Paracetamol-Induced Renal Tubular Injury: A Role for ER Stress
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
J Am Soc Nephrol 15: 380–389, 2004
Paracetamol-Induced Renal Tubular Injury: A Role for ER
Stress
CORINA LORZ,* PILAR JUSTO,* ANA SANZ,* DOLORES SUBIRÁ,†
JESÚS EGIDO,* and ALBERTO ORTIZ*
*Laboratory of Experimental Nephrology and Vascular Pathology, Fundación Jiménez Díaz, Universidad
Autónoma de Madrid, Madrid, Spain; and †Unidad de Hematología, Fundación Jiménez Díaz, Madrid, Spain.
Abstract. Paracetamol (also known as acetaminophen) causes caspase-dependent process that involves activation of
acute and chronic renal failure. While the mechanisms leading caspase-9 and caspase-3 in the absence of cytosolic cyto-
to hepatic injury have been extensively studied, the molecular chrome c or Smac/DIABLO. The authors also detected induc-
mechanisms of paracetamol-induced nephrotoxicity are poorly tion of endoplasmic reticulum (ER) stress, characterized by
defined. Paracetamol induced cell death with features of apo- GADD153 upregulation and translocation to the nucleus, as
ptosis in murine proximal tubular epithelial cells. While parac- well as caspase-12 cleavage. Interestingly, after treatment of
etamol increased the expression of the death receptor Fas on murine tubular cells with paracetamol and calpain inhibitors,
the cell surface, the Fas pathway was not involved in the the caspase-12 cleavage product was still detectable, and cal-
paracetamol-induced apoptosis of tubular cells. The mitochon- pain inhibitors were unable to protect tubular cells from parac-
drial pathway was not activated during paracetamol-induced etamol-induced apoptosis. The results suggest that induction of
apoptosis; there was no dissipation of mitochondrial potential apoptosis may underlie the nephrotoxic potential of paraceta-
or release of apoptogenic factors such as cytochrome c or mol and identify ER stress as a therapeutic target in
Smac/DIABLO. However, paracetamol-induced apoptosis is a nephrotoxicity.
Paracetamol, also known as acetaminophen, is widely used as active form of cell death that offers the opportunity for thera-
an analgesic and antipyretic drug. An acute paracetamol over- peutic intervention (11,12). Paracetamol has been shown to
dose can lead to potentially lethal liver and kidney failure in promote hepatocyte apoptosis (13–15). However, the mode of
humans and experimental animals (1– 4) and in severe cases to renal cell death during paracetamol nephrotoxicity and the
death. Paracetamol is a phenacetin metabolite (5). Phenacetin mechanisms involved are obscure. Indeed, there is evidence
was considered one of the most nephrotoxic analgesics and has that the molecular basis of nephrotoxicity may differ from
now been withdrawn from the market in most countries (6). A those of hepatotoxicity, as N-acetyl-cysteine protects from the
chronic nephrotoxic effect of therapeutic dosing of paraceta- latter, but has been shown not to protect from nephrotoxicity
mol is suggested by case-control studies (7–9). These findings (4).
have led to the recommendation that paracetamol be used only Fas belongs to the tumor necrosis factor receptor family of
in limited amounts and for limited time periods (10). Research proteins and plays a critical role in the normal development and
into the biologic basis of paracetamol nephrotoxicity has been homeostasis of T cells (16). However, inappropriate or exces-
recently encouraged by a National Kidney Foundation Ad Hoc sive Fas-mediated apoptosis has been implicated in a number
Committee (10). of pathologic conditions. In the context of hepatotoxicity, Fas
Tubular cell loss is a characteristic feature of both acute expression is increased in the liver of animals treated with
renal failure and chronic renal disease (11) and is observed paracetamol. The severity of liver damage is reduced by oli-
when cell death predominates over mitosis. Apoptosis is an gonucleotide-mediated suppression of Fas expression, demon-
strating a role for Fas in paracetamol toxicity in the liver (17).
Lethal intracellular proteins include the caspases, a family of
Corina Lorz’s present affiliation: Weston Laboratory, IRDB, Department of 14 proteases widely expressed in a variety of tissues and cell
PO&G, Imperial College of Sciences, Technology and Medicine, Hammer- types, that play a central role in promoting apoptosis (18).
smith Campus, London, UK. Studies in human hepatic cells have shown that paracetamol-
Received July 25, 2003. Accepted November 16, 2003. induced apoptosis is caspase-dependent and that mitochondria
Correspondence to: Alberto Ortiz, Laboratorio de Nefrología Experimental y
are a primary target (15).
