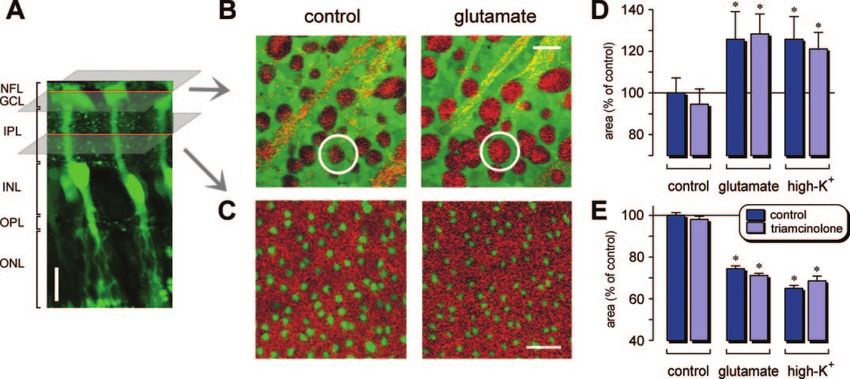
Fig. 1. Acute application of glutamate or high-K⫹ evokes morphological alterations in whole-mount guinea pig retinas that are not inhibited by
triamcinolone. The effects of glutamate (1 mM) and high (50 mM) K⫹ were determined after 10 and 5 min of exposure, respectively. Triamcinolone
(100 M) was applied either alone or simultaneously with glutamate or high K⫹. A, in an acutely isolated retinal slice, Mitotracker Orange (10 M)
selectively stained Müller glial cells (green). The morphological alterations were recorded in whole mounts at two focal planes. GCL, ganglion cell
layer; INL, inner nuclear layer; IPL, inner plexiform layer; NFL, nerve fiber layer; ONL, outer nuclear layer; OPL, outer plexiform layer. B, application
of glutamate (1 mM) caused swelling of neuronal cell bodies in the GCL. The Müller cell end feet are green-stained, and the unstained neuronal cell
bodies reflect light (red). Yellow-appearing elongated structures are nerve fiber bundles. C, green-stained Müller cell profiles in the IPL decreased
their thickness in response to glutamate; the synaptic structures reflect red light. Scale bars, 20 m. D, mean cross-sectional area of the neuronal cell
bodies in the GCL. E, mean cross-sectional area of the Müller cell profiles in the IPL. The bars represent values obtained in 20 to 103 cells. Significant
differences versus control: ⴱ, P ⬍ 0.001.Triamcinolone Inhibits Glial Cell Swelling 1039
Glial Cell Swelling. As shown earlier (Pannicke et al., H2O2. The swelling-inducing effect of H2O2 was dose-depen-
2004), the somata of Müller glial cells in slices of 3-day dent, with a half-maximal effect at 4 M (Fig. 3D, inset).
postischemic retinas responded to acute hypotonic stress by When triamcinolone (100 M) was applied simultaneously
an increase in their cross-sectional area by 10 to 20%, with the hypotonic solution, the soma swelling evoked by
whereas cell somata in control retinas did not increase their H2O2 was fully inhibited (Fig. 3E).
size (Fig. 2A). A similar osmotic soma swelling was observed It is known that arachidonic acid evokes vasogenic and
in retinal slices isolated at 7 days after intravitreal applica- cytotoxic brain edema (Chan et al., 1983) and swelling of
tion of LPS (Fig. 2D) (Pannicke et al., 2005), as well as in cultured astrocytes (Staub et al., 1994). Metabolites of ara-
control retinas when the K⫹ channels were blocked by extra- chidonic acid such as prostaglandins (especially PGE2) are
cellular Ba2⫹ ions (Fig. 3A). The osmotic swelling of the cells implicated in the development of retinal edema during ocular
in diseased retinas has been shown to be caused by the inflammation. To determine whether arachidonic acid or
down-regulation of distinct K⫹ channels, which impairs the PGE2 may cause glial cell swelling, both substances were
extrusion of K⫹ ions under hypotonic conditions and can be tested acutely in slices of control rat retinas. Arachidonic acid
mimicked by closure of K⫹ channels in control cells in the (Fig. 4A) and PGE2 (Fig. 4B) evoked swelling of glial cell
presence of the K⫹ channel blocker Ba2⫹ (Pannicke et al., somata when the substances were applied simultaneously
2004). with the hypotonic solution. An involvement of endogenous
Although triamcinolone (100 M) had no effect on the size arachidonic acid and PGE2 in the osmotic soma swelling in
of glial cell somata when the slices of 3-day postischemic rat postischemic retinas is suggested by the blocking effects of
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
retinas were perfused with an isotonic extracellular solution, the inhibitor of the phospholipase A2, 4-bromophenacyl bro-
it fully inhibited the soma swelling evoked by perfusion of a mide (Fig. 4, C and D), and of the cyclooxygenase inhibitor,
hypotonic solution (P ⬍ 0.001) (Fig. 2, A and B). In contrast, indomethacin (Fig. 4, E and F), respectively. Triamcinolone
acetazolamide (100 M), a carbonic anhydrase blocker that is inhibited the soma swelling evoked by arachidonic acid or
used clinically to resolve macular edema, did not reverse the PGE2 (Fig. 4, A and B).
