Small Vessel Disease-Related Dementia: An Invalid Neurovascular Coupling? - MDPI
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below

International Journal of
Molecular Sciences
Review
Small Vessel Disease-Related Dementia: An Invalid
Neurovascular Coupling?
Rita Moretti * and Paola Caruso
Neurology Clinic, Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste,
Italy; paolacaruso83@gmail.com
* Correspondence: moretti@units.it
Received: 11 January 2020; Accepted: 4 February 2020; Published: 7 February 2020
Abstract: The arteriosclerosis-dependent alteration of brain perfusion is one of the major determinants
in small vessel disease, since small vessels have a pivotal role in the brain’s autoregulation.
Nevertheless, as far as we know, endothelium distress can potentiate the flow dysregulation
and lead to subcortical vascular dementia that is related to small vessel disease (SVD), also being
defined as subcortical vascular dementia (sVAD), as well as microglia activation, chronic hypoxia and
hypoperfusion, vessel-tone dysregulation, altered astrocytes, and pericytes functioning blood-brain
barrier disruption. The molecular basis of this pathology remains controversial. The apparent
consequence (or a first event, too) is the macroscopic alteration of the neurovascular coupling. Here,
we examined the possible mechanisms that lead a healthy aging process towards subcortical dementia.
We remarked that SVD and white matter abnormalities related to age could be accelerated and
potentiated by different vascular risk factors. Vascular function changes can be heavily influenced by
genetic and epigenetic factors, which are, to the best of our knowledge, mostly unknown. Metabolic
demands, active neurovascular coupling, correct glymphatic process, and adequate oxidative and
inflammatory responses could be bulwarks in defense of the correct aging process; their impairments
lead to a potentially catastrophic and non-reversible condition.
Keywords: small vessel disease; vascular damage; endothelium; neurovascular coupling; inflammation;
oxidative response; redox; brain’s autoregulation
1. Introduction
Cerebral small vessel disease (SVD) primarily distresses the small perforating arteries, being
defined as vessels with less than 50 µm diameters, also defined as “all the vessels within the brain
parenchyma plus the vessels with a diameter less than 500 µm in the leptomeningeal space” supplying
the deep brain structures [1,2]. Nevertheless, general increased arterial stiffness is associated with an
increased white matter lesion burden [3]. Therefore, while the microvasculature is the primary target
of SVD, the contribution of larger arteries should not be immediately discounted. SVD is the most
important and common cause of vascular dementia, leading to 45% of dementia, and it accounts for
about 20–30% of all strokes worldwide, 25% of ischemic (or lacunar strokes). Moreover, it significantly
increases the risk of future stroke [4]. Often, SVD lesions are clinically insidious and they act as “silent”
lesions. Thus, SVD is a dynamic pathology, lesions progress over time, and the long-term outcome and
impact on brain damage vary [5]. In sporadic cerebral SVD, sporadic aging and hypertension are listed
as the most critical risk factors. However, different hereditary forms of cerebral SVD have also been
described [6]. In the latter forms, several pathological changes to the vasculature in small arterioles
(like vascular muscle dysfunction, lipohyalinosis, vascular remodeling, and the deposition of fibrotic
material) have been identified. Venous structures are also affected [7]. These facts are shared in both
forms, with time of onset beng the only difference.
