Poster Abstracts - Poster Abstracts - Penn State
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Poster Abstracts Poster Abstracts
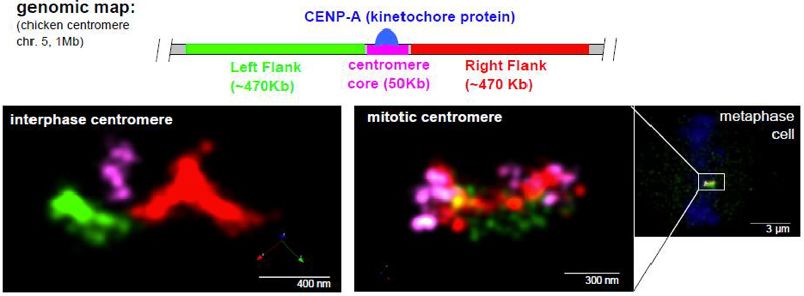
1 2 Evolutionary studies of nucleosome recognition by BAH Structure of the Human Core Centromeric Nucleosome domains. Complex Stephen Abini-Agbomson*, Pablo De Ioannes*, Victor A. Leon, Praveen Kumar Allu1,2, Jennine M. Dawicki-McKenna1,2, Trevor Zheng Kuang, Miao Wang, Jef D. Boeke, Andreas Hochwagen Van Eeuwen1, Moriya Slavin3, Merav Braitbard3, Chen Xu4, Nir & Karim-Jean Armache Kalisman3, Kenji Murakami1, and Ben E. Black1,2* 1 The Origin Recognition Complex (ORC) is essential for Department of Biochemistry and Biophysics, Perelman School of replication, heterochromatin formation, telomere maintenance, Medicine, University of Pennsylvania, Philadelphia, PA 19104, and genome stability in eukaryotes. Here we present the structure USA. 2 of the yeast Orc1 BAH domain bound to the nucleosome core Epigenetics Institute, Perelman School of Medicine, University particle. Our data reveal that unlike its close homolog involved of Pennsylvania, Philadelphia, PA 19104, USA. 3 in gene silencing Sir3, Orc1 does not appear to discriminate Department of Biological Chemistry, The Alexander Silberman between acetylated and non-acetylated lysine 16, modifications Institute of Life Sciences, Hebrew University of Jerusalem, in the H4 tail that specify open and closed chromatin Jerusalem 91904, Israel. 4 respectively. We elucidate the mechanism for this unique Department of Biochemistry and Molecular Pharmacology, feature of Orc1 and hypothesize that its ability to interact with University of Massachusetts Medical School, Worcester, MA nucleosomes regardless of K16 modification state enables it to 01605, USA. perform critical functions in both hetero- and euchromatin. We also present our data on the evolution of BAH domains and Centromeric nucleosomes are at the interface of the chromosome discuss how their nucleosome recognition regulates their and the kinetochore that connects to spindle microtubules in function in heterochromatin formation across species. mitosis. The core centromeric nucleosome complex (CCNC) harbors the histone H3 variant, CENP-A and its binding proteins, Email: stephen.abini-agbomson@nyulangone.org CENP-C and CENP-N. Here, we find that one copy of CENP-N is lost for every two copies of CENP-C just prior to kinetochore formation. We present the structures of symmetric and asymmetric forms of the CCNC that vary in CENP-N stoichiometry. Our structures explain how the central domain of CENP-C achieves its high specificity for CENP-A nucleosomes and how CENP-C and CENP-N sandwich the histone H4 tail. The natural centromeric DNA path in our structures generates symmetric surfaces for CCNC assembly, deviating from what is observed in prior structures using artificial sequences. At mitosis, we propose that CCNC asymmetry accommodates its asymmetric connections at the chromosome/kinetochore interface. Email: kumarpr@pennmedicine.upenn.edu Poster Abstracts
3 4 Cryo-EM structure of Dot1L-H2BK120ub nucleosome NELF establishes a 5’ cap protection checkpoint distinct from complex illustrates trans-histone epigenetic crosstalk RNA polymerase II pause-release Cathy J. Anderson, Matthew R. Baird, Allen L. Hsu, Emily H. Yuki Aoi,1,2 Edwin R. Smith,1,2 Patrick A. Ozark,1,2 Ashley R. Barbour, Yuka Koyama, Mario J. Borgnia, Robert K. McGinty Woodfin,1,2 Fei X. Chen,1,2 Emily J. Rendleman,1,2 Stacy A. Post-translational modification of histone proteins plays a major Marshall,1,2 Avani P. Shah,1,2 Ramin Shiekhattar,3 and Ali role in epigenetic regulation by providing binding sites for Shilatifard1,2 numerous effector proteins that tune gene expression. Still, relatively little structural information is available describing the 1Simpson Querrey Center for Epigenetics, Feinberg School of interactions between the proteins that regulate these modifications and their nucleosome substrate. Methylation of histone H3 lysine Medicine, Northwestern University, Chicago, IL 60611, USA. 79 (H3K79me) is an epigenetic modification enriched at actively 2Department of Biochemistry and Molecular Genetics, Feinberg transcribed genomic regions, and its presence at oncogenic HOX School of Medicine, Northwestern University, Chicago, IL 60611, genes is implicated in the development of mixed lineage leukemia. USA. 3Department of Human Genetics, Sylvester Comprehensive H3K79me is deposited by the methyltransferase Dot1L, and this Cancer Center, University of Miami Miller School of Medicine, methylation activity is upregulated by ubiquitylation of histone Miami, Florida 33136, USA H2B lysine 120 (H2BK120ub) in a trans-histone crosstalk pathway. A lack of high-resolution structural information RNA polymerase II (Pol II) is generally paused at promoter- describing Dot1L interactions with either the nucleosome or proximal regions in most metazoans, and based on in vitro studies, ubiquitin has left molecular details of this crosstalk and its role in this function has been attributed to negative elongation factor leukemia unknown. Here we report the cryo-EM structure of (NELF). Such in vitro studies indicate that release of paused Pol Dot1L bound to its H2BK120ub nucleosome substrate. We II is triggered by dissociation of NELF from Pol II, however, capture Dot1L bound to the nucleosome with its active site demonstrating the validity of this model in vivo has been positioned above H3K79, in a state poised for methylation. Dot1L challenging. Here, using acute and rapid depletion of NELF and binds to the nucleosome disk face at the edge of the nucleosome precision run-on sequencing (PRO-seq), we find that the NELF- acidic patch, making critical contacts through variant arginine Pol II complex is paused at the entrance of the +1 nucleosome (1st anchors in a Dot1L-nucleosome interaction loop. Our structure pause), and upon NELF loss, Pol II fails to enter gene bodies further reveals that Dot1L interacts directly with ubiquitin through stopping instead around the +1 nucleosomal dyad-associated hydrophobic interactions that drive the activation of Dot1L. This regions (2nd pause). Furthermore, the transition of paused Pol II structure paves the way for targeted studies of Dot1L methylation to 2nd pause sites upon NELF loss appears to be independent of by providing the tools to determine the ubiquitin-dependent P-TEFb/SEC. Using this in vivo system, we show that NELF loss activity of Dot1L in normal and leukemia cells. impairs recruitment of cap-binding complex, and concomitantly, results in aberrant accumulation of the 5’ RNA exonuclease XRN2 Email: catson4@email.unc.edu at promoters. The heat shock response, where most genes become more paused and a few genes are rapidly induced, remains robust in the absence of NELF. Our results demonstrate that in vivo, NELF is not responsible for the overall phenomena of promoter- proximal pausing but has a critical role in establishing the site of pausing to enable a 5'-end cap protection checkpoint for Pol II. Email: yuki.aoi@northwestern.edu Poster Abstracts