osmotic swelling of glial cell somata in postischemic retinas Mechanism of the Inhibitory Effect of Triamcino-
(Fig. 2C). Triamcinolone (100 M) also inhibited the osmotic lone on Glial Swelling. It is known that adenosine (which
swelling of glial cell somata in slices obtained at 7 days after is rapidly released within the retina upon ischemia; Roth et
intravitreal application of LPS (Fig. 2D), as well as in control al., 1997) exerts a protective effect against ischemic injury in
retinas in the presence of Ba2⫹ (1 mM) (Fig. 3B). The inhib- the retina by the activation of A1 receptors (Larsen and
itory effect of triamcinolone on the Ba2⫹-evoked hypotonic Osborne, 1996; Ghiardi et al., 1999). A swelling-inhibiting
glial cell swelling was dose-dependent (Fig. 3C). The concen- effect of adenosine may contribute to this protective effect in
tration estimated to evoke half-maximal inhibition was 1.3 ischemic injury. To determine whether adenosine inhibits
M. Vehicle (DMSO) alone had no effect (Fig. 3F). the osmotic swelling of glial cell somata, we applied adeno-
Oxidative stress is a major cause of retinal injury after sine simultaneously with the hypotonic solution to retinal
ischemia (Osborne et al., 2004). To determine whether oxi- slices. Adenosine inhibited the osmotic swelling of glial cell
dative stress induces swelling of glial cell somata, slices of somata in postischemic retinas (Fig. 5A) and in control reti-
control rat retinas were perfused with H2O2. As shown in Fig. nas in the presence of extracellular Ba2⫹ (Fig. 5B) or arachi-
3D, H2O2 did not induce swelling of glial cell somata at donic acid (Fig. 5C). This effect of adenosine was mediated by
isotonic conditions. However, hypotonic challenge evoked activation of A1 receptors since a selective A1 receptor ago-
soma swelling in the presence (but not in the absence) of nist, CPA, also inhibited the swelling, and since the effect of
Fig. 2. Effect of triamcinolone on the osmotic soma swelling of Müller glial cells in slices from 3-day postischemic retinas and from endotoxin-treated
eyes, respectively. Swelling was induced by changing the perfusate into a hypotonic solution (60% of control osmolarity). A, hypotonic challenge
resulted in soma swelling in the case of postischemic retinas, whereas Müller cells in control retinas did not increase the cross-sectional area of their
somata. Application of triamcinolone (100 M) simultaneously with the hypotonic solution fully inhibited the soma swelling. The insets show original
records of two dye-filled somata in a postischemic retina, before (left) and during (right) exposure of hypotonic medium. Scale bar, 5 m. B and C, mean
cross-sectional area of Müller cell somata in slices of postischemic retinas, in dependence on the recording conditions. D, mean soma area in retinal
slices derived from eyes at 7 days after intravitreal injection of LPS. Triamcinolone (100 M) or acetazolamide (100 M) were applied simultaneously
with the iso- or hypotonic solution, as indicated. The values were measured after 4-min perfusion with the iso- or hypotonic solution and are expressed
as percentage of the control value measured before application of the test solution (100%). The bars represent values obtained in 4 to 19 cells.
Significant differences versus control (100%): ⴱ, P ⬍ 0.05; ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍ 0.001. Significant blocking effects: †, P ⬍ 0.05; †††, P ⬍ 0.001.1040 Uckermann et al.
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Fig. 3. Triamcinolone inhibits the osmotic soma swelling of Müller glial cells in slices of control retinas. The swelling was induced by changing the
perfusate into a hypotonic solution (60% of control osmolarity) in the presence of extracellular Ba2⫹ (1 mM) or H2O2. A, in the presence of Ba2⫹,
hypotonic challenge evoked a reversible swelling of Müller cell somata in control retinas. The mean cross-sectional area of glial cell somata was
measured in dependence on the recording time. B, triamcinolone (100 M) inhibited the osmotic swelling of Müller cell somata in the presence of Ba2⫹.
Mean cross-sectional area of the somata in dependence on the recording conditions. C, dose dependence of the inhibiting effect of triamcinolone on the
Ba2⫹-evoked osmotic soma swelling. The curve was fitted with the Boltzmann equation and a half-maximal inhibitory concentration of 1.3 M. D, H2O2
evoked dose dependently soma swelling under hypotonic conditions. Inset, data points were fitted with the Boltzmann equation, with a half-maximal
effect at 4 M. E, triamcinolone (100 M) inhibited the osmotic soma swelling in the presence of H2O2 (200 M). F, vehicle (DMSO; 0.1%) had no effect
on the soma swelling in the presence of Ba2⫹ (1 mM) or H2O2 (200 M). Triamcinolone was applied simultaneously with the iso- or hypotonic solutions.