Int. J. Mol. Sci. 2020, 21, 1095; doi:10.3390/ijms21031095 www.mdpi.com/journal/ijms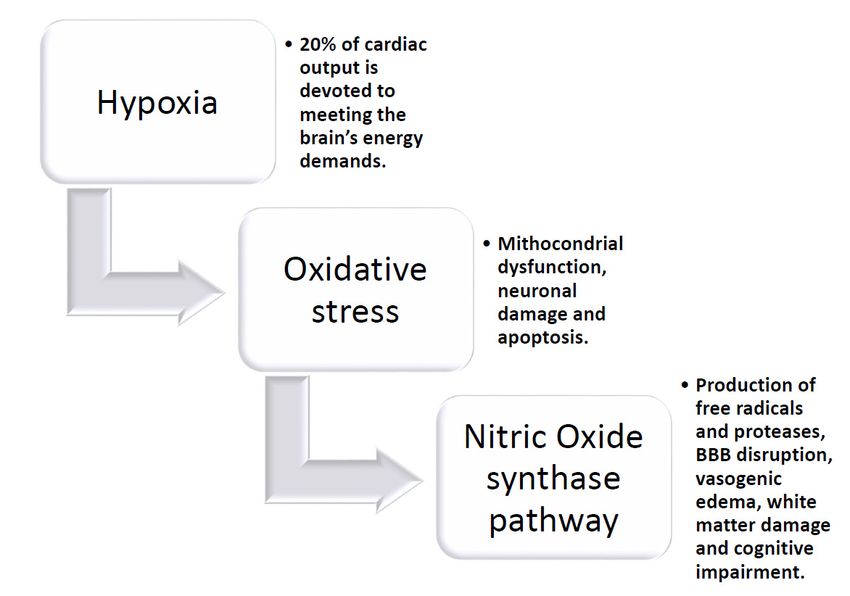
Int. J. Mol. Sci. 2020, 21, 1095 2 of 35
Cerebral amyloid angiopathy (CAA) is a common form of cerebral SVD and it refers to the
deposition of amyloid b-peptide (Ab) in the cerebral leptomeningeal and parenchymal arteries and
arterioles walls. The incidence of CAA increases with age. More often, CAA is related to hemorrhagic
stroke. Additionally, in this case cause, structural variations, such as concentric splitting, loss of smooth
muscle cells, and fibrinoid necrosis, which may increase the propensity for vessel rupture and, thus,
hemorrhage, have been seen [8,9].
2. Vascular Dementia and Small Vessel Disease-Related Dementia
The diagnosis of vascular dementia should be easy due to the temporal correlation between an
acute vascular brain lesion and the onset of cognitive and behavioral problems. Nonetheless, consensus
criteria for vascular cognitive impairment are still under debate, since 1983, when NINDS-AIREN
Criteria had first been written [10,11], and the ICD-10 had been debated [12]. Subsequently, different
validations have been proposed, and many criteria have been written, but the current clinical diagnostic
criteria for vascular dementia are still argued, even from a neuropathological perspective. Nowadays,
we accept the generic definition of genetic vascular dementia (CADASIL or CARASIL), macrovascular
dementia (multi-infarct dementia or strategic infarct dementia), or microvascular dementia (subcortical
vascular dementia or more appropriately, small vessel disease-related dementia) [13–15]. Very recently,
DSM V [16,17] and the STRIVE Consortium (Standards for reporting Vascular changes on Neuroimaging)
conditioned the diagnostic criteria to specific neuroimaging studies [5,18]. In particular, the diagnostic
criteria for the small vessel disease should include, in a conventional MRI, recent subcortical infarcts,
white matter hyperintensities, lacunes, prominent perivascular spaces, and cerebral microbleeds [5,18].
Therefore, we take small vessel disease (SVD) into account, which is the consequence of the different
damages to the small penetrating arteries and arterioles in the pial and lepto-meningeal circulation,
along with penetrating and parenchymal arteries and arterioles, pericytes, capillaries, and venules [19].
SVD prevalence increases exponentially with aging. Around Europe, the prevalence rates of SVD related
dementia, between ages 65–69 to 80+ years, ranged from 2.2 to 16.3% [20–23]. As aforesaid, aging is the
most critical risk factor in developing the small-vessel disease, due to the loss of arterial elasticity, and a
consequent reduction of arterial compliance [24]. The loss of arterial compliance is the major determinant
of the altered autoregulation capacities, which leads to the deep sensitiveness of the brain of SVD
patients to brisk decreases of blood pressure [25–27]. Moreover, apart from the reduction of elasticity, it
should be considered that aging also causes a low-level functioning of the autonomic nervous system,
with direct and endothelium-mediated altered baroreflex activity [28–31]. Pathological expressions of
SVD are the arteriolosclerosis process and cerebral amyloid angiopathy (CAA) [32–35]. After that, even
if debated, SVD could affect the integrity of the medial cholinergic pathway, for the hypoperfusion
preferred localization, in the deep white matter capsule, [36], or, due to the multiple lacunar infarcts,
the basal forebrain cholinergic bundle could be deafferentated from the tubero-mamillary tracts [37,38].