5 6 Oxidative Stress-induced Global Stalling of RNA Polymerase Characterizing RNA Aptamers as tools to dissect the II distinct molecular functions of HSF1 Nitika Badjatia, Matthew J. Rossi, Alain R. Bataille, William Lina Bagepalli1, Fabiana Duarte2, John T. Lis K.M. Lai, B. Franklin Pugh Cornell University, Department of Molecular Biology and Center for Eukaryotic Gene Regulation, Department of Genetics1, Harvard University2 Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA The Heat Shock Response is a highly conserved protective mechanism that is regulated at the transcriptional level by Transcriptional plasticity in response to environmental stimuli is transcription factors called Heat Shock Factors (HSFs). When essential in order for cells to adapt and survive in changing activated by high temperatures or proteotoxic stress, HSFs conditions. Acute environmental stress reprograms genomes, strongly induce the expression of Heat Shock genes (HS genes), which is manifested by a change in protein-DNA interactions that encode Heat Shock Proteins (HSPs). Of the family of HSFs genome-wide. These changes can be global (affecting most genes) in mammals, HSF1 is the functional homolog of the single HSF whereas others may be gene specific. We used acute exposure to in yeast and fruit fly. HSF1 consists of three major domains; the oxidative stress (0.3 mM H2O2 for 6 min) to probe genome-wide DNA Binding Domain (DBD), Trimerization Domain (TD) and binding events related to gene regulation in the budding yeast the Trans-Activation Domain (TAD) which function to Saccharomyces cerevisiae. We found that a major consequence of coordinate HSF1’s ability to bind to its target DNA elements oxidative stress is accumulation of RNA polymerase (Pol) II near and trans-activate HS genes under HS conditions. To elucidate the 5’ ends of genes. We term this phenomenon stalling, as no the mechanistic roles of each domain in living cells, we are molecular relationship with similar events like pausing are using the novel approach of blocking domains with RNA implied. The general transcription factors and promoter-proximal aptamers. RNA aptamers are short, single stranded RNAs that phosphorylation state of serine5 and serine7 of Pol II CTD bind specifically and with high affinity to their selected target, remained mostly unaffected, indicating that this Pol II stalling was whether it is a protein domain, transcription factor or small occurring downstream of these events. In contrast, elongation molecule. We have thousands of selected candidate aptamers factors that associate with Pol II downstream, loaded at their for HSF1, however, we needed to identify those that have normal position along gene bodies; but accumulated to an extent domain specific binding in order to test a domain specific effect. consistent with the underlying stalled Pol II. Thus, a failure to load Initially, to identify the binding affinities of the selected RNA these elongation factors could not explain the stalling. Depletion Aptamers, we used Electrophoretic Mobility Shift Assays of Spt4 or Spt5, partially phenocopied Pol II stalling, suggesting (EMSA). It appears that the initial selection yielded many high that the stalling might be related to down-regulation of Spt4/5. affinity aptamers, however this method of characterization does This study suggests that Pol II normally passes through a Spt4/5- not identify where the aptamers bind HSF1. Using a higher directed elongation checkpoint that becomes rate- limiting during resolution, biochemical, UV crosslinking approach we have oxidative stress. This may provide a means by which genes are identified that many of the selected aptamers bind to the DBD- globally down- regulated post-initiation during acute stress. TD portion of HSF1. To test whether these aptamers inhibit DNA binding, we performed competition assays with a Heat Email: nxb30@psu.edu Shock Element (HSE) and found at least one candidate aptamer that inhibits the DNA binding capacity of HSF1. Future work includes expressing few selected inhibitory aptamers in mammalian cells to identify the extent to which they inhibit HSF1 induced transcription in vivo. We will measure the primary effects of aptamer expression on genome- wide transcription using Precision Run-On sequencing (PRO-seq) which allows for base-pair resolution mapping of actively transcribing RNA Pol II and measure the effect of aptamer expression on HSF1 binding using CUT&RUN. HSF1 Aptamers have the potential to delineate the domain-specific mechanisms by which HSF1 functions. Email: lrb93@cornell.edu Poster Abstracts
7 8 RNA Polymerase II Analysis in Ssu72 Using Crosslinking Human cytomegalovirus IE2 differentially modulates Mass Spectrometry transcription of select viral transcription units during late infection Dominique A. Baldwin, Jose F. Victorino, Amber L. Mosley Christopher B. Balla, Ming Lia,b, Kyle A. Nilsonc, Jeffery L. Department of Biochemistry and Molecular Biology, Indiana Meierb, and David H. Pricea University School of Medicine, Indianapolis, Indiana 46202 a Department of Biochemistry, University of Iowa b RNA polymerase II (RNAPII) is the protein complex responsible Department of internal Medicine, University of Iowa Veterans for transcribing DNA into messenger RNA. For the regulation of Affairs Health Care System c RNAPII, Rpb1, the largest subunit of RNAPII, has a C-terminal Department of Biochemistry and Molecular Biology, Penn State domain (CTD) which contains a repeating heptad of amino acids. This repeat is dynamically phosphorylated to modulate protein- Human cytomegalovirus (HCMV) is the leading infectious cause protein interactions and activity. Properly conducted transcription of birth defects in the United States and presents a significant is necessary for the survival of all living organisms. Perturbations threat to the health of immunocompromised individuals. HCMV within transcription can lead to a host of complications that can utilizes the host cell RNA polymerase II (Pol II) transcriptional contribute to diseases. Because of this, studying RNAPII apparatus for its gene expression. The HCMV major immediate- transcription and its regulation can prove to be useful in the early (MIE) protein, IE2 p86, is required for HCMV replication elucidation of many disease states. and controls transcription of the viral genome. Its expression is regulated through a negative feedback loop that involves IE2 In an effort to uncover more information about transcription binding to a cis-repression sequence (crs) located between the termination, we chose to conduct a comparative analysis between MIE promoter’s TATA box and transcription start site. Two Ssu72-2 temperature sensitive mutant and wildtype smaller isoforms of IE2, p40 and p60, both contain the DNA Saccharomyces cerevisiae strains. Ssu72 is a RNAPII CTD binding domain of p86 and are expressed from downstream phosphatase, as well as a subunit of the cleavage and promoters late in infection. To determine if the IE2 proteins polyadenylation factor (CPF). Previous work has provided regulate transcription of other viral genes in late infection, we evidence that the Ssu72-2 mutation causes differing activity and adopted a new approach using a degradation tag to quickly genomic density of RNAPII. Our hypothesis is that perturbation eliminate IE2 and determined the effect on the position and level of Ssu72 activity will lead to differing composition of RNAPII of nascent transcripts across the viral genome using precision subunits and interactions with other transcription regulatory nuclear run-on sequencing (PRO-Seq). Tagged IE2 proteins were partners. In order to define changes in protein- protein