B to F, values were measured after 4-min perfusion of the iso- or hypotonic solution and are expressed as percentage of the control value measured
before application of the solution (100%). The bars represent values obtained in 4 to 45 cells. Significant difference versus control (100%): ⴱ, P ⬍ 0.05;
ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍ 0.001. Significant blocking effect: †, P ⬍ 0.05; ††, P ⬍ 0.01; †††, P ⬍ 0.001.
adenosine was blocked by the selective antagonist of A1 triamcinolone, selective antagonists of A1 and A2a receptors,
receptors, DPCPX (Fig. 5B). In contrast, the swelling-inhib- DPCPX and CSC, respectively, were tested. As shown in Fig.
iting effect of adenosine was not modified by a selective A2a 6, B and C, DPCPX fully blocked the swelling-inhibiting
receptor antagonist, CSC (Fig. 5B). effect of triamcinolone, whereas CSC had no effect. On the
To determine whether triamcinolone inhibits the osmotic other hand, MPEP and LY341495, antagonists of subtype 5
swelling of glial cell somata via stimulation of the release of of groups I and II metabotropic glutamate receptors, did not
endogenous adenosine and subsequent stimulation of aden- block the effect of triamcinolone (Fig. 6B). The data suggest
osine receptors, it was applied in the presence of antagonists that the effect of triamcinolone is mediated by a release of
of adenosine receptors and transporters. Theophylline, a endogenous adenosine and subsequent activation of A1 re-
blocker of A1 and A2 receptors, fully reversed the effect of ceptors. Thus, we tried to determine whether this adenosine
triamcinolone (Fig. 6A). To determine the subtype of adeno- was released from intracellular compartments via stimula-
sine receptors that mediates the swelling-inhibiting effect of tion of nucleoside transporters or generated by degradationTriamcinolone Inhibits Glial Cell Swelling 1041
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Fig. 4. Arachidonic acid (AA) and PGE2 induce swelling of Müller cell somata at hypotonic conditions, which are inhibited by triamcinolone. A,
triamcinolone (100 M) inhibits the AA (10 M)-evoked swelling in control retinas. B, triamcinolone (100 M) decreases the soma swelling evoked by
PGE2 (30 nM) in control retinas. C, the inhibitor of the phospholipase A2, 4-bromophenacyl bromide (300 M), reduces the osmotic soma swelling in
postischemic retinas and in control retinas in the presence of Ba2⫹ (1 mM) (D). E, the cyclooxygenase inhibitor, indomethacin (10 M), reversed the
osmotic swelling in postischemic retinas and F, in control retinas in the presence of Ba2⫹. The test substances were simultaneously applied with the
hypotonic solution. The bars represent values obtained in 4 to 15 cells. Significant differences versus control (100%): ⴱ, P ⬍ 0.05; ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍
0.001. Significant blocking effect: ††, P ⬍ 0.01; †††, P ⬍ 0.001.
Fig. 5. Adenosine inhibits the osmotic swelling of Müller cell somata via activation of A1 receptors. Retinal slices were perfused with a hypotonic
solution, and the relative soma size was measured before (100%) and after 4-min perfusion of the solution. A, soma size in control and postischemic
retinas, in the absence and presence of adenosine (10 M). B, soma size in control retinas in the presence of Ba2⫹ (1 mM). The swelling-inhibiting effect
of adenosine (10 M) was largely blocked by the selective antagonist of A1 receptors, DPCPX (100 nM), and remained unaltered in the presence of the
A2a receptor antagonist CSC (200 nM). The A1 receptor agonist CPA (100 nM) mimicked the swelling-inhibiting effect of adenosine. C, the osmotic
soma swelling induced by AA (10 M) was largely inhibited by adenosine (10 M). The test substances were simultaneously applied with the hypotonic
solution; blocking substances were preincubated for 10 min. The bars represent values obtained in 4 to 35 cells. Significant differences versus control
(100%): ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍ 0.001. Significant blocking effect: ††, P ⬍ 0.01; †††, P ⬍ 0.001. Significant inhibition of the agonist effect: EEE, P ⬍ 0.001.