Patología Vascular, Fundación Jiménez Díaz, Av Reyes Católicos 2l, 28040
Madrid, Spain. Phone: 34-915504940; Fax: 34-915494764; E-mail: Caspase-12 is specifically localized on the cytoplasmic side
aortiz@fjd.es of the endoplasmic reticulum (ER) and connects ER stress to
1046-6673/1502-0380 the caspase activation cascade (19). Because caspase-12 is
Journal of the American Society of Nephrology expressed at high levels in the kidney and specifically in renal
Copyright © 2004 by the American Society of Nephrology tubular epithelial cells, the cells affected during paracetamol
DOI: 10.1097/01.ASN.0000111289.91206.B0 nephrotoxicity, we examined the role of caspase-12 and ER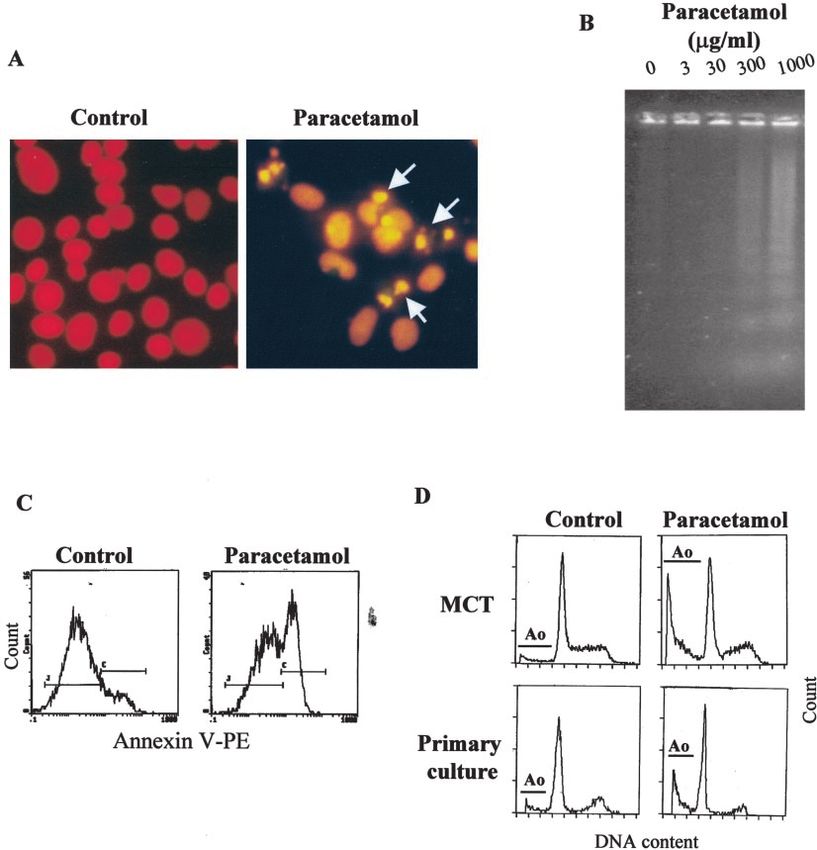
J Am Soc Nephrol 15: 380–389, 2004 Paracetamol: A Role for ER Stress 381
stress in renal tubular epithelial cell death after paracetamol DNA fragmentation ladder was demonstrated with ethidium bromide
treatment. staining of the gel as described previously (23).
While the mechanisms leading to hepatic injury have been
extensively studied (14,15,17), there are virtually no data on Cell Survival MTT Assay
the molecular mechanisms of paracetamol-induced nephrotox- The methylthiazoletetrazolium (MTT) assay relies on the conver-
icity. We have now investigated the ability of paracetamol to sion of MTT to colored formazan by succinate dehydrogenase in
induce apoptosis of cultured mouse renal tubular epithelial metabolically active cells and provides a measurement of cell viabil-
cells and the participation of the death receptor Fas, ER stress, ity. For viability experiments, 5000 MCT cells/well were placed into
96-well tissue culture plates; after 24 h, the medium was changed to
and caspases in the process.
RPMI-0% and cells were treated with paracetamol for defined periods
of time. Cells were then washed and allowed to grow for 48 h in
Materials and Methods RPMI-10%. At the end of the experiment, cell viability was measured
Cell Lines and Cell Culture by MTT assay as described previously (24). Results are expressed as
MCT cells are a cultured line of proximal tubular epithelial cells percent viability.
harvested originally from the renal cortex of SJL mice (20). The cells
were maintained in culture as described previously (20). Primary Western Blot
cultures of murine tubular epithelial cells were obtained as previously Western blot analyses were performed as described previously
published from kidneys of balb/c mice (21). For experiments on the (21). Primary antibodies were: anti-Fas, anti-cytochrome c, and anti-
effect of paracetamol, cells were plated and then grown for 24 h in GADD153 (all from Santa Cruz Biotechnology, Santa Cruz, CA);
RPMI with 10% fetal calf serum (RPMI-10%). Then the medium was anti-caspase-3, anti-caspase-9, and anti-caspase-12 (all from Cell Sig-
replaced with fresh serum-free RPMI (RPMI-0%), and the cells were naling, Hertfordshire, UK). Antibodies were diluted in 5% milk PBS/
grown with the indicated stimuli. Tween. The appropriate horseradish peroxidase-conjugated secondary
For the experiments with recombinant FasL or blocking FasL antibody (1:2000; Amersham, Aylesbury, UK) was used. Blots were
antibodies, cells in RPMI-0% medium were treated for 24 h with 100 then probed with anti-tubulin antibody to detect differences in load-
ng/ml recombinant FasL (Alexis, Switzerland) or 10 g/ml FasL ing. Each experiment was performed at least three independent times.
blocking antibody (MFL3, Pharmingen, San Diego, CA) in the pres-
ence or absence of 300 g/ml paracetamol. MCT cells readily un-
dergo apoptosis when stimulated with this concentration of recombi-
Flow Cytometry Analysis of Fas Expression
nant FasL following priming with TNF␣ ⫹ IFN␥ ⫹ LPS, a mixture For cytofluorography, cells were cultured in the presence of control
that upregulates Fas expression (21). Paracetamol (Sigma, St Louis, medium or paracetamol. After washing the culture with PBS, adherent
MO) was dissolved in 100% ethanol (vehicle). Final concentration of cells were detached with 2.2 mM EDTA, 0.2% BSA in PBS. Single
ethanol in culture did not modulate cell death. cell (5 ⫻ 105) suspensions were incubated in PBS/BSA for 30 min at
The pan-caspase inhibitory peptide benzyloxycarbonyl-Val-Ala- 4°C with 20 g/ml of Jo-2 or 20 g/ml of a control Ig. FITC
DL-Asp-fluoromethylketone (zVAD-fmk) was obtained from anti-hamster IgG (dilution, 1:100) was used as a secondary antibody
Bachem (Bubendorf, Switzerland). The caspase-8 inhibitory peptide (all from Pharmingen). For analysis, dead cells and debris were
benzyloxycarbonyl-Ile-Glu(OMe)-Thr-Asp(OMe)-fluoromethylk- excluded by selective gating on the basis of anterior and right angle
etone (zIETD-fmk) and calpain inhibitors I and II were from Sigma. scatter. At least 10,000 events were collected from each sample, and
They were dissolved in DMSO. Final concentration of DMSO in data were displayed on a logarithmic scale of increasing green fluo-
culture was 0.1% maximum. This concentration did not modulate cell rescence intensity. Mean cell fluorescence was calculated using LY-
death (22). N-Acetyl-Cysteine (Sigma) was added to the cell culture SIS II software.