interactions eliminated over a 6 h timeframe in human foreskin fibroblasts within RNAPII or interactions between regulatory partners and infected for 96 h with HCMV. Depletion of the lE2 proteins RNAPII, we employed crosslinking mass spectrometry (XL-MS) greatly increased transcription of the IE1 and IE2 genes. This was with disuccinimidyl sulfoxide (DSSO). accompanied by a pronounced decrease in MIE enhancer transcription and both sense and antisense transcription of the DSSO is a MS-cleavable crosslinker that attaches closely upstream UL genes, UL128-UL145. RT-qPCR corroborated these positioned proteins together to allows for co-analysis of findings. Promoter-proximal read density at the upstream UL interacting proteins. Our preliminary XLMS data suggests promoters was reduced or abolished. The loss of IE2 also affected additional RNAPII protein-protein interactions in the Ssu72-2 several distally located transcription units. Promoter-proximal strain, compared to wildtype, which could suggest mechanisms of read density was increased at the RNA 5.0 and US29 promoters compensation at the site of transcription. Additional XL-MS and decreased at many other promoters, suggesting IE2 repressor analysis with an optimized methodology are required to better and activator functions, respectively. In several regions, IE2 characterize the preliminary protein interactions defined by appears to cause bidirectional accumulation of engaged crosslinks. polymerases that is lost upon depletion of IE2. These apparent barriers to elongation coincide with putative IE2 binding sites. Email: dombaldw@iu.edu These data affirm a role for IE2 in regulating transcription of the MIE promoter and reveal other novel functions for IE2 in controlling transcription across the HCMV genome. Email: christopher-ball-1@uiowa.edu Poster Abstracts
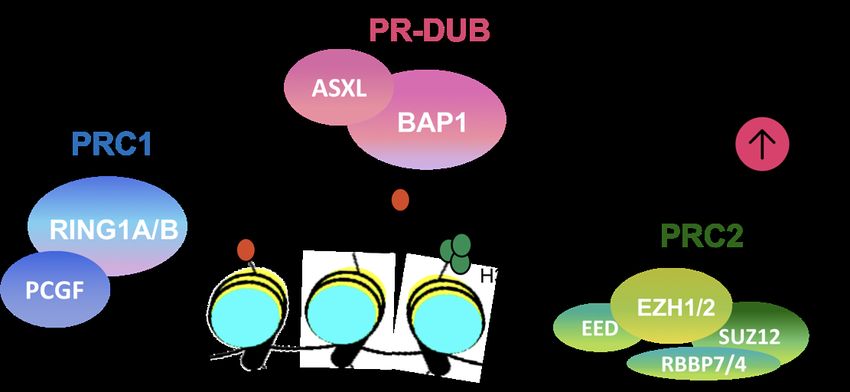
9 10 Interplay of YY1 and CtBP in the recruitment of PcG Regional control of Polycomb Repressive Complexes by long complexes to DNA. noncoding RNAs and CpG island DNA Abigail Avila, Madeline Derleth, Amber Qureshi, Frank Keean C.A. Braceros, Megan D. Schertzer, J. Mauro Calabrese Wilkinson, and Arindam Basu Polycomb Repressive Complexes (PRCs) are highly conserved Recruitment of Polycomb Group (PcG) proteins to DNA is vital chromatin-modifying enzymes required for embryonic for normal development. PcG proteins regulate expression of development, stem cell differentiation, and adult tissue homeotic genes that are essential for axial body patterning during homeostasis. The two major forms of PRCs, PRC1 and PRC2, development. Earlier work showed that YAF2 is responsible for deposit repressive histone marks and organize chromatin to PcG recruitment to DNA, which is mediated by YY1 DNA promote gene silencing. Across the genome, PRCs occupy broad binding. Our goal is to understand the role of CtBP, YAF2 and domains in a non-uniform fashion. Though much remains to be YY1 in context of recruitment of Polycomb complexes to DNA. discovered about how PRCs target specific regions of the genome Using a co-immunoprecipitation (CoIP) approach, different as large domains, increasing evidence suggests that long protein-protein interactions are studied using protein-specific noncoding RNAs (lncRNAs) play a big role. This is best antibodies. This approach will allow us to recognize binding exemplified on the inactive X chromosome in female mammals, interactions between specific proteins. Moreover, our preliminary where the lncRNA Xist recruits and spreads PRCs to silence the results show that CtBP might interact directly with the C- terminal entire chromosome. In addition to Xist, a handful of other end of YY1 to recruit PcG proteins. CtBP represses transcription lncRNAs, including Airn and Kcnq1ot1, recruit PRCs for gene in a histone deacetylase-dependent or -independent manner, repression within their respective silenced domains. We recently depending on the promoter context. Based on this information, we found evidence to suggest that Xist, Airn, and Kcnq1ot1 induce are investigating the modulation of transcriptional activity of YY1 the spread of PRCs from specific CpG islands (CGIs) that bind upon interaction with CtBP using luciferase-based assays. In PRCs autonomously, independent of the lncRNA. Furthermore, conclusion, our studies will provide information regarding the in the Airn target region, we found that a subset of CGIs is interplay between CtBP, YAF2, and YY1 and their overall frequently in spatial proximity to the lncRNA gene locus, and that relationship in recruitment of PcG proteins to DNA. this proximity is enhanced by the presence of Airn. Lastly, prior works have shown that PRCs are recruited to CGIs by a number Email: aub54@psu.edu of accessory factors, and that DNA methylation blocks this recruitment. Taken together, these data suggest that genome architecture, CGIs, and the factors that recruit PRCs to them control the spread of PRCs induced by lncRNAs on a region-by- region basis. We are now in the process of testing this hypothesis through convergent approaches. Specifically, using mouse trophoblast stem cells as our model system, we will map chromatin conformations, profile CGI methylation, and identify factors that recruit PRCs to CGIs in the Xist, Airn, and Kcnq1ot1 target regions. We aim to rigorously vet a new model that explains how DNA elements work in concert with lncRNAs to control the regional spread of PRCs over chromatin. Our results may suggest new and translationally relevant ways to selectively reactivate subsets of target genes in complex PRC- and lncRNA-silenced domains. The principles we identify from the interaction between lncRNAs and CGIs should apply to other regulatory elements, such as enhancers and promoters. Email: keeanb@email.unc.edu Poster Abstracts
11 12 Dynamic Nucleosome Unwrapping by SWI/SNF Dynamics of H1 During Transcription Factor Binding Remodelers Facilitate Transcription- Factor Binding and Regulation of H1 Function by H1 PTMs Sandipan Brahma1 and Steven Henikoff1,2 Nathaniel L. Burge, Ziyong Z. Hong, Jenna L. Thuma, Jennifer J. Otteson, Michael G. Poirier 1 Basic Sciences Division, Fred Hutchinson Cancer Research Histone H1 is integral for compaction of chromatin into higher Center, Seattle, WA 98109, USA order structures and regulates the binding of transcription 2 Howard Hughes Medical Institute, USA factors (TF) and other proteins to chromatin. H1 binding to nucleosomes reduces accessibility of DNA to TFs by reducing Gene regulation requires the controlled access of DNA DNA unwrapping from the histone octamer core. The dynamics sequence-specific transcription factors (TFs) to their sites in a of H1 during a TF binding event, however, is not well chromatin landscape dominated by nucleosomes. Nucleosomes understood. We have developed FRET constructs with are refractory to factor-binding, and must be removed from fluorophores on the DNA, histone octamer, and H1 that allow regulatory regions for most TFs to gain access. However, us to monitor nucleosome dynamics, H1 dynamics, and TF regulatory elements such as promoters and enhancers are not binding. Measuring FRET between H1 and either the histone always nucleosome-free, and in fact, TFs and nucleosomes octamer or linker DNA, we observed H1 dynamics during TF compete for de novo binding to newly- replicated DNA. Then binding. Our data, in combination with