of extracellular ATP. For this purpose, various blocking sub- tion. The swelling-inhibiting effects of triamcinolone and of
stances were tested. The inhibitor of nucleoside transporters, adenosine remained unaltered in the presence of the mem-
NBTI, blocked the effect of triamcinolone (Fig. 6D). On the brane-permeable Ca2⫹ chelator BAPTA/AM (Fig. 7, A and B),
other hand, the ecto-ATPase inhibitor, ARL-67156, and the suggesting that an intracellular Ca2⫹ response was not in-
ectonucleotidase inhibitor, AOPCP, did not block the swell- volved in the effects of both agents. On the other hand,
ing-inhibiting effect of triamcinolone (Fig. 6E). These data application of a cAMP-enhancing cocktail that contained
suggest that the effect of triamcinolone was mediated by pCPT-cAMP (a membrane-permeable cAMP analog), forsko-
stimulation of transporter-mediated release of adenosine lin (a direct activator of adenylyl cyclase), and IBMX (a
from intracellular compartments but not by extracellular phosphodiesterase inhibitor), reduced the osmotic glial soma
formation of adenosine via degradation of ATP. Tetrodotoxin swelling by approximately two-thirds in postischemic retinas
failed to interfere with the effect of triamcinolone (Fig. 6F), (Fig. 7C) and in control retinas in the presence of Ba2⫹ (1
suggesting that it is independent of the activation of retinal mM) (Fig. 7D). To explore whether the effects of triamcino-
neurons. lone and adenosine were mediated by an activation of the
Finally, we tried to identify the intracellular signaling adenylyl cyclase and a subsequent activation of protein ki-
mechanisms that inhibit swelling after A1 receptor stimula- nase A, blockers of both enzymes were tested. 2,3-Dideoxy-1042 Uckermann et al.
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Fig. 6. The swelling-inhibiting effect of triamcinolone is mediated by endogenous release of adenosine and activation of A1 receptors. Osmotic swelling
of Müller cell somata was induced by hypotonic challenge in slices of control retinas in the presence of Ba2⫹ (1 mM). A, the swelling-inhibiting effect
of triamcinolone (100 M) was blocked by the nonselective antagonist of adenosine receptors, theophylline (250 M). B, the effect of triamcinolone (100
M) was blocked by DPCPX (100 nM) and remained unaltered in the presence of MPEP (50 M) and LY341495 (100 M). C, CSC (200 nM) did not
block the swelling-inhibiting effect of triamcinolone (100 M). D, NBTI (10 M) reversed the swelling-inhibiting effect of triamcinolone (100 M). E,
ARL-67156 (50 M) or AOPCP (250 M) had no effects on the swelling-inhibiting action of triamcinolone (100 M). F, the effect of triamcinolone (100
M) remained unaltered in the presence of tetrodotoxin (TTX; 1 M). The bars represent values obtained in 3 to 19 cells. Significant differences versus
control (100%): ⴱ, P ⬍ 0.05; ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍ 0.001. Significant blocking effects: †, P ⬍ 0.05; ††, P ⬍ 0.01; †††, P ⬍ 0.001. Significant inhibition
of the agonist effects: E, P ⬍ 0.05.
adenosine, an inhibitor of the adenylyl cyclase, blocked the Discussion
effect of triamcinolone (Fig. 7E). Likewise, an inhibitor of the
protein kinase A, H-89, reduced the effects of triamcinolone Various ischemic and inflammatory ocular diseases are
(Fig. 7E) and adenosine (Fig. 7F) by approximately two associated with the development of macular edema that may
thirds. be caused by vascular leakage (vasogenic, extracellular
Arachidonic acid has been shown to inhibit the efflux of edema) and/or by swelling of retinal cells (cytotoxic, intracel-
osmolytes such as amino acids and Cl⫺ from cultured astro- lular edema). It has been suggested that swelling of Müller
cytes, an effect that may contribute to osmotic swelling glial cells contributes to macular edema (e.g., in cases with-
(Sanchez-Olea et al., 1995). Thus, the inhibition of cell swell- out significant angiographic vascular leakage; Fine and
ing may involve the opening of extrusion pathways for os- Brucker, 1981; Yanoff et al., 1984) and that a dysregulation
molytes. To determine whether adenosine inhibits osmotic of retinal fluid absorption, normally carried out by pigment
swelling by opening of such extrusion pathways for K⫹ epithelial and Müller glial cells (Bringmann et al., 2004), is
and/or Cl⫺, the K⫹ channel blocker, quinine, and the Cl⫺ likely to be crucial for the formation of chronic edema (Mori
channel blockers, flufenamic acid and NPPB, were tested. et al., 2002). Since Müller cells mediate the water transport
Indeed, these substances were found to block the effect of between the inner retinal tissue and the blood and vitreous
adenosine (Fig. 7G). Taken together, our data suggest that (Bringmann et al., 2004), Müller cell swelling, when it occurs
the inhibitory effect of triamcinolone on osmotic Müller cell in vivo, may reflect such a dysregulated fluid clearance.