1 h before paracetamol. Staurosporine (STS, from Sigma) was used at
a concentration of 100 nM. Tunicamycin (Sigma) was used at a Examination of Mitochondrial Transmembrane
concentration of 1 g/ml. Potential
Changes in mitochondrial transmembrane potential (⌬⌿m) were
Assessment of Apoptosis determined by staining the cells with JC-1 (Molecular Probes Europe
BV) before flow cytometry analysis, as described previously (25).
Apoptosis was quantified by flow cytometry analysis of DNA
Data analyses were performed using Cell Quest software by measur-
content in permeabilized, propidium iodide-stained cells, as described
ing both the green (530 ⫾ 15 nm) and red (585 ⫾ 21 nm) JC-1
previously (23). The percentage of cells with decreased DNA staining
fluorescence. The loss in ⌬⌿m is seen as a shift to lower JC-1 red
(A0) comprising apoptotic cells with fragmented nuclei was counted.
fluorescence accompanied by an increase in JC-1 green fluorescence.
To assess for the pyknotic nuclear changes seen in apoptosis, cells
At least 10,000 events were collected per sample. The proton trans-
were plated onto Labtek slides (Nunc Inc, Napersville, IL) in RPMI-
locator carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone
10%. After 24 h, the medium was changed to RPMI-0% and then
(CCCP 150 M) was used as positive controls because it disrupts the
grown for an additional 48 h in the presence of paracetamol or vehicle.
mitochondrial electrochemical gradient.
The cells were fixed in 10% buffered formalin and stained with
propidium iodide as described previously (23).
To identify apoptosis versus necrosis, Annexin V-FITC/7-amino- Assay of Cytochrome c and Smac/Diablo Release from
actinomycin D (7-AAD) staining was performed using the Apotosis Mitochondria
Detection Kit I (Pharmingen), and samples were analyzed by flow Release of cytochrome c from mitochondria to cytosol was mea-
cytometry within 30 min. sured by Western blot analysis. Cells (5 ⫻ 106) were harvested,
For assessment of internucleosomal genomic DNA fragmentation, washed once with ice-cold PBS, and gently lysed for 6 min in ice with
low–molecular weight DNA was separated in a 1.5% agarose gel. The 100 l of lysis buffer (250 mM sucrose, 80 mM KCl, 500 g/ml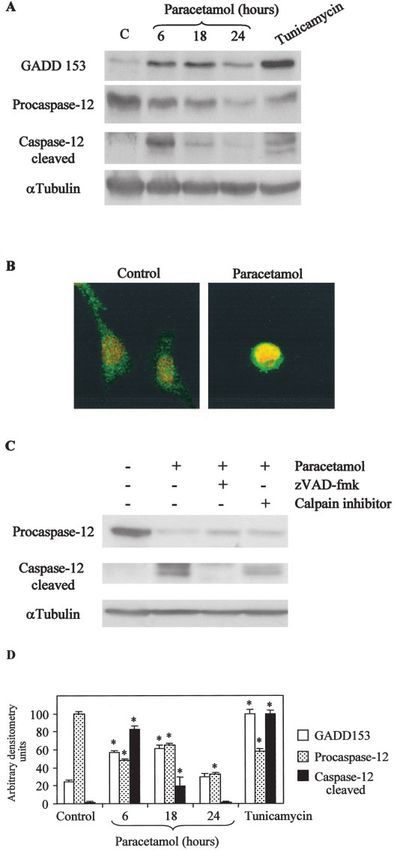
382 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 380–389, 2004
digitonin, 1 mM DTT, 0.1 mM PMSF, protease inhibitors, in PBS). Paracetamol-induced apoptosis increased with time (Figure
Lysates were centrifuged at 12, 000 ⫻ g at 4°C for 5 min to obtain the 2A) and dose (Figure 2B). Paracetamol plasma concentrations
supernatants (cytosolic extract free of mitochondria) and the pellets of 10 to 20 g/ml are associated with antipyretic activity (26).
(fraction that contains the mitochondria). Supernatants (50 g) and Although the therapeutic analgesic plasma concentration for
pellets (50 g) were electrophoresed on 15% polyacrylamide gels and
paracetamol is not well defined, new protocols are using higher
then analyzed by Western blot as described above. Cytochrome c
antibody clone 7H8.2C12 was from Pharmingen, and mouse specific
concentrations, which result in mean paracetamol plasma lev-
anti-Smac/DIABLO clone 9H10 form Kamiya Biomedical (Seattle, els of 30 g/ml (27). An increased rate of apoptosis was
WA). The mitochondrial enzyme cytochrome oxidase subunit IV observed with as little as 30 g/ml paracetamol (P ⬍ 0.05
(Molecular Probes, Leiden, The Netherlands) is not released from versus control), a concentration that can be reached during
mitochondria during apoptosis and was used as control for the therapeutic dosing (27). For further experiments we chose a
technique. concentration of 300 g/ml of paracetamol, which induces
about a 50% of the cells to undergo apoptosis in 24 h (Figure
Cytochrome c and GADD153 Immunostaining 2A). This concentration of paracetamol can be reached during
For Cytochrome c and GADD153 immunostaining, cells were clinical paracetamol toxicity (28).
plated onto Labtek slides in RPMI-10%. After 24 h, the medium was Cell survival studies showed that at 8 h of paracetamol
changed to RPMI-0% and cells were incubated from 1 to 24 h with the incubation 50% of the cells were committed to die (Figure 2C).
indicated stimuli. Then cells were fixed in 4% paraformaldehyde and Even though, at this time point the rate of apoptosis was NS in
permeabilized in 0.2% Triton X-10 in PBS for 10 min each. After the cell cultures compared with control cells (Figure 2A).
washing in PBS, cells were incubated overnight at 4°C with anti- In vivo treatment of paracetamol toxicity with N-acetylcys-
cytochrome c (clone 6H2.B4, Pharmingen) or anti-GADD153 fol- teine can prevent hepatic damage in most cases (29,30). Nev-
lowed by 1 h incubation with a FITC-labeled anti-IgG. Cell nuclei
ertheless, relatively high doses of N-acetylcysteine (10 mM)
were counterstained with DAPI or propidium iodide.
failed to prevent apoptosis of MCT cells induced by 300 g/ml
paracetamol at 24 h (Paracetamol 56 ⫾ 2%, N-acetylcysteine
Caspase-3 Activity Assay ⫹ paracetamol 57 ⫾ 2% apoptotic cells, NS).