previous published how do TFs overcome the nucleosome barrier? Our study results, suggests H1 maintains contacts between the suggests that ATP-dependent SWI/SNF family remodelers nucleosome dyad and linker DNA while the DNA unwraps generate unwrapped nucleosomal intermediates at promoters during TF binding. We hypothesize the globular domain of H1 and enhancers to expose TF-binding sites and facilitate TF remains bound to the dyad while the ~100 amino acid long, binding. We have recently developed a novel genome-wide disordered C terminal maintains contacts to the linker DNA and approach for mapping protein co-occupancies in single particles accommodates unwrapping. Work is in progress using by combining CUT&RUN which is an in situ immuno-targeted additional FRET constructs to investigate this model. nuclease cleavage and release method, with native ChIP-seq (CUT&RUN.ChIP). We applied this approach to directly profile Using the FRET constructs above, we are also investigating two co-occupancy of remodeling complexes and TFs with H1 PTMs hypothesized to affect chromatin condensation using nucleosomal and subnucleosomal histones genome-wide. We fully synthetic H1 containing either phosphorylation in the C find that the essential RSC nucleosome-remodeling complex in terminal domain or citrullination in the globular domain. These yeast is associated with partially-unwrapped nucleosomes ongoing studies provide insight into the dynamics of H1 during containing active histone marks at promoters where a dynamic TF binding, the role of H1 PTMs in regulating H1 binding and micrococcal nuclease (MNase)-sensitive nucleosome (often function, and regulation of transcription. described as “fragile” nucleosomes) is found over binding sites for general regulatory TFs (GRFs). The GRFs Abf1 and Reb1 Email: burge.71@osu.edu also bind to partially-unwrapped nucleosomes at these promoters but not at other promoters, suggesting that RSC-remodeling causes nucleosomes to unravel and facilitates binding of GRFs. Similarly, we find that in mouse embryonic stem cells (mESC), the SWI/SNF remodeling complex BAF is associated with unwrapped nucleosomal intermediates at sites for the binding of pluripotency TFs (such as Oct4, Sox2, Nanog, and Klf4). Our study reveals the interplay between remodelers, unwrapped nucleosomes, and TFs that sets up the dynamic chromatin landscape at promoters and enhancers. Email: sbrahma@fredhutch.org Poster Abstracts
13 14 Chromatin digestion by the chemotherapeutic agent Effect of pericentric satellite RNA on nuclear protein Bleomycin produces nucleosome & TF footprinting patterns distribution and cell division similar to MNase Dawn Carone Joshua Stolz, Ronak Dave, Benjamin Carone Swarthmore College Rowan University, Glassboro, NJ Within the pericentric regions of human chromosomes reside large Bleomycin (BLM), a glycopeptide antibiotic commonly used in arrays of tandemly repeated satellite sequences. Expression of the chemotherapeutic treatments, has been shown to produce single human pericentric satellite HSATII) is normally prevented by and double stranded DNA breaks. Subsequent analysis of DNA extensive heterochromatin-mediated silencing within pericentric fragmentation patterns has demonstrated preferential digestion of regions in normal cells, yet in cancer cells, HSATII RNA is chromatin in the TSS of active genes and the ability to produce aberrantly expressed and accumulates in large nuclear foci. nucleosome-sized fragments within intact chromatin. Expression and aggregation of HSATII RNA in cancer cells is Nucleosome positioning plays a critical role in the regulation of concomitant with recruitment and sequestration of key chromatin gene activation. Currently, micrococcal nuclease (MNase) is used regulatory proteins including MeCP2, which binds to HSATII as the standard for mapping the position of nucleosomes in the transcripts. While HSATII expression has been observed in a genome. In order to identify whether BLM can be used as an wide variety of cancer cell lines and tissues, the effect of its effective nucleosome-mapping agent, BLM was used to digest misregulation within the nuclear environment is unknown. Due to chromatin in S. cerevisiae, followed by Next Generation both its localization within pericentric regions and the Sequencing of paired-end DNA fragments. Our results observations that HSATII RNA accumulates in cis and binds key demonstrate comparable DNA fragmentation patterns for both chromatin regulatory proteins, we tested the effect of stable nucleosomes as well as other DNA-protein interactions and expression of HSATII RNA within cells that do not normally furthermore explain the propensity for BLM to digest within the express HSATII. Stable ectopic expression of HSATII leads to promoters of active genes. Finally, we show that BLM can be used focal accumulation of HSATII RNA in cis and recruitment of to identify genome-wide nucleosome and Transcritption factor MeCP2 to nuclear HSATII RNA bodies. Further, long-term footprints as an effective alternative for MNase and additionally ectopic expression of HSATII RNA leads to cell division defects lacks the strong sequence biases of MNase digestion. including lagging chromosomes, chromatin bridges, and other chromatin defects. Email: carone@rowan.edu Email: dcarone1@swarthmore.edu Poster Abstracts
15 16 Characterization of SWI/SNF complex in response to Hypoxia Retroviral Gag Proteins Interact with Chromatin-Associated Proteins Tomali Chakravarty1, Kathleen Tran1, Dinesh Dhamecha2, Jyothi Menon2 and Arnob Dutta1 Jordan Chang1, Kevin Tuffy1, Breanna Rice1, Rebecca Kaddis Maldonado1, Alan Cochrane3, Leslie Parent1,2 Department of 1Department of Cell and Molecular Biology, 2College of Medicine1 and Microbiology & Immunology2, Penn State Pharmacy, University of Rhode Island Kingston, Rhode Island- College of Medicine Department of Molecular Genetics3, 02881, USA University of Toronto Chromatin remodeling is one of the mechanisms essential in Retroviruses possess a single-stranded RNA genome that must dynamic regulation of gene expression. In eukaryotic cells, this is first be reverse transcribed to form a proviral DNA intermediate. performed by a number of different proteins/protein complexes. Upon integration of the proviral DNA into the host cell SWI/SNF is one such chromatin remodeling complex that was first chromosome, retroviruses utilize host transcription and translation identified in yeast. Studies have shown that about 20% cancers in machinery to produce its own viral protein. All retroviruses, such humans harbor mutations in SW1/SNF, with Arid1a being among as Rous sarcoma virus (RSV) and human immunodeficiency virus most commonly mutated subunits of this complex. Since the (HIV), encode three conserved genes: gag, pol, and env. The gag microenvironment of core of a tumor is hypoxic, and given the role encodes the Gag polyprotein that selects the unspliced genomic of SWI/SNF in regulating stress responsive genes, we wanted to RNA for packaging into new virus particles. It was previously investigate the role and composition of SWI/SNF under hypoxia thought that retroviral Gag proteins functioned only in the and upon loss of Arid1a. This was achieved by characterizing the cytoplasm. However, using confocal microscopy, we observed composition of SWI/SNF in colorectal cancer cells HCT116 (WT) that Gag proteins of RSV and HIV-1 form