swelling involves the release of endogenous adenosine and It has been shown that experimental ischemia reperfusion
subsequent activation of A1 receptors. A1 receptor activation of the rat retina evokes a biphasic water accumulation in the
results in enhancement of the intracellular cAMP level and retina; the retinal water content increases strongly during
activation of the protein kinase A, which finally causes a the ischemic episode and increases again slowly within days
compensatory efflux of K⫹ and Cl⫺ ions. after reperfusion (Stefánsson et al., 1987). The edema thatTriamcinolone Inhibits Glial Cell Swelling 1043
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
Fig. 7. The swelling-inhibiting effect of triamcinolone is largely mediated by activation of the adenylyl cyclase and protein kinase A and by opening
of K⫹ and Cl⫺ channels. Osmotic swelling of Müller cell somata was induced by hypotonic challenge in slices of 3-day postischemic retinas or of control
retinas in the presence of Ba2⫹ (1 mM). A, preincubation of the slices with BAPTA/AM (100 M) for 30 min did not block the swelling-inhibiting effects
of triamcinolone (100 M) or of B, adenosine (10 M). C, application of a cAMP-enhancing cocktail that contained pCPT-cAMP (100 M), forskolin (10
M), and IBMX (100 M) reduced the osmotic soma swelling in postischemic retinas as well as in control retinas (D). E, 2,3-dideoxyadenosine (20 M)
and H-89 (1 or 30 M), respectively, reduced the swelling-inhibiting effect of triamcinolone (100 M) in control retinas. F, H-89 (1 or 30 M) reduced
the effect of adenosine (10 M). G, the swelling-inhibiting effect of adenosine (10 M) was blocked by quinine (200 M) and by the Cl⫺ channel blockers
flufenamic acid (500 M) and NPPB (100 M), respectively. The bars represent values obtained in 4 to 26 cells. Significant differences versus control
(100%): ⴱⴱ, P ⬍ 0.01; ⴱⴱⴱ, P ⬍ 0.001. Significant blocking effects: †, P ⬍ 0.05; ††, P ⬍ 0.01; †††, P ⬍ 0.001. Significant inhibition of the agonist effects:
EE, P ⬍ 0.01; EEE, P ⬍ 0.001.
develops during the ischemic period is likely caused by neu- osmotic swelling characteristics of Müller glial cells in the rat
ronal cell swelling and is mediated, at least in part, by retina (Pannicke et al., 2004, 2005). During hypotonic stress,
overstimulation of ionotropic glutamate receptors, which al- Müller cells in postischemic retinas (Fig. 2, A and B) or in
lows strong ion fluxes into retinal neurons, accompanied by retinas obtained from eyes displaying ocular inflammation
water uptake (Uckermann et al., 2004b). The edema that (Fig. 2D) increase their soma volume, whereas cells in control
develops after reperfusion may be caused, at least in part, by retinas do not swell (Fig. 2A). The experimental model of
glial cell swelling (Pannicke et al., 2004). A similar pattern of acute hypotonic challenge used in the present study may be
neuronal and glial cell edema was described in the ischemic representative for pathological in vivo conditions. Hypotonic
retina of the rabbit; during ischemic episodes, both plexiform challenge mimics the osmotical conditions at the glio-vascu-
layers and the cytoplasm of neuronal cells become edema- lar and glio-vitreal interfaces during pathological states,
tous, whereas the Müller glial cells appear to be unaffected. such as ischemia, in which the retinal parenchyma becomes
However, in the postischemic tissue, the Müller glial cells hyperosmotic in comparison with the blood and vitreous.
become edematous, whereas neural elements degenerate When the Müller glial cells take up excess osmolytes (e.g., K⫹
(Johnson, 1974). and Na⫹/glutamate during periods of pathological neuronal
Experimental ischemia reperfusion or endotoxin-evoked hyperexcitation) and are impaired in their ability to redis-
ocular inflammation cause a significant alteration of the tribute these osmolytes into the blood and vitreous, an in-1044 Uckermann et al.