These aspects affect the normally-accurate cerebral flow regulation and they can further disturb
the “retrograde vasodilatation system” with necessary consequences in neurovascular coupling [39].
Cerebral small vessel disease includes a neuroimaging and pathological descriptions, which comprise
different imaging changes in the white matter and subcortical grey matter, including small subcortical
infarct, lacunes, white matter hyperintensities (WMHs), prominent perivascular spaces (PVS), cerebral
microbleeds (CMBs), and atrophy. Moreover, an associated hypoperfusion progression characterized
SVD, causing incomplete ischemia of the deep white matter [7,40–42] accompanied by inflammation,
diffuse rarefaction of myelin sheaths, axonal disruption, and astrocyte gliosis [35]. In small vessel
disease, the occlusion of deep periventricular-draining veins is also evident [43], together with a
disruption of the blood-brain barrier; the two factors together causing a severe leakage of fluid and
plasma cells to potentiate the inflammatory cascade, which seem to happen in the course of chronic
hypoperfusion, by collecting multifactorial causes for white matter alterations [44–46]. Cerebral small
vessel disease is what has been described as “a progressive disease” [35]. Lesions progress over
time, and the long-term outcome and impact on brain damage vary, even not knowing why or how;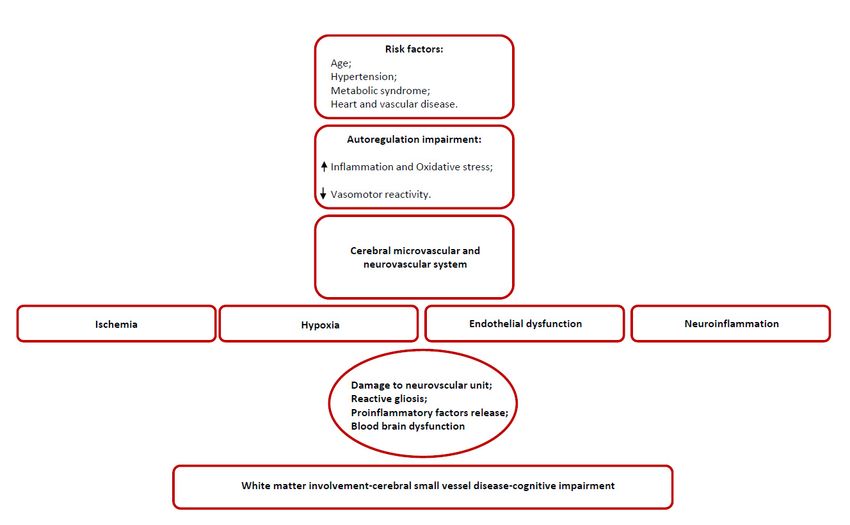
Int. J. Mol. Sci. 2020, 21, 1095 3 of 35
been described as “a progressive disease” [35]. Lesions progress over time, and the long-term
Int. J. Mol. Sci.
outcome and 2020, 21, 1095on
impact brain damage vary, even not knowing why or how; reasonably, it should 3 of 35
be
said that the most rapid and confluent progression of the isolated white matter hyperintensities could
be considered
reasonably, as being
it should the most
be said relevant,
that the to theand
most rapid best of knowledge,
confluent predictor
progression of theofisolated
the fatalwhite
progression
matter
of SVD [47–50]. Of course, the total amount of lacunes and profound white
hyperintensities could be considered as being the most relevant, to the best of knowledge, predictor matter alterations relate
oftothe
thefatal
degree of cognitive
progression impairment
of SVD [47–50]. [51–53].
Of course, Thethepreferred location
total amount of the lesions
of lacunes is placedwhite
and profound along
with the frontal and prefrontal-thalamus-basal forebrain networks, [54,55], directly
matter alterations relate to the degree of cognitive impairment [51–53]. The preferred location of the implying the so-
called cortical-deafferentation. Additionally, lesions due to SVD are specific
lesions is placed along with the frontal and prefrontal-thalamus-basal forebrain networks, [54,55], to the caudate nucleus
(the most
directly precociously
implying affected
the so-called region), the putamen, Additionally,
cortical-deafferentation. insula, precentral gyrus,
lesions due toinferior
SVD are frontal gyrus,
specific to
and middle frontal gyrus. The higher metabolic request of these regions (more
the caudate nucleus (the most precociously affected region), the putamen, insula, precentral gyrus, than 20%) at steady
state than
inferior other
frontal brainand
gyrus, areas fullyfrontal
middle explains the pathology
gyrus. The higher [56–63].