discreet nuclear foci. and cells lacking Arid1a (mutant) by growing them as Subnuclear fractionation experiments revealed that RSV and HIV monocultures in 1% O2. Additionally, in order to truly replicate Gag localize to the both euchromatin and heterochromatin the tumor microenvironment, we used tumourspheres formed from fractions. This led us to question the role of chromatin association these cells. We noticed alterations in composition of SWI/SNF of retroviral Gag proteins in viral replication. Because RSV and complex when cells were subjected to hypoxia, along with HIV-1 undergo the same type-C virus particle assembly pathway, changes in subcellular localization of some subunits of the we sought to compare and contrast the function of both Gag complex. Furthermore, we have identified subunits of SWI/SNF proteins in this study. that are critical for survival under hypoxia specifically on loss of Arid1a. A better understanding of these changing dynamics of Previous studies in our laboratory demonstrated RSV Gag nuclear SWI/SNF subunits will have significant potential in development trafficking is important for efficient genomic RNA packaging. To of therapeutic interventions. determine whether we could visualize the association of Gag with unspliced vRNA in the nucleus, we employed two independent Email: tchakravarty@uri.edu methods, RNA fluorescent in situ hybridization (FISH) and the MS2 aptamer system, to label vRNA. Co-localization of RSV and HIV Gag with their respective vRNAs was detected in both fixed and living cells. Using live-cell imaging, we observed RSV Gag:vRNA complexes at transcription sites that subsequently trafficked through the nuclear envelope into the cytoplasm. To identify chromatin-associated factors that interact with RSV and HIV Gag proteins, mass spectrometry analysis of affinity purifications was performed. Gene ontology was used to functionally characterize the identified proteins. Among the nuclear proteins identified, we observed a statistically significant enrichment in transcription-related and RNA processing proteins, suggesting Gag modulates transcription, splicing, or RNA processing during its capture of nascent vRNAs. Taken together, we hypothesize that HIV-1 and RSV Gag both enter the nucleus and interact with chromatin-associated host factors to select the unspliced vRNA for packaging. Email: jchang3@pennstatehealth.psu.edu Poster Abstracts
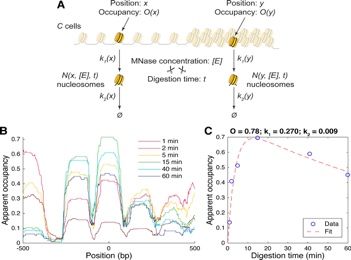
17 18 Chromatin insulator-mediated epigenetic regulation of Quantitative measurement of nucleosome occupancy and neuronal maturation DNA accessibility Dahong Chen, Cameron Palmer, Elissa P. Lei Răzvan V. Chereji1, Terri D. Bryson2, Steven Henikoff2 The nervous system undergoes dramatic remodeling to become 1NICHD, National Institutes of Health, Bethesda, MD, USA, fully mature. This conserved process consists of precisely 2Fred Hutchinson Cancer Research Center, Seattle, WA, USA. regulated degeneration of juvenile-specific neurons and projections, followed by regeneration of adult-specific neuronal MNase is typically used to digest free DNA in order to map projections. Many transcription factors have been identified as nucleosome positions. Unfortunately, MNase has a strong DNA essential for proper neuronal remodeling, but their actions sequence specificity, and different levels of digestion releases underlying neuronal maturation at the chromatin level remain different sets of nucleosomes [1,2]. Mild digestions release from poorly understood. chromatin the more accessible nucleosomes, while extensive digestions destroy the former ones and release the less accessible Chromatin insulators are DNA-protein complexes that play a nucleosomes. Recently, we have developed a new nucleosome central role in the regulation of chromatin 3D structure, thus they mapping method that does not use MNase, and eliminates the are prime candidates for controlling transcriptional changes during biases introduced by this nuclease [3]. Nevertheless, MNase-seq development. We recently found that loss of a conserved insulator remains the most used method of mapping nucleosomes, although protein CTCF in Drosophila led to enlarged adult brains that its results are not quantitative and are strongly affected by the level retained larval-specific neurons, indicating incomplete of digestion. Using a rigorous mathematical treatment of all the degeneration. Interestingly, CTCF depletion also rescued steps involved in DNA digestion by nucleases, we have developed behavioral defects and regeneration defects of neurons caused by a quantitative framework to model the nucleosome occupancy and depletion of Shep, an RNA-binding protein essential for neuronal DNA accessibility genome- wide, at the single-nucleosome level. regeneration that also inhibits a distinct class of chromatin We show that the coverage obtained in traditional MNase- seq by insulator. Together these results suggest that Shep antagonizes stacking the undigested DNA fragments obtained at any level of CTCF to promote neuronal regeneration during maturation. RNA- digestion is not proportional to the real nucleosome occupancy, seq analysis was further performed with fluorescence-sorted and therefore is an inaccurate measure of the fraction of cells neurons and showed that CTCF depletion rescued expression of containing a nucleosome at a given position. Using a series of 60 among 888 genes that were dysregulated upon Shep depletion. digestions, we determine the real fraction of cells containing a Subsequent genetic and behavioral assays indicated that these nucleosome at each position, DNA accessibility, and the rates of rescued genes played key roles in neuronal regeneration, MNase digestion corresponding to each nucleosome in confirming that Shep antagonizes CTCF to regulate neuronal Drosophila. maturation. [1] Chereji RV et al. - Nucleic Acids Research 44, 1036 (2016) [2] Chereji RV et al. - Molecular Cell 65, 565 (2017) To specifically monitor transcription, nascent RNA-seq in CNS- [3] Chereji RV et al. - Genome Biology 19, 19 (2018) derived BG3 cells was also performed and our results indicated that CTCF depletion rescued transcription of 1,154 among 3,998 genes that were transcriptionally dysregulated by Shep depletion. Subsequent ChIP-seq analyses indicated that these rescued genes are highly enriched for chromatin binding targets of Shep and CTCF, but depleted for CP190 targets, an essential cofactor of the CTCF insulator protein. We are in the process of testing the hypothesis that Shep antagonizes the CTCF insulator by restricting CP190 recruitment. Notably, we observed that the mouse Shep ortholog, RBMS1, has enriched expression primarily in maturing Figure: (A) Nucleosomes release by MNase depends on the retinal ganglion cells. We are currently investigating whether fraction of cells that contain a nucleosome (occupancy, ), DNA RBMS1 antagonism of CTCF during neuronal maturation is accessibility to MNase (characterized by rate #), and the rate of conserved in mammals, using the mouse retina as our model nucleosome decay due to MNase overdigestion ( $). (B) Examples system. By providing the first report of regulation of insulator of apparent occupancy profiles obtained for different levels of function during neuronal remodeling, our results significantly digestion and (C) the fit obtained using the kinetic theory of enrich our understanding of molecular mechanisms underlying chromatin digestion, which provides us with the real occupancy of neuronal maturation. the given nucleosome. Email: dahong.chen@nih.gov Email: razvan.chereji@nih.gov Poster Abstracts