creased intracellular osmolarity may constitute an osmotical triamcinolone selectively stimulates the transporter-medi-
driving force for water movement from the blood and vitreous ated release of adenosine from retinal cells, whereas it has no
into the Müller cells, resulting in cell swelling (Pannicke et effect on the extracellular formation of adenosine via degra-
al., 2004). However, the experimental model of hypotonic dation of ATP. It is conceivable that triamcinolone stimulates
stress used in the present study may also reflect the condi- the water clearance function of Müller cells in situ by the
tions at the glio-synaptic interface since it is known that glial activation of ion secretion via Müller cell end feet into the
uptake of excess K⫹ causes a reduction of the extracellular blood and vitreous. Similar rapid effects of triamcinolone
osmolarity (Dietzel et al., 1989). Ion fluxes from the extra- (within minutes) have been described for other cell systems,
cellular space into the synapses through ionotropic receptors e.g., for the remodeling of the nuclear envelope structure in
may contribute to the fall of extracellular osmolarity during Xenopus oocytes (Shahin et al., 2005). Our results suggest
neuronal activity. Therefore, experimental hypotonic stress that stimulation of A1 receptors by adenosine results in an
may mimic the osmotic gradients during neuronal hyperex- enhancement of the intracellular cAMP level. Although in
citation that occurs during ischemic insults. most cell systems activation of A1 receptors causes a de-
The present results suggest that inflammatory mediators crease of the cAMP level, there are observations that (in
and/or oxidative stress (that is one factor causing neuronal dependence on the density of the receptors expressed by the
degeneration during reperfusion; Osborne et al., 2004) medi- cells) A1 receptors may couple also to stimulating G proteins,
ate the alteration of the osmotic swelling characteristics of resulting in an enhancement of the intracellular cAMP level
Müller glial cells. Müller cell swelling in the postischemic (Cordeaux et al., 2000). However, the inhibitory effect of
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
retina may be induced by inflammatory mediators, due to the cAMP elevation on cell swelling was only partial (Fig. 7, C
activation of phospholipase A2 and cyclooxygenase by osmotic and D). Similarly, blockade of the adenylyl cyclase or of the
stress. The mechanisms how arachidonic acid, PGE2, or H2O2 protein kinase A resulted in a partial inhibition of the effects
induce hypotonic glial cell swelling are unclear. It has been of triamcinolone and adenosine (Fig. 7, E and F), suggesting
shown that arachidonic acid inhibits distinct outwardly di- that a second intracellular pathway may contribute to the
rected K⫹ currents in isolated Müller cells (Bringmann et al., signaling from A1 receptors to ion channels.
1998), and it is conceivable (but remains to be proven) that In the present study, we used triamcinolone in a concen-
arachidonic acid and PGE2 close plasma membrane path- tration of 100 M (i.e., 43 g/ml), unless stated otherwise. In
ways that are involved in osmolyte extrusion out of the cells. human patients, a vitreal dosage of 1 mg/ml is used regularly
A failure in osmolyte extrusion upon hypotonic challenge (4 mg of triamcinolone in 4 ml of human vitreous volume),
would cause water movement into the cells and, thus, cell and peak concentrations between 2 and 7 g/ml in aqueous
swelling. The observation that blockade of K⫹ and Cl⫺ chan- humor samples were described to occur within days after a
nels inhibits the effect of adenosine (Fig. 7G) strongly sup- single 4-mg intravitreal injection of triamcinolone (Beer et
ports the view that a closure of ion release pathways is al., 2003). We observed a half-maximal effect of triamcino-
involved in the induction of osmotic swelling. It is known that lone at 1.3 M, i.e., 0.56 g/ml (Fig. 3C), which is well in the
arachidonic acid induces both vasogenic and cytotoxic edema range of concentration observed in human subjects.
in the brain (Chan et al., 1983; Wahl et al., 1993) and evokes In summary, we show that triamcinolone does not inhibit
swelling of cultured astrocytes (Staub et al., 1994), and it has glutamate- and high-K⫹-evoked swelling of retinal neurons
been suggested that the swelling-inducing effect of arachi- but inhibits the osmotic swelling of Müller glial cells during
donic acid is mediated by a direct inhibition of the efflux of retinal ischemia reperfusion and inflammation. This inhibi-
osmolytes such as amino acids and Cl⫺ (Sanchez-Olea et al., tory effect of triamcinolone on cytotoxic glial cell swelling
1995). may contribute to the fast edema-resolving effect of this
Corticosteroids such as triamcinolone are used clinically to substance observed in human patients. This view is sup-
resolve macular edema (Ip et al., 2003; Massin et al., 2004). ported by the observation that triamcinolone also rapidly
The mechanisms of the triamcinolone effects are incom- resolves cystoid macular edema in patients without signifi-
pletely understood; present research is focused on the inhi- cant angiographic vascular leakage. Inhibition of glial cell
bition of vascular edema. It has been shown recently that swelling may represent also a component of the protective
triamcinolone induces a very rapid reduction of macular effect of corticosteroids used in the treatment of brain edema.
edema in patients with retinal vein occlusion and diabetic
References
retinopathy, which is evident as early as 1 h after treatment Ando N, Sen HA, Berkowitz BA, Wilson CA and de Juan E Jr (1994) Localization and
(Miyamoto et al., 2005). However, the regulation of tight quantification of blood-retinal barrier breakdown in experimental proliferative
junctional and VEGF protein expression by triamcinolone is vitreoretinopathy. Arch Opthalmol 112:117–122.