metabolic Onrequest
the other hand,
of these SVD usually
regions (more
than 20%) at steady state than other brain areas fully explains the pathology [56–63]. On the[64,65];
implies a reduced metabolic rate of oxygen (estimated of about 35% in white matter) other
metabolic
hand, SVD incongruity
usually impliesbetween the brain
a reduced oxygen rate
metabolic supply and its consumption
of oxygen (estimated of has aboutbeen35% described
in whitein
SVD, which
matter) determines
[64,65]; metabolicanincongruity
altered neurovascular
between thecoupling and altered
brain oxygen supply vasomotor reactivity [35,66–
and its consumption has
71]. Neuropsychological pattern profiles of dementia that are
been described in SVD, which determines an altered neurovascular coupling and altered related to SVD are related to the
vasomotor
subcortical-cortical
reactivity [35,66–71].loops deafferentation and
Neuropsychological they are
pattern distinguished
profiles of dementia by poor
that executive
are related function,
to SVD poor are
planning, working memory alterations, loss of inhibition, reduced mental flexibility,
related to the subcortical-cortical loops deafferentation and they are distinguished by poor executive multitasking
procedures
function, poorinvalidation, and decrease
planning, working memoryspeed of executive
alterations, process
loss [72–78].reduced
of inhibition, Any specific
mental treatment has
flexibility,
been discovered,
multitasking either as
procedures pathogenicand
invalidation, or highly
decreasestandard
speed recommended
of executive processfor this condition.
[72–78]. Any specific
Insert Figure 1 appr. here:
treatment has been discovered, either as pathogenic or highly standard recommended for this condition.
Insert Figure 1 appr. here:
Figure 1. A synopsis of the possible superimposing factors conditioning the progression of SVD.
Figure 1. A synopsis of the possible superimposing factors conditioning the progression of SVD.
3. Anatomical and Structural Weaknesses in Small Vessel Disease
3. Anatomical and Structural
SVD is considered Weaknesses
to be the in Small Vessel
major contributing Disease
factor or the sole responsible for the “generic
defined” dementia-syndrome worldwide [79]. The small
SVD is considered to be the major contributing factor or the vessels represent its principal
sole responsible fortarget, which
the “generic
include
defined” pial and small penetrating
dementia-syndrome arteries,[79].
worldwide smallThe intra-parenchymal arterioles
small vessels represent (with smooth
its principal muscle
target, which
cells), perivascular spaces, astrocytic endfeet, cerebral capillaries and veins, and
include pial and small penetrating arteries, small intra-parenchymal arterioles (with smooth musclevenules. There is
wide
cells),speculation
perivascularon spaces,
all the structures
astrocytic involved, to establish
endfeet, cerebral a potential
capillaries role inand
and veins, the venules.
development
Thereofis
the chronic ischemic-hypoxic state, which is the final responsibility for the SVD, even if
wide speculation on all the structures involved, to establish a potential role in the development of the the principal
SVD-model is the arterioles damage-based,
chronic ischemic-hypoxic state, which isand the even
finalifresponsibility
we do not know formuch abouteven
the SVD, perivascular spaces.
if the principal
The pathophysiological
SVD-model role ofdamage-based,
is the arterioles PVS, their functionand andeveninteraction withknow
if we do not cerebral
muchmicrocirculation, has
about perivascular
not been established yet. There is a broad consensus that PVS forms a network of spaces around
cerebral microvessels, acting as a canal for fluid transport, the exchange between cerebrospinal fluidInt. J. Mol. Sci. 2020, 21, 1095 4 of 35
(CSF), and interstitial fluid (ISF) and the clearance of catabolites from the brain. The perivascular
compartment contains several cell types, like perivascular macrophages, pial cells, mast cells, nerve
fibers, and collagen fibers [80]. Usually, as arterioles penetrate deeper into the brain, the glial
membrane, the pericyte membrane fuse together and then obliterate the perivascular spaces [80,81],
but it has been proposed that either in humans either in animals, the perivascular space could act as a
brain lymphatic system, also being defined as “para-arteriolar”, “para-venular”, “paravascular”, or
“glymphatic” [82]. This system has many complex functions (further in the review, we will explain
it regarding neurovascular coupling), but it seems likely to exert the drainage work of the brain.