Caspase-3 activity was measured following the manufacturer’s
instructions (Biomol, Plymouth, PA). In brief, cell extracts (30 g of The Fas Pathway Is Not Involved in Paracetamol-
protein) were incubated in half-area 96-well plates for 3 h at 37°C
Induced Apoptosis in Tubular Cells
with 200 M Asp-Glu-Val-Asp-pNA (DEVD-pNA) peptide in a total
volume of 50 l. The color of free pNA, generated as a result of
A direct role for Fas in paracetamol toxicity has been pre-
cleavage of the aspartate-pNA bond, was monitored continuously over viously shown in the liver (17). Figure 3A shows that parac-
3 h by measuring absorbance at 405 nm in a spectrophotometer plate etamol increased Fas protein expression in MCT cells and that
reader. The emission from each well was plotted against time, and the receptor was expressed on the cell surface (Figure 3B).
linear regression analysis of the initial velocity (Vmax) for each curve These results raised the possibility that, as in the liver, in the
yielded the activity. As a control, caspase-3 activity in the samples tubular epithelium Fas is involved in the apoptotic process
was inhibited with Ac-Asp-Glu-Val-Asp-CHO (Ac-DEVD-CHO). induced by paracetamol. To test this possibility, we cultured
MCT cells with recombinant FasL or a FasL blocking anti-
Statistical Analyses body, in the presence of paracetamol (Figure 3C). The addition
Results are expressed as mean ⫾ SD. Significance at the 95% level of recombinant FasL did not increase the amount of paraceta-
was established using one-way ANOVA and t test. The presence of mol-induced cell death. Likewise, blocking FasL with an anti-
significant differences between groups was examined by a post hoc FasL blocking antibody did not protect against apoptosis
test (Bonferroni method) by means of the SigmaStat statistical soft- caused by paracetamol. Furthermore, the caspase-8 inhibitor,
ware (Jandel, San Rafael, CA). zIETD-fmk, was unable to protect tubular cells from paraceta-
mol-induced apoptosis (Figure 3D), and no caspase-8 activity
Results was detected on activity assays (data not shown). Together,
Paracetamol Induces Apoptosis in Primary Cultures of these results indicate that, contrary to the findings in liver,
Renal Tubular Epithelial Cells and in the Tubular paracetamol does not induce apoptosis of renal tubular epithe-
Epithelial Cell Line MCT lial cells through the Fas pathway.
Cell death of tubular epithelium caused by paracetamol has
features of apoptosis. These include characteristic nuclear mor- Paracetamol Does Not Induce Cytochrome c Release
phology (Figure 1A) and internucleosomal DNA degradation or Changes in the Mitochondrial Transmembrane
(Figure 1B). Significantly, MCT cells treated with paracetamol Potential of Tubular Epithelial Cells
were positive for annexin-V but did not show uptake of the Mitochondrial changes that lead to apoptosis include release
vital dye 7-AAD (Figure 1C). This indicates that, at the con- of caspase-activating proteins and/or loss of mitochondrial
centration used in our experiments, paracetamol induced apo- transmembrane potential (⌬⌿m). We investigated whether
ptosis and not necrosis of tubular epithelial cells. In addition, paracetamol treatment of renal tubular epithelial cells induced
decreased DNA content was present in paracetamol-treated changes in the mitochondria. While the proton translocator
murine tubular epithelial MCT cells and in primary cultures of CCCP caused a significant membrane depolarization, no loss
murine tubular epithelial cells (Figure 1D). of ⌬⌿m was detected in the tubular epithelial cells treated withJ Am Soc Nephrol 15: 380–389, 2004 Paracetamol: A Role for ER Stress 383 Figure 1. Paracetamol-induced tubular cell death has features of apoptosis. (A) Characteristic shrunk, pyknotic-fragmented nuclei (arrows) are present among fixed, propidium iodide-stained, paracetamol-treated tubular epithelial MCT cells, but not among control cells. (B) Internu- cleosomal DNA degradation in MCT cells treated with increasing concentrations of paracetamol for 24 h. (C) Nonpermeabilized MCT cells treated with paracetamol are positive for annexin-V, but they do not show 7-AAD staining. (D) Presence of apoptotic, hypodiploid (A0) cells among MCT cells and primary cultures of murine tubular epithelial cells as shown by flow cytometry. Except otherwise stated, cells were treated under serum-free conditions with 300 g/ml paracetamol for 24 h. paracetamol (Figure 4). Cytochrome c release from the mito- Paracetamol-Induced Cell Death Is Caspase- chondria was studied by Western blot and by immunofluores- Dependent cence (Figures 5 and 6). We did not detect any cytochrome c Paracetamol induced processing of caspase-3 starting at 6 h, release from the mitochondria of MCT cells treated with parac- as shown by the appearance of caspase-3 cleavage product in etamol, even at time points when around 50% of the cells were Western blots (Figure 7A) and by the presence of caspase-3– undergoing apoptosis and caspases had already been activated like activity in cell extracts treated with paracetamol (Figure (Figure 7). Cytochrome c release per se, however, is functional 7B), with maximal activity detected at 6 to 8 h. Caspase-3 is in MCT cells, because treatment with staurosporine (an inducer mainly activated by caspase-8 or caspase-9. Caspase-8 does of the mitochondrial pathway) induced apoptosis at a similar not play a role in paracetamol-induced cell death; we therefore level of lethality than paracetamol with a significant release of studied caspase-9 cleavage by Western blot. Paracetamol in- cytochrome c. Similar findings were observed with the cellular duced caspase-9 processing starting at 6 h (Figure 7A). Two distribution of Smac/DIABLO. Taken together, these results bands of 39 kD and 37 kD, corresponding to the cleaved suggest that in MCT cells treated with paracetamol mitochon- fragments of caspase-9, were observed on Western blots. Tu- dria do not suffer evident alterations. nicamycin, an inducer of ER stress, also induced cleavage of
384 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 380–389, 2004
death triggered by ER stress (32). GADD153 expression was
upregulated in tubular cells treated with paracetamol similarly
to cells treated with the ER stress-inducer tunicamycin (Figure
9A). Consistent with its role as a transcription factor, we
detected translocation of GADD153 from the cytosol to the
nucleus in cells treated with paracetamol (Figure 9B).