19 20 Interactions Between H2A.Z and H1 During Budding Yeast Discriminative histone imputation using transcription (dHIT) Meiosis reveals a complex and dynamic relationship between Lorencia Chigweshe, Amy MacQueen*, Scott Holmes* transcription and histone modification Zhong Wang, Alexandra G. Chivu, Lauren A. Choate, Edward J. *Wesleyan University, Department of Molecular Biology and Rice, Tinyi Chu, John T. Lis, and Charles G. Danko Biochemistry Cornell University & Baker Institute for Animal Health, Ithaca, Statement of the Problem/Background: New York Meiosis in Saccharomyces cerevisiae is associated with unique chromosome choreography and chromatin reorganization. In Transcription by RNA polymerase II (Pol II) has a complex and vegetative cells, the histone variant H2A.Z, encoded by the HTZ1 dynamic relationship with post- translational modifications to core gene, has a significant role in gene expression, DNA repair, histones. Decades of work using tools like chromatin chromosome segregation and maintenance of euchromatin- immunoprecipitation (ChIP) have described numerous heterochromatin boundaries, however, its function in meiosis is correlations between histone modifications and transcriptionally unclear. H1, the ‘linker’ histone encoded in budding yeast by the active or quiescent regions of the genome. These correlations are HHO1 gene has a role in global gene repression and genome often interpreted as evidence that histone modifications play a compaction. Published results demonstrate that H1 is not required causal role in regulating transcription by Pol II. However, while for meiosis, however, H1 was found to promote early meiotic gene histone modifications are undoubtedly critical for a variety of repression. In addition, H1 was found to promote genome cellular processes, whether histone modifications have a direct compaction in late meiosis. Interestingly, studies in our lab found role in regulating transcription remains largely unknown. that deletion of the HHO1 gene in vegetative cells knocked out for the HTZ1 gene rescues some of the defects associated with the loss Here we report a new machine learning tool that quantitively of H2A.Z. Based on these observations, it is clear there is a genetic describes the relationship between histone modifications and interaction between H2A.Z and H1 histones in the regulation of transcription in mammalian cells. Our strategy, called chromatin structure. Nonetheless, the mechanisms behind the discriminative histone imputation using transcription (dHIT), uses interactions between H2A.Z and H1 are currently unknown. a support vector regression classifier to predict the abundance of histone marks using PRO-seq data as input. We trained dHIT to Research Question/Hypothesis: impute the level of 10 different histone modifications using PRO- Given that the role of H2A.Z in meiosis is not fully defined and a seq data in K562 cells (H3K27ac, H3K122ac, H3K9ac, genetic collaboration has been observed between H2A.Z and H1, H3K4me1, H3K4me2, H3K4me3, H3K36me3, H3K27me3, our work aims to investigate the interactions between H2A.Z and H4K20me1, H3K9me3). We found that dHIT predicts active H1 during budding yeast meiosis. histone modifications with an accuracy that approaches data collected from independent ChIP-seq experiments in cell types Results/Summary of the Investigation: that were held out during model training (e.g., Pearson Our findings show that H2A.Z is not required for wild type levels correlations: H3K4me1: 0.69, H3K4me2: 0.85). At individual of sporulation. Strains lacking H2A.Z have reduced spore viability regulatory loci, dHIT described a rich and complex relationship and have recombination efficiencies that are not significantly between the location of Pol II and the location of nucleosomes that altered from those of the wild type strain. In agreement with carry active histone marks. Finally, dHIT also identified broad published data, we found that cells lacking H1 have wild type domains of H3K27me3, the PRC2-dependent mark associated levels of sporulation efficiency and spore viability. Moreover, the with transcriptional repression, with remarkably high accuracy. hho1∆ homozygote exhibits no changes in recombination Our findings describe a complex correlation structure that hints at efficiency. Interestingly, the loss of H1 can suppress some of the rich and biologically interpretable relationships between Pol II and decrease in spore viability seen in the htz1∆ strains. Intriguingly, histone modifications. the loss of H2A.Z and H1 compromised efficiency of synaptonemal complex assembly. The correlation between active histone modifications and transcription implies that either histone modifications play a Interpretation/Conclusion of the Investigation: causal role in regulating transcription or that the disruption of In summary, our findings suggest H2A.Z has a role in proper chromatin during transcription could influence the placement of execution of meiosis and together with H1 regulates meiotic active histone marks. To determine the direction of causality, we chromosome structure. Our future work will aim to understand perturbed transcription and checked for the redistribution of how H2A.Z and H1 collaborate to influence the synaptonemal histone marks. We used triptolide, an epoxide that impedes complex structure assembly and integrity at a molecular level. TFIIH’s capacity to unwind the DNA, to block transcription initiation. We verified this blockage by PRO-seq in parallel with This work was supported by Wesleyan University ChIP-seq measurements of five active histone marks: H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K79me3, and one Email: lchigweshe@wesleyan.edu repressive mark: H3K27me3. Surprisingly, preliminary analysis showed that triptolide decreased signal for all active marks by 1 Poster Abstracts
21 hour of treatment, and redistributed marks along the genome by 4 Cross-species transcription factor binding prediction using hours. This data supports a transcription-centric model in which neural networks the placement of histone modifications is partially a consequence, rather than a cause, of active transcription. Kelly Cochran1, Divyanshi Srivastava1, Akshay Balsubramani2, Anshul Kundaje2, Shaun Mahony1 Email: agc94@cornell.edu 1 Department of Biochemistry & Molecular Biology, Center for Eukaryotic Gene Regulation, Penn State University, University Park, PA 2 Departments of Genetics and Computer Science, Stanford University, Stanford, CA The binding patterns of highly conserved transcription factors (TFs) appear to diverge significantly across closely related species. Experiments measuring protein-DNA binding (such as ChIP-seq) for the same TFs in the same tissues show strikingly few binding sites shared across mammals. This divergence persists even in the conserved regulatory regions for highly homologous genes. However, the DNA binding preferences (motifs) of these TFs appear to be strongly conserved, and the same cohorts of TFs appear to drive regulatory activities similarly in the same tissues across mammalian species. Therefore, despite this binding site divergence, we expect the general features of tissue-specific