Therefore, modification of this system produces deleterious effects, whose results are an accumulation
of catabolites and toxic substances, together with a pronounced neural starvation [83,84]. In SVD,
this system is invalid; one of the SVD hallmarks is the enlargement and widening of PVS, due to an
obstructive process that is maintained by catabolites, proteins, and cell debris [82]. In small vessel
disease, the occlusion of deep periventricular-draining veins is also evident [43], together with the
disruption of the blood-brain barrier (BBB). All these facts together lead a consequent leakage of fluid
and plasma cells, which eventually might potentiate the perivascular inflammation, and all of the
cascades of the inflammatory/obstructive/stagnation-induced process [44–46,85]. The immobility of
the fluid drainage can support PVS’s role in different diseases: the possible explanation of the PVS
involvement in SVD, is the argued relationship demonstrated between an altered cerebrovascular
reactivity (CVR), which is the change in cerebral blood flow in response to a vaso-active stimulus in
the so-called neurovascular coupling, the found BBB dysfunction, and the correspondent perivascular
inflammation [86]. Therefore, a lacuna should not indicate an enlarged perivascular space, as it is, still
nowdays; it should never be the correspondent of the CSF-filled cavities on brain MRI or residual
lesion of a small hemorrhage [82,87–92]. Nowadays, it should be more appropriate for the definition
“lacuna of presumed vascular origin” to replace the term ”lacuna” [20,93–96].
3.1. Arteriolosclerosis as a Functional Model for SVD
Arterioles are the best studied target for SVD, starting from the pathological process that they
undergo, the arteriolosclerosis. Arteriolosclerosis occurs in two primary histological forms, the
hyperplastic and the hyaline arteriolosclerosis [97,98]. The hyperplastic is the most common lesion,
principally due to the chronic state of hypertension. It begins with the hypertrophy of the smooth
muscle in the media, and it is accompanied by the reduplication of elastic laminae, the growth of new
cells in the intima, and the deposition of collagen, which progressively substitutes the muscle cells
(onion skin arteries) and severely obliterates the lumen [97]. Hyaline sclerosis is another change in
the vessels of hypertensive patients: the vessel wall becomes thickened with collagen [99]. Arterioles
undergo a progressive deposition of hyaline material throughout the entire circumference of the vessel
and which extends through the media [100]. The hyaline material is a consequence of the leakage of the
plasma proteins, mainly the inactive form of complement (C3b) through the endothelium, and also by
an increment of the basement membrane components by the smooth muscle cells [100]. Healthy aging
implies the loss of the Windkessel effect and the loss of arterial elasticity, which reflects an anticipated
and precocious return of the so-called wave reflection. Healthy aging also determines an increase of
the systolic and a decrease of diastolic pressure, with a loss of resting flow effect through the Willis,
which decrements the usual high perfusion pressure towards the most profound small arteries of the
brain [101–103], thus provoking a loss of brain flow autoregulation. Arteriolosclerosis perpetuates the
hypo-perfusion in the profound territories that are irrigated by penetrating arteries.