We detected caspase-12 cleavage in MCT cells treated with
paracetamol starting at 6 h (Figure 9A). Caspase-12 cleavage
product was not present in cells treated with the caspase
inhibitor zVAD-fmk before paracetamol incubation (Figure
9C). Calpains are a family of cysteine proteases that are acti-
vated by elevated intracellular calcium. They have been in-
volved in the activation of caspase-12 upon disturbance of
intracellular calcium homeostasis (33). Nevertheless,
caspase-12 cleavage product was still detectable in tubular
cells that had been treated with paracetamol and calpain inhib-
itors (Figure 9C). Calpain inhibitors I and II did not protect
tubular cells from paracetamol-induced apoptosis (data not
shown).
Discussion
Paracetamol overdose causes acute renal failure, and chronic
exposure to paracetamol has been linked to chronic renal
failure (9). While the mechanisms of paracetamol-induced
hepatotoxicity have been extensively studied (14,15,17,34 –
37), information about the specific molecular pathways that
lead to apoptosis of tubular cells during nephrotoxic injury is
incomplete (11). The mechanisms involved in paracetamol-
induced apoptosis in nephrotoxicity may differ to those during
hepatotoxicity, as suggested by the fact that N-acetylcysteine
can prevent in vivo paracetamol hepatic damage (29,30) but did
not prevent apoptosis of tubular cells. Because tubular cell
death is a feature of both acute and chronic renal failure, we
examined the ability of paracetamol to induce cell death in
murine proximal tubular cells and the intracellular mechanisms
Figure 2. Paracetamol-induced apoptosis is time-dependent and con-
centration-dependent. Cell death was assessed by flow cytometry after
involved.
culture in the presence of 300 g/ml paracetamol for different time Paracetamol induced a mild degree of tubular cell apoptosis,
periods (A) and in cells cultured in the presence of different concen- even at concentrations found during therapeutic dosing. These
trations of paracetamol for 72 h (B) under serum-free conditions. *P findings are consistent with the chronic long-term toxicity of
⬍ 0.05 versus control. (C) After 8 h of paracetamol treatment, 50% of the drug (7–9). To explore the molecular mechanisms of parac-
the cells are committed to die. MCT cells were cultured for the etamol-induced apoptosis, we examined the effect of a con-
indicated times with 300 g/ml of paracetamol; they were then centration of paracetamol that is reached in humans during
allowed to recover for 48 h in 10% FCS medium. Cell viability was acute paracetamol toxicity (28). Upon treatment with paraceta-
measured by MTT assay. Results are expressed as percent viability. mol, primary cultures of murine tubular epithelial cells and the
murine proximal tubular cell line MCT showed morphologic
changes associated with apoptosis, such as chromatin conden-
caspase-3 and caspase-9 in MCT cells. zVAD-fmk is a irre- sation and internucleosomal DNA fragmentation. Moreover,
versible, broad-spectrum inhibitor of caspases (31). zVAD-fmk the loss of membrane asymmetry observed after paracetamol
afforded complete protection against nuclear features of apo- treatment as detected by annexin-V staining without loss of
ptosis induced by paracetamol (Figure 8) and inhibited parac- membrane integrity suggests that apoptosis is the primary
etamol-induced caspase-3 activity at the concentrations used. mode of cell death in tubular cells treated with paracetamol.
A variety of cytototoxins such as chemotherapeutic agents
Paracetamol Treatment Induces ER Stress and (38), toxic bile salts (39), and paracetamol (17) may induce
Caspase-12 Cleavage in Tubular Epithelial Cells apoptosis by upregulating the death receptor Fas expression.
To determine whether paracetamol induced ER stress in This prompted the study of the Fas pathway during paraceta-
murine tubular cells, we studied the expression and localization mol nephrotoxicity. We found that Fas expression was in-
of GADD153, a transcription factor that is induced during cell creased in tubular cells upon paracetamol treatment. The FasJ Am Soc Nephrol 15: 380–389, 2004 Paracetamol: A Role for ER Stress 385 Figure 3. Paracetamol-induced apoptosis of tubular cells does not involve the Fas pathway. (A) Western blot studies revealed that Fas expression increases in MCT cells treated with increasing doses of paracetamol for 24 h. (B) Fas is expressed in the surface of cells treated with paracetamol (24 h, 300 g/ml) compared with untreated cells. Flow cytometry of nonpermeabilized cells. Control IgG–stained cells display a peak that overlies that of the untreated cells. (C) Incubation of paracetamol-treated cells with recombinant FasL did not increase drug-induced cell death. Also paracetamol-induced apoptosis of MCT cells was not prevented in the presence of a FasL blocking antibody. (D) MCT cells were treated for 2 h with the caspase-8 inhibitor zIETD-fmk (200 M) before paracetamol incubation. Cell death was assessed by flow cytometry after 24 h. Figure 4. Changes in mitochondrial transmembrane potential (⌬⌿m) were analyzed by JC-1 staining. The loss in ⌬⌿m is seen as a shift to lower JC-1 red fluorescence. Results show the percentage of cells with reduced ⌬⌿m. MCT cells were treated with 300 g/ml paracetamol for 24 h. As a positive control, cells were treated for 4 h with 150 M CCCP. receptor could theoretically be activated by autocrine FasL, as pathways converge on the mitochondria to induce dissipation tubular epithelium constitutively expresses FasL (21). Never- of mitochondrial membrane potential and release of proteins theless, neither Fas receptor activation by recombinant FasL that are normally strictly confined to the mitochondrial inter- nor FasL neutralization significantly modified the rate of cell membrane space, such as cytochrome c and Smac/DIABLO. death induced by paracetamol treatment alone. Together with Cytochrome c stimulates the cytosolic assembly of the apop- the fact that we did not detect caspase-8 activation in treated tosome by binding to Apaf-1. This leads to caspase-9 oli- cells, and caspase-8 inhibitors were unable to protect from gomerization and activation and to caspase-3 cleavage. Cyto- paracetamol-induced apoptosis, we can conclude that, contrary chrome c release from mitochondria followed by caspase-9 and to paracetamol hepatotoxicity, the Fas receptor pathway is not caspase-3 cleavage has been reported during paracetamol tox- involved in paracetamol-induced cell death in murine proximal icity in human hepatic cells (15). Paracetamol treatment of tubular cells. renal tubular epithelial cells, at the concentrations used in our Numerous pro-apoptotic signal transduction and damage studies, did not induce loss of mitochondrial transmembrane
386 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 380–389, 2004
Figure 5. Mitochondria do not release cytochrome c during paraceta-
mol treatment. Western blot analyses of cytochrome c and Smac/
DIABLO in cytosolic and mitochondrial extracts of MCT cells treated
for different time with 300 g/ml paracetamol or 100 nM staurospor-
ine (STS). Cytochrome oxidase subunit IV and ␣ Tubulin are controls
for fraction separation and loading.