Antcliff RJ and Marshall J (1999) The pathogenesis of edema in diabetic maculopa-
suggested to occur at the gene expression level that needs thy. Semin Ophthalmol 14:223–232.
several hours. Therefore, it has been suggested that triam- Becker MD, Nobiling R, Planck SR, and Rosenbaum JT (2000) Digital video-imaging
of leukocyte migration in the iris: intravitreal microscopy in a physiological model
cinolone acts, at least additionally, via activation of signaling during the onset of endotoxin-induced uveitis. J Immunol Methods 240:23–37.
cascades (Miyamoto et al., 2005). Here, we present data Beer PM, Bakri SJ, Singh RJ, Liu W, Peters GB, and Miller M (2003) Intraocular
concentration and pharmacokinetics of triamcinolone acetonide after a single
supporting the idea that triamcinolone inhibits Müller cell intravitreal injection. Ophthalmology 110:681– 686.
swelling in situ via stimulation of endogenous adenosine Bringmann A, Reichenbach A, and Wiedemann P (2004) Pathomechanisms of cystoid
macular edema. Ophthalmic Res 36:241–249.
signaling. A1 receptor stimulation after release of endoge- Bringmann A, Skatchkov SN, Biedermann B, Faude F, and Reichenbach A (1998)
nous adenosine causes an activation of the adenylyl cyclase Alterations of potassium channel activity in retinal Müller glial cells induced by
arachidonic acid. Neuroscience 86:1291–1306.
and of the protein kinase A that results in a stimulation of Brooks HL Jr, Caballero S Jr, Newell CK, Steinmetz RL, Watson D, Segal MS,
compensatory ion release by Müller glial cells. The lack of Harrison JK, Scott EW, and Grant MB (2004) Vitreous levels of vascular endo-
thelial growth factor and stromal-derived factor 1 in patients with diabetic reti-
effects of inhibitors of the ecto-ATPase and the ectonucleoti- nopathy and cystoid macular edema before and after intraocular injection of
dase on the effect of triamcinolone (Fig. 6E) suggests that triamcinolone. Arch Ophthalmol 122:1801–1807.Triamcinolone Inhibits Glial Cell Swelling 1045
Chan PH, Fishman RA, Caronna J, Schmidley JW, Prioleau G, and Lee J (1983) Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, and Melena J (2004)
Induction of brain edema following intracerebral injection of arachidonic acid. Ann Retinal ischemia: mechanisms of damage and potential therapeutic strategies.
Neurol 13:625– 632. Prog Retin Eye Res 23:91–147.
Cordeaux Y, Briddon SJ, Megson AE, McDonnell J, Dickenson JM, and Hill SJ Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P,
(2000) Influence of receptor number on functional responses elicited by agonists Wolburg H, Reichenbach A, and Bringmann A (2004) A potassium channel-linked
acting at the human adenosine A1 receptor: evidence for signaling pathway- mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci
dependent changes in agonist potency and relative intrinsic activity. Mol Phar- 26:493–502.
macol 58:1075–1084. Pannicke T, Uckermann O, Iandiev I, Wiedemann P, Reichenbach A, and Bringmann
Derevjanik NL, Vinores SA, Xiao WH, Mori K, Turon T, Hudish T, Dong S, and A (2005) Ocular inflammation alters swelling and membrane characteristics of rat
Campochiaro PA (2002) Quantitative assessment of the integrity of the blood- Müller glial cells. J Neuroimmunol 161:145–154.
retinal barrier in mice. Investig Ophthalmol Vis Sci 43:2462–2467. Penfold PL, Wen L, Madigan MC, Gillies MC, King NJ, and Provis JM (2000)
Dietzel I, Heinemann U, and Lux HD (1989) Relations between slow extracellular Triamcinolone acetonide modulates permeability and intercellular adhesion mol-
potential changes, glial potassium buffering and electrolyte and cellular volume ecule-1 (ICAM-1) expression of the ECV304 cell line: implications for macular
changes during neuronal hyperactivity in cat brain. Glia 2:25– 44. degeneration. Clin Exp Immunol 121:458 – 465.
Duke-Elder S and Dobree JH (1967) System of Ophthalmology, Vol. X. Diseases of the Roth S, Rosenbaum PS, Osinski J, Park SS, Toledano AY, Li B, and Moshfeghi AA
Retina, Mosby, St. Louis, MO. (1997) Ischemia induces significant changes in purine concentration in the retina-
Fine BS and Brucker AJ (1981) Macular edema and cystoid macular edema. Am J choroid in rats. Exp Eye Res 65:771–779.
Ophthalmol 92:466 – 481. Sanchez-Olea R, Morales-Mulia M, Moran J, and Pasantes-Morales H (1995) Inhi-
Gass JDM, Anderson DR, and Davis EB (1985) A clinical, fluorescein angiographic bition by polyunsaturated fatty acids of cell volume regulation and osmolyte fluxes
and electron microscopic correlation of cystoid macular edema. Am J Ophthalmol in astrocytes. Am J Physiol 269:C96 –C102.