regulatory architecture to be conserved. Several recently published models have predicted TF binding across cell types based on genomic sequence and various epigenetic assays. Typically, these models are trained and tested on data generated in multiple tissues from a single species. An assumption underlying these methods is that a generalized logic of TF binding learned from training cell types can transfer effectively into a previously unseen cell type. We hypothesize that an analogous form of transfer learning may be possible across species, controlling for cell type. A model capable of predicting TF binding across species could allow us to both investigate binding site divergence and infer TF binding sites in multiple species without the need to perform expensive ChIP-seq experiments. Here we assess the ability of a neural network to perform transfer learning of TF binding logic across species. First, we designed a network that takes in the sequence from a given genomic window and predicts whether a ChIP-seq peak indicating TF binding is present within that window. We used ChIP-seq data from the same cell type, liver, in multiple species. First, we trained this model using the peaks and genomic sequence from one species; then we assessed its performance on data from another species. We quantified the performance differential between models trained and tested within the same species vs. across different species. Next, we modified our standard network architecture to perform training-integrated domain adaptation to discourage the network from learning any species-specific sequence features. Our domain- adaptive model is augmented with a loss gradient reversal layer followed by a secondary output tasked with predicting the origin species of the input genomic window. We hypothesize that these modifications should improve cross-species model predictions by forcing the model to learn a more species-generic representation of binding logic as it trains on single-species data. Poster Abstracts
22 We find that while our standard models are able to generalize How can you mend a broken heart? Investigating SMYD1 across species, we also measure a persistent cross-species gap in lysine methyltransferase substrate selectivity performance. Through investigation of genomic sites differentially predicted by within- and cross-species models, we Eric Cordeiro-Spinetti, Lucas Chan, Joseph Faski, Jens show that this performance gap is driven by false positive Forsberg, Robert Vaughan, Bradley Dickson, Stefan Jovinge, predictions made by cross-species models on sites containing Evan Cornett, Scott Rothbart repeats unique to the test species. We then demonstrate that our domain-adaptive model offers superior performance by Center for Epigenetics, Van Andel Research Institute, Grand decreasing the false positive rate for species-unique repeats. Thus, Rapids, Michigan, USA. our results indicate that transfer learning of TF binding logic across species is possible, but that model training designed to Indispensable for heart formation and function, SMYD1 is an address species-unique sequences is essential to achieving evolutionarily conserved SET domain-containing lysine comparable performance to within-species models. Our work methyltransferase (KMT) expressed by cardiac cells since additionally provides a framework for further investigation into embryogenesis. In mouse models, Smyd1 knockout is embryonic the mechanisms behind TF binding divergence across species. lethal due to impaired ventricular chamber formation, and cardiomyocyte-specific deletion in adults causes cardiac Email: kxc1032@psu.edu hypertrophy and heart failure. Interestingly, a de novo mutation of SMYD1 was reported in a patient with hypertrophic cardiomyopathy. Furthermore, patients diagnosed with end-stage heart failure express high levels of SMYD1. The currently reported substrates for SMYD1 KMT activity have not been directly linked to these cardiac phenotypes. Since the addition of a methyl group to a substrate modifies its interactome, stability, and function, we hypothesized that SMYD1 acts through its KMT activity to regulate proteins involved in heart development and myofibril assembly. To test this hypothesis, we recently developed a functional proteomics platform to reveal the sequence determinants of KMT substrate selectivity using a lysine-oriented peptide library (K-OPL). Screening with recombinant human SMYD1 revealed a preference for basic (K and R) and aromatic residues (F, Y and W) in key positions surrounding a target lysine. Acidic residues (D and E) were unfavorable in any position. Based on this substrate profile, we informatically linked back to human proteins containing optimal sequence composition for SMYD1 activity. Further informatic analysis narrowed in on several proteins known to share similar phenotypes as SMYD1 in the regulation of cardiac function. Data will be presented related to the functional validation of newly identified SMYD1 substrates. Collectively, this work further validates the K-OPL platform as a robust screening tool for substrate identification and reveals key functions for lysine methylation signaling in the regulation of cardiac development and function. Email: eric.spinetti@vai.edu Poster Abstracts
23 24 RSC controls hypertranscription during quiescence exit Probing Chromatin Dynamics With a DNA Origami Nanocaliper Christine Cucinotta, Keean Braceros, and Toshio Tsukiyama Michael A. Darcy1*, Jenny V. Le2 , Kyle Crocker1, Ralf Quiescence is a conserved stage by which cells can reversibly exit the cell cycle for long-term survival. The reversibility of Bundschuh1, Carlos E. Castro2,3 Michael G. Poirier1,2 quiescence is crucial to organismal health, as it is essential for differentiation, tissue regeneration, stem cell renewal, and immune 1Biophysics Graduate Program, 2Department of Physics, and cell activation. Quiescent chromatin is compact, highly MNase- 3Department of Mechanical and Aerospace Engineering, The resistant, and broadly hypoacetylated. Additionally, nucleosome- Ohio State University, Columbus OH 43214. depleted regions (NDRs) are narrow and histone occupancy is increased, resulting in a chromatin environment that is refractory The organization of eukaryotic DNA into nucleosomes and to DNA-templated processes. The mechanisms by which cells chromatin undergoes dynamic structure changes to regulate overcome this severely repressive chromatin environment to exit genome processing, including transcription and DNA repair. quiescence are undefined. Interestingly, we observed widespread transcriptional activation occurring within a few minutes of Critical chromatin rearrangements occur over a wide range of nutrient repletion. Our data show that many of the genes activated distances including the mesoscopic length scale of tens of upon quiescence exit have RNA Polymerase II and the SWI/SNF nanometers. However, there is a lack of methodologies that probe family chromatin remodeling enzyme RSC bound to the 5’-ends changes over this mesoscopic length scale within chromatin. We during quiescence. RSC in quiescent cells primarily localizes to have designed, constructed, and implemented a DNA-based the NDRs of early exit genes and RSC depletion in quiescent cells nanocaliper that probes this mesoscopic length scale. We developed an approach of integrating nucleosomes into our causes NDRs to remain narrow