Figure 7. Paracetamol induces caspase-3 and caspase-9 activation in
tubular cells. (A) Western blot analyses of the processing of caspase-3
and caspase-9 during paracetamol-induced apoptosis. The migration
position of the caspase-9 cleavage products is indicated. (B) Caspase-
3–like activity of extracts of tubular cells treated with paracetamol
(300 g/ml) or STS (100 nM) for the indicated times. Ac-DEVD-
CHO was used to inhibit caspase-3 like activity.
potential or release of the proapoptotic factors cytochrome c
and Smac/DIABLO into the cytosol. Nevertheless, paraceta-
mol-induced apoptosis is a caspase-dependent process, as
shown by the fact that zVAD-fmk protects against features of
apoptosis induced by paracetamol. Indeed, paracetamol treat-
ment leads to activation of caspase-9 and caspase-3 in renal
tubular epithelial cells.
Paracetamol hepatotoxicity is a process characterized by
calcium deregulation (13,34 –37). Recently, the endoplasmic
reticulum has been shown to participate in the initiation of
apoptosis in response to calcium signaling (19). There is in-
creasing evidence to suggest that the ER stress apoptotic path-
way is important in the kidney, specifically in tubular epithelial
cells. Tunicamycin has been reported to induce ER stress-
mediated apoptosis in renal proximal tubules in mice, followed
by the development of a histologic picture similar to the human
condition known as acute tubular necrosis (32). We found that
the expression of GADD153, a marker of ER stress, is in-
creased in tubular epithelial cells during paracetamol-induced
apoptosis. GADD153 is a transcription factor that promotes
apoptosis (40). Consistent with its role as a transcription factor,
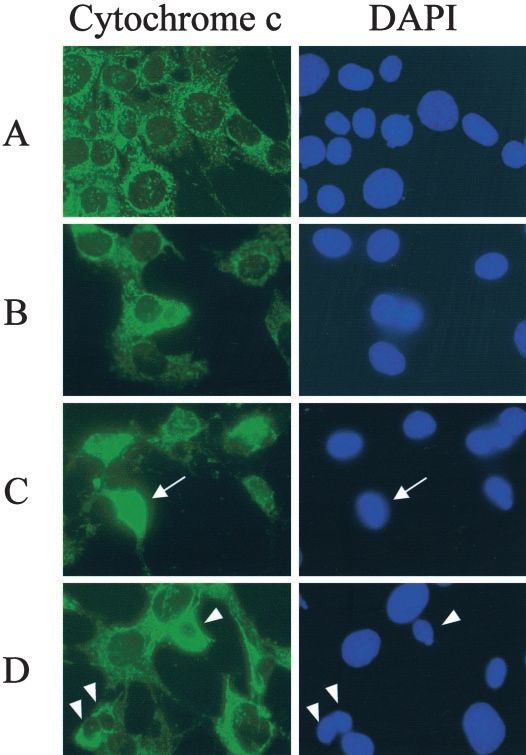
Figure 6. Cytochrome c immunostaining and the corresponding DAPI
we detected GADD153 translocation to the nucleus in tubular
staining of MCT cells treated with: (A) vehicle 6 h; (B) paracetamol
cells treated with paracetamol, both in cell culture and in the
6 h; (C) STS 6 h; (D) paracetamol 24 h. Cytochrome c labeling
appears punctuate in control cells, where it is localized at the mito- whole animal (unpublished observation).
chondria. When cytochrome c is released from the mitochondria, the Caspase-12 is ubiquitously expressed in mouse tissues, and
labeling becomes diffuse (arrow in C). We could not detect cyto- it is expressed at high levels in kidney, specifically in renal
chrome c release, even in late paracetamol treated apoptotic cells proximal tubular epithelial cells, but not in the glomerulus.
(arrowheads in D). Caspase-12 is activated by ER stress, but apparently not byJ Am Soc Nephrol 15: 380–389, 2004 Paracetamol: A Role for ER Stress 387
Figure 8. Effect of the pan-caspase inhibitor zVAD-fmk on apoptosis
induced by 300 g/ml paracetamol for 24 h. (A) Apoptosis quantified
as hypodiploid cells in permeabilized, propidium iodide–stained cells.