100:82– 86. Shahin V, Ludwig Y, Schafer C, Nikova D, and Oberleithner H (2005) Glucocorti-
Ghiardi GJ, Gidday JM, and Roth S (1999) The purine nucleoside adenosine in coids remodel nuclear envelope structure and permeability. J Cell Sci 118:2881–
retinal ischemia-reperfusion injury. Vision Res 39:2519 –2535. 2889.
Guex-Crosier Y (1999) The pathogenesis and clinical presentation of macular edema Staub F, Winkler A, Peters J, Kempski O, Kachel V, and Baethmann A (1994)
in inflammatory diseases. Doc Ophthalmol 97:297–309. Swelling, acidosis and irreversible damage of glial cells from exposure to arachi-
Ip M, Kahana A, and Altaweel M (2003) Treatment of central retinal vein occlusion donic acid in vitro. J Cereb Blood Flow Metab 14:1030 –1039.
Downloaded from jpet.aspetjournals.org at ASPET Journals on February 7, 2015
with triamcinolone acetonide: an optical coherence tomography study. Semin Stefánsson E, Wilson CA, Lightman SL, Kuwabara T, Palestine AG, and Wagner HG
Ophthalmol 18:67–73.
(1987) Quantitative measurements of retinal edema by specific gravity determi-
Johnson NF (1974) Effects of acute ischaemia on the structure of the rabbit retina.
nations. Investig Ophthalmol Vis Sci 28:1281–1289.
Trans Ophthalmol Soc U K 94:394 – 405.
Tamura H, Miyamoto K, Kiryu J, Miyahara S, Katsuta H, Hirose F, Musashi K, and
Kimelberg HK (1995) Current concepts of brain edema. J Neurosurg 83:1051–1059.
Yoshimura N (2005) Intravitreal injection of corticosteroid attenuates leukostasis
Larsen AK and Osborne NN (1996) Involvement of adenosine in retinal ischemia:
and vascular leakage in experimental diabetic retina. Investig Ophthalmol Vis Sci
studies on the rat. Investig Ophthalmol Vis Sci 37:2603–2611.
46:1440 –1444.
Marmor MF (1999) Mechanisms of fluid accumulation in retinal edema. Doc Oph-
Tso MOM (1982) Pathology of cystoid macular edema. Ophthalmology 89:902–915.
thalmol 97:239 –249.
Uckermann O, Iandiev I, Francke M, Franze K, Grosche J, Wolf S, Kohen L,
Massin P, Audren F, Haouchine B, Erginay A, Bergmann JF, Benosman R, Caulin C,
and Gaudric A (2004) Intravitreal triamcinolone acetonide for diabetic diffuse Wiedemann P, Reichenbach A, and Bringmann A (2004a) Selective staining by
macular edema: preliminary results of a prospective controlled trial. Ophthalmol- vital dyes of Müller glial cells in retinal wholemounts. Glia 45:59 – 66.
ogy 111:218 –224. Uckermann O, Vargova K, Ulbricht E, Klaus C, Weick M, Rillich K, Wiedemann P,
Matsuda S, Gomi F, Oshima Y, Tohyama M, and Tano Y (2005) Vascular endothelial Reichenbach A, Sykova E, and Bringmann A (2004b) Glutamate-evoked alter-
growth factor reduced and connective tissue growth factor induced by triamcino- ations of glial and neuronal cell morphology in the guinea-pig retina. J Neurosci
lone in ARPE19 cells under oxidative stress. Investig Ophthalmol Vis Sci 46:1062– 24:10149 –10158.
1068. Wahl M, Schilling L, Unterberg A, and Baethmann A (1993) Mediators of vascular
Miyamoto N, Iossifov D, and Behar-Cohen F (2005) Intravitreal triamcinolone ace- and parenchymal mechanisms in secondary brain damage. Acta Neurochir Suppl
tonide early effects on macular edema. Investig Ophthalmol Vis Sci 46:ARVO 57:64 –72.
E-Abstract 1428. Yanoff M, Fine BS, Brucker AJ, and Eagle RC (1984) Pathology of human cystoid
Mori F, Hikichi T, Takahashi J, Nagaoka T, and Yoshida A (2002) Dysfunction of macular edema. Surv Ophthalmol 28:S505–S511.
active transport of blood-retinal barrier in patients with clinically significant
macular edema in type 2 diabetes. Diabetes Care 25:1248 –1249. Address correspondence to: Dr. Andreas Bringmann, Department of Oph-
Nauck M, Karakiulakis G, Perruchoud AP, Papakonstantinou E, and Roth M (1998) thalmology and Eye Clinic, University of Leipzig, Liebigstrasse 10-14, D-04103
Corticosteroids inhibit the expression of the vascular endothelial growth factor Leipzig, Germany. E-mail: bria@medizin.uni-leipzig.de
gene in human vascular smooth muscle cells. Eur J Pharmacol 341:309 –315.You can also read