upon exit, indicating that RSC is nanocaliper at two attachment points with over 50% efficiency. poised for rapid NDR restoration during exit. In the absence of Here, we focused on attaching the two DNA ends of the RSC, Pol II remains at the 5’-ends of genes during exit, suggesting nucleosome to the ends of the two nanocaliper arms, so the hinge a role for RSC in enhancing transcription elongation. Together, angle is a readout of the nucleosome end-to-end distance. We these results imply a poised transcriptional program controlled by found that the nanocaliper angle is a sensitive measure of RSC that prepares cells for large-scale rapid responses despite a globally repressive chromatin environment. nucleosome disassembly and can read out transcription factor (TF) binding to its target site within the nucleosome. Furthermore, Email: ccucinot@fredhutch.org fluorescent reporter molecules located between the arms allow us to distinguish between closed and open hinges in ensemble and in solution rather than by electron microscopy. Interestingly, the nanocaliper not only detects TF binding within a bound nucleosome, but it significantly increases the probability of TF occupancy at its site by partially unwrapping the nucleosome. Extending these transmission electron microscopy studies, we report data on the dynamics of hinges as measured by the fluorescent reporters. These studies demonstrate the feasibility of using DNA nanotechnology to both detect and manipulate nucleosome structure, which provides a foundation of future mesoscale studies of nucleosome and chromatin structural dynamics. Email: michaeladarcy@gmail.com Poster Abstracts
25 26 Characterization of the Novel Nucleic Acid Binding Activity A putative condensin loading factor that controls yeast of the Polybromo-1 Bromodomains chromosome III architecture Saumya M. De Silva1, Nicholas J. Schnicker2, Catherine A. Manikarna Dinda1, Ryan D. Fine1, Mikngguang Li1,2, and Musselman3 Jeffrey S. Smith1 1 Department of Chemistry, College of Liberal Arts and 1 Department of Biochemistry and Molecular Genetics, Sciences, The University of Iowa, Iowa City, Iowa. University of Virginia School of Medicine, Charlottesville, VA 2 Protein and Crystallography Facility, Carver College of 22908. 2Department of Laboratory Medicine, Jilin Medical Medicine, The University of Iowa, Iowa City, Iowa. University, 132013, China. 3 Department of Biochemistry, Carver College of Medicine, The Condensin plays an important and evolutionarily conserved role University of Iowa, Iowa City, Iowa in mitotic chromosome condensation, three-dimensional genome DNA in eukaryotic cells is packaged into chromatin, which is organization and regulation of gene expression. Condensin is a made up of repeats of the basic subunit, the nucleosome. Each multi-subunit complex that has natural affinity for the promoters nucleosome is composed of ~147 base pairs of DNA wrapped of highly transcribed genes, and also associates with specific around an octamer of histone proteins that contains two copies of transcription factors. However, a general mechanism for each H2A, H2B, H3, and H4. Modulation of chromatin structure functional loading of the complex onto chromatin remains elusive. is critical for all DNA templated processes and occurs through In a recent study, our lab demonstrated that budding yeast several mechanisms including post-translational modifications condensin and Sir2 (a histone deacetylase) are both recruited to a of histones and remodeling of the nucleosome core. The recombination enhancer (RE) element located on the left arm of remodeling of nucleosomes is mediated by several families of chromosome III that directs proper donor preference of mating- ATP- dependent remodeling complexes. These complexes must type switching. This targeting to the RE is specific for MATa navigate a diverse chromatin landscape such that the proper haploid cells and occurs on the promoter of an embedded small chromatin structure is formed both spatially and temporally. gene of unknown function called RDT1, which appears to function as a locus control region (LCR). Sir2 locally regulates PBAF (Polybromo-associated BRG1 or hBRM-associated transcription of RDT1, while condensin contributes to the 3- factors), is a remodeling complex that belongs to the SWI/SNF dimensional organization of chromosome III, as well as donor family of remodeling complexes. The PBAF complex is preference (BioRxiv, doi: https://doi.org/10.1101/558494). In our distinguished from BAF (Bromo-associated BRG1 or hBRM- current study, we further characterized the mechanism of associated factors) by the presence of three unique subunits, one condensin recruitment to the RDT1 promoter LCR, and uncovered of which is Polybromo-1 (PBRM1). PBRM1 contains six tandem a critical role for a non-meiotic version of the monopolin complex, bromodomains (BDs) and is thought to be important in known as cohibin (Lrs4 and Csm1 subunits), similar to its known nucleosome recognition by the PBAF complex. Bromodomains role in recruiting condensin to the rDNA locus. To test if cohibin are a highly conserved domain of ~110 amino acids, composed functioned more generally in condensin loading, we performed of a bundle of four alpha-helices linked by two loops. Canonical ChIP-seq for genome-wide condensin subunit binding sites in WT BDs associate with acetylated lysines on histone tails and BD1 and lrs4∆ strains. Numerous Brn1 peaks (including RDT1 and the through BD5 of the PBRM1 have been shown to associate with rDNA) were eliminated or significantly reduced by lrs4∆, histone H3 peptides containing single acetylations.1 Recently, consistent with the condensin loader hypothesis. Micro-C XL was our laboratory demonstrated that certain BDs can also associate then used to characterize the general effects of defective condensin with DNA, and have predicted that some of the PBRM1 BDs recruitment on genomic conformation, in a lrs4∆ mutant, or when harbor DNA binding activity.2 From our work with PBRM1 BDs the Brn1 condensin subunit was depleted using an auxin-inducible we found that four of the six BDs associate with DNA and RNA degron system. Alterations were observed on multiple with different affinities. Here, we present the interactions of the chromosomes, though the most severe changes occurred on PBRM1 BDs with both DNA and RNA using gel shift assays, chromosome III, which had significant negative effects on the NMR spectroscopy, and Bio-Layer Interferometry. efficiency of mating-type switching, as well as donor preference. We therefore hypothesize that cohibin (Lrs4/Csm1) is indeed a [1] Chandrasekaran, R. and Thompson, M., 2007. Polybromo-1- condensin loader at the RDT1 promoter region, where it bromodomains bind histone H3 at specific acetyl-lysine establishes chromosome III conformation through a loop extrusion positions. Biochemical and biophysical research mechanism. Experiments to examine a possible role for cohibin in communications, 355(3), pp.661-666. chromosome condensation are ongoing. [2] Morrison, E.A., Sanchez, J.C., Ronan, J.L., Farrell, D.P., Varzavand, K., Johnson, J.K., Gu, B.X., Crabtree, G.R. and Email: md9tp@virginia.edu Musselman, C.A., 2017. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nature communications, 8, p.16080. Email: saumya-desilva@uiowa.edu Poster Abstracts
You can also read

















