For the expression of specific protection, apoptosis in the presence of
paracetamol alone was considered to be 100%, and apoptosis in the
presence of the caspase inhibitor and paracetamol was expressed as a
percentage of this. (B) Internucleosomal DNA degradation in MCT
cells treated with 300 g/ml paracetamol in the presence or absence
of 200 M zVAD-fmk for 24 h.
death receptor-mediated or mitochondria-targeted apoptotic
signals (19). We detected caspase-12 cleavage in tubular cells
treated with paracetamol; this cleavage was prevented by Figure 9. Paracetamol induces ER stress and caspase-12 cleavage in
zVAD-fmk. Caspase-12 has been reported to be cleaved by tubular cells. (A) Western blot analyses of the expression of
calpains (33), a family of cysteine proteases that are activated GADD153 and caspase-12 cleavage during paracetamol-induced ap-
by elevated intracellular calcium. Nevertheless, after treatment optosis. (B) Confocal images of GADD153 (green) immunostaining
of murine tubular cells with paracetamol and calpain inhibitors, of MCT cells treated with 300 g/ml paracetamol for 24 h. In red
the caspase-12 cleavage product was still detectable, and cal- nuclei stained with propidium iodide. (C) Western blot analyses of
pain inhibitors were unable to protect tubular cells from parac- caspase-12 cleavage during paracetamol-induced apoptosis in the
etamol-induced apoptosis. presence of caspase inhibitor zVAD-fmk or calpain inhibitor I. Sim-
ilar results were obtained with calpain inhibitor II. (D) Densitometric
The present study shows that paracetamol induces apoptosis
analyses of at least three independent experiments for the Western
of cultured murine tubular epithelial cells through a caspase-
blots shown on panels A and C. *P ⬍ 0.005 versus control.
mediated mechanism that involves caspase-9 and caspase-3 in388 Journal of the American Society of Nephrology J Am Soc Nephrol 15: 380–389, 2004
a cytochrome c and Smac/DIABLO–independent manner. 12. Savill J: Apoptosis and the kidney. J Am Soc Nephrol 5: 12–21,
Caspase-12 has been reported to cleave caspase-9 in vitro in 1994
the absence of cytochrome c (41); this raises the possibility that 13. Ray SD, Kamendulis LM, Gurule MW, Yorkin RD, Corcoran
caspase-12 is the apical caspase in paracetamol-induced apo- GB: Ca2⫹ antagonists inhibit DNA fragmentation and toxic cell
death induced by acetaminophen. FASEB J 7: 453– 463, 1993
ptosis in tubular epithelial cells. Nevertheless, we cannot ex-
14. Ray SD, Jena N: A hepatotoxic dose of acetaminophen modu-
clude the possibility that other factors (released or not from the
lates expression of Bcl-2, Bcl-xL, and Bcl-xS during apoptotic
mitochondria) are responsible for paracetamol-induced and necrotic cell death of mouse liver cells in vivo. Arch Toxicol
caspase-9 activation. Paracetamol causes ER stress in tubular 73: 594 – 606, 2000
cells, leading to GADD153 upregulation and translocation to 15. Boulares HA, Zoltoski AJ, Stoica BA, Cuvillier O, Smulson ME:
the nucleus, as well as caspase-12 cleavage. Our results suggest Acetaminophen induces a caspase-dependant and Bcl-xL sensi-
that induction of apoptosis may underlie the nephrotoxic po- tive apoptosis in human hepatoma cells and lymphocytes. Phar-
tential of paracetamol and identify ER stress as a therapeutic macolgy & Toxicology 90: 38 –50, 2002
target in nephrotoxicity. 16. Nagata S, Goldstein P: The Fas death factor. Science 267: 1449 –
1455, 1995
Acknowledgments 17. Zhang H, Cook J, Nickel J, Yu R, Stecker K, Myers K, Dean
This work was supported by grants FISSS 01/0199, Comunidad de NM: Reduction of liver Fas expression by an antisense oligonu-
Madrid (08.2/0030/2000), Sociedad Española de Nefrología, Instituto cleotide protects mice from fulminant hepatitis. Nat Biotech 18:
Reina Sofia de Investigaciones Nefrológicas, and EU project QLG1- 862– 867, 2000
CT-2002– 01215. PJ was supported by Fondo de Investigaciones 18. Thornberry NA, Lazebnik Y: Caspases: Enemies within. Science
Sanitarias. AS was supported by Conchita Rábago de Fundación 281: 1312–1316, 1998
Jiménez Díaz. CL was supported by Ministerio de Educación, Ciencia 19. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA,
y Deporte. Yuan J: Caspase-12 mediates endoplasmic-reticulum-specific
apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98 –103,
References 2000
1. Rumack BH, Peterson RC, Koch GG, Amara IA: Acetaminophen 20. Haverty TP, Kelly CJ, Hines WH, Amenta PS, Watanabe M,
overdose: 662 cases with evaluation of oral acetylcysteine treat- Harper RA, Kefalides NA, Neilson EG: Characterization of a
ment. Arch Intern Med 14: 380 –385, 1981 renal tubular epithelial cell line which secretes the autologous
2. Emeigh Hart SG, Wyand DS, Khairallah EA, Cohen SD: Acet- target of autoimmune experimental interstitial nephritis. J Cell
aminophen nephrotoxicity in the CD-1 mouse II. Protection by Biol 107: 1359 –1368, 1988
probenecid and AT-125 without diminution of renal covalent 21. Lorz C, Ortiz A, Justo P, González-Cuadrado S, Duque N,
binding. Toxicol Appl Pharmacol 136: 161–169, 1996 Gómez-Guerrero C, Egido J: Proapoptotic Fas ligand is ex-
3. Eguia L, Materson BJ: Acetaminophen-related acute renal failure pressed by normal kidney tubular epithelium and injured glomer-
without fulminant liver failure. Pharmacotherapy 17: 363–370, uli. J Am Soc Nephrol 11: 1266 –1277, 2000
1997 22. Ortiz A, Lorz C, Catalán MP, Ortiz A, Coca S, Egido J: Cyclo-