3.2. Hypoperfusion and Neuroinflammation
It is intriguing enough that chronic cerebral hypoperfusion defeats the traditional and acknowledged
way of the anatomical thinking, with regards to the preponderance of cortical neurons on the other
brain structures. Nowadays, it is well accepted that 10 min. of transient global ischemia in rat-brains
determines a precocious sufferance of the perineural spaces, and then of the white matter, along withInt. J. Mol. Sci. 2020, 21, 1095 5 of 35 the internal and external capsule; one day after the ischemia, oligodendrocytes die [104–106], and the neuronal death occurs several days after the initial damage [107,108]. Moreover, as experimentally demonstrated, ischemia occurs in the brain (rat, mouse, and rabbit) [109,110], it seems evident that there is an induction of significant microglial activation, with significant regional variability [109]. When measured the time of onset, it has been described that microglial activation firstly appears in the hippocampus, but the activation does not last more than 48 h [109]. In the meantime, from 48 to 72 h after the ischemia, there is increased activation of the microglia. It occurs throughout the white matter, and the thalamus (from the second day after up to the fourth day). From the fourth day, the activation occurs through the cortex, protracting until 30 days after the initial ischemia [111]. Besides, microglia tend to retract their branches after ischemia, leading to a reduction in the total length and the total number of microglial processes [112]. The loss of blood flow in the peri-infarct region results in marked de-ramification and amoeboid transformation of soma [113]. Microglial activation is believed to be involved in the pathological progression of ischemic tissue. However, the function of activated microglia in ischemic events remains not entirely understood [113–115]. The experimental models seem to validate the hypothesis of two-step sequential microglia activation: the first one, mostly dependent on M1 type activation, with the production of oxidative species, proinflammatory cytokines, and lysosomal cascades [116]; soon after, there is an M2 activation, which seems to be reparative and blocking the inflammatory cascade of events [113,116]. It has been supposed that when the ischemic event is not an acute one, but there is chronic ischemia, like in SVD, there is a preponderance of M1 activation, with minimal M2 action [117–120]. Chronic ischemia determines a severe oligodendrocyte degeneration; soon after, it causes microglial activation and it is further associated with an increase of apoptosis processes that are associated with an elevation of caspase 3 RNA, and of matrix-metalloprotease 2 (MMP-2) expression [121,122]. Astrocytes react to the chronic ischemic condition, as a result of the length and severity of the insult. In the early ischemic period, the astrocytes respond with a remarkable proliferation, but, in the case of persistent hypoperfusion, with their degeneration and death [123–125]. It has been argued that astrocytes act in response to the ongoing modification of the neurons metabolic changed requests, possibly through glutamate signaling [126], and contribute to the regulation of blood flow modifying capillary permeability, by stretching out their endfeet to the microvessel, establishing a proximal connection with the capillary. Their death, due to chronic hypoperfusion, leads to an expanding, and auto-potentiating system of neuronal death, due to a misleading neurovascular coupling. The deterioration of astrocytic function at the late stages of white matter hyperintensities also supports the progressive character of SVD, as shown in a recent clinicopathological study [127]. The more actual histological works discovered collagenous pouches and tubes around small vessels, now referred to as vascular bagging, suggested as a possible biological marker of SVD [128]. Ultrastructural studies have found the splitting, branching, and thickening of the capillary basement membrane and perivascular deposition of collagen, also called microvascular fibrosis, in the brains of aged rats [129] and rhesus monkeys [130]. Frosberg et al. [128] showed vascular bagging in the frontoparietal and temporal control deep white matter. The Authors found that plasma proteins fill the vascular bagging, and argued that SVD should be characterized by a porous endothelium and an altered basement membrane [128,131]. Frosberg et al. [128] showed that, in SVD with diffuse white matter alteration, smaller basal ganglia vessels, including pre-capillary arterioles and capillaries, revealed vascular bags with COLL4-positive walls [132–134]. Post-capillary venules also showed vascular bagging in SVD, but they cannot be distinguished from capillaries, merely based on vessel diameters, deformed erythrocytes squeezing through vessels, or the presence of pericytes [135]. While the pericytes that were found in the Frosberg et al. work [128] were located outside the vascular bags, these cells and their processes are enclosed by two layers of the basement membrane [130], and therefore their degeneration might contribute to a splitting of the basement membrane, supporting what we have afore-described. Frosberg et al. [128] also reported the presence of string vessels in SVD. String vessels are thin connective tissue strands, remnants of capillaries, with no endothelial cells and without the primary function of blood transport [129]. String vessels suggest
Int. J. Mol. Sci. 2020, 21, 1095 6 of 35
the precise location of the originally normal-functioning vessels, and after significant events (abrupt
or chronic ischemia, aging, but also neurodegenerative disorders), they gradually disappear [136].
Many events induce their regression, which is probably due to a converging two-vias: an induced
apoptotic phenomenon associated with the destruction of endothelial cells, attached by macrophages.
Frequently the regression might be triggered by the loss of the vascular endothelium grogth factor
(VEGF) [136]. In their work, Frosberg et al. [