4. Blakely P, McDonald BR: Acute renal failure due to acetamin- sporine A induces apoptosis in murine tubular epithelial cells:
ophen ingestion: A case report and review of the literature. J Am Role of caspases. Kidney Int suppl 68: S25–S29, 1998
Soc Nephrol 6: 48 –53, 1995 23. Ortiz A, Gonzalez-Cuadrado S, Lorz C, Garcia del Moral R,
5. Prescott LF: Kinetics and metabolism of paracetamol and phen- O’Valle F, Egido J: Cytokines and Fas regulate apoptosis in
acetin. Br J Clin Pharm 10: 2915–2985, 1989 murine renal interstitial fibroblasts. J Am Soc Nephrol 8: 1845–
6. De Broe ME, Elseviers MM: Analgesic nephropathy. N Engl 1854, 1997
J Med 338: 446 – 452, 1998 24. Hansen M, Nielsen S, Berg K: Re-examination and further
7. McLaughlin JK, Lipworth L, Chow WH, Blot WJ: Analgesic use development of a precise and rapid dye method for measuring
and chronic renal failure: A critical review of the epidemiologic cell growth/cell kill. J Immunol Methods 119: 203–210, 1989
literature. Kidney Int 54: 679 – 686, 1998 25. Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S: Presence
8. Perneger TV, Whelton PK, Klag MJ: Risk of kidney failure of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10
associated with the use of acetaminophen, aspirin, and nonste- in the mitochondrial fraction of jurkat cells. EMBO J 18: 2040 –
roidal antiinflammatory drugs. N Engl J Med 331: 1675–1679, 2048, 1999
1994 26. Berde CB, Sethna NF: Analgesics for the treatment of pain in
9. Fored CM, Ejerblad E, Lindblad P, Fryzek JP, Dickman PW, children. N Engl J Med 3;347: 1094 –103, 2002
Signorello LB, Lipworth L, Elinder CG, Blot WJ, McLaughlin 27. Bolton P, Bridge HS, Montgomery CJ, Merrick PM: The anal-
JK, Zack MM, Nyren O: Acetaminophen, aspirin, and chronic gesic efficacy of preoperative high dose (40 mg ⫻ kg(⫺1)) oral
renal failure. N Engl J Med 345: 1801–1808, 2001 acetaminophen after bilateral myringotomy and tube insertion in
10. Henrich WL, Agodoa LE, Barrett B, Bennett WM, Blantz RC, children. Paediatr Anaesth 12: 29 –35, 2002
Buckalew VM Jr., D’Agati VD, DeBroe ME, Duggin GG, 28. Dargan PI, Ladhani S, Jones AL: Measuring plasma paracetamol
Eknoyan G: Analgesics and the kidney: Summary and recom- concentrations in all patients with drug overdose or altered
mendations to the Scientific Advisory Board of the National consciousness: Does it change outcome? Emerg Med J 18: 178 –
Kidney Foundation from an Ad Hoc Committee of the National 182, 2001
Kidney Foundation. Am J Kidney Dis 27: 162–165, 1996 29. Kozer E, Koren G: Management of paracetamol overdose: Cur-
11. Ortiz A, Lorz C, Catalan MP, Justo P, Egido J: Role and rent controversies. Drug Saf 24: 503–512, 2001
regulation of apoptotic cell death in the kidney. Y2K update. 30. Flanagan RJ, Meredith TJ: Use of N-acetylcysteine in clinical
Front Biosci 5: D735–D749, 2000 toxicology. Am J Med 91: 131S–139S, 1991J Am Soc Nephrol 15: 380–389, 2004 Paracetamol: A Role for ER Stress 389
31. Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, 37. Shen W, Kamendulis LM, Ray SD, Corcoran GB: Acetamino-
Thornberry NA: Inhibition of human caspases by peptide-based phen-induced cytotoxicity in cultured mouse hepatocytes: Ef-
and macromolecular inhibitors. J Biol Chem 273: 32608 –32613, fects of Ca(2⫹)-endonuclease, DNA repair, and glutathione de-
1998 pletion inhibitors on DNA fragmentation and cell death. Toxicol
32. Zinszner H, Kuroda M, Wang XZ, Batchvarova N, Lightfoot RT, Appl Pharmacol 112: 32– 40, 1992
Remotti H, Stevens JL, Ron D: CHOP is implicated in pro- 38. Muller M, Strand S, Hug H, Heinemann EM, Walczak H, Hof-
grammed cell death in response to impaired function of the mann WJ, Stremmel W, Krammer PH, Galle PR: Drug-induced
endoplasmic reticulum. Gene Dev 12: 982–995, 1998 apoptosis in hepatoma cells is mediated by the CD95 (APO-1/
33. Nakagawa T, Yuan J: Cross-talk between two cysteine protease Fas) receptor/ligand system and involves activation of wild-type
families: Activation of caspase-12 by calpain in apoptosis. J Cell p53. J Clin Invest 99: 403– 413, 1997
Biol 150: 887– 894, 2000
39. Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ,
34. Ray SD, Sorge CL, Raucy JL, Corcoran GB: Early loss of large
Svingen PA, Kaufmann SH, Gores GJ: Toxic bile salts induce
genomic DNA in vivo with accumulation of Ca2⫹ in the nucleus
rodent hepatocyte apoptosis via direct activation of Fas. J Clin
during acetaminophen-induced liver injury. Toxicol Appl Phar-
macol 106: 346 –351, 1990 Invest 103: 137–145, 1999
35. Ray SD, Sorge CL, Tavacoli A, Raucy JL, Corcoran GB: Ex- 40. McCullouh KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ:
tensive alteration of genomic DNA and rise in nuclear Ca2⫹ in Gadd153 sensitizes cells to endoplasmic reticulum stress by
vivo early after hepatotoxic acetaminophen overdose in mice. down-regulation of Bcl-2 and perturbing the cellular redox state.
Adv Exp Med 283: 699 –705, 1991 Mol Cell Biol 21: 1249 –1259, 2001
36. Shen W, Kamendulis LM, Ray SD, Corcoran GB: Acetamino- 41. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko
phen-induced cytotoxicity in cultured mouse hepatocytes: Cor- Y: An endoplasmic reticulum stress-specific caspase cascade in
relation of nuclear Ca2⫹ accumulation and early DNA fragmen- apoptosis. Cytochrome c-independent activation of caspase-9 by
tation with cell death. Toxicol Appl Pharmacol 111: 242–254, 1991 caspase-12. J Biol Chem 277: 34287–34294, 2002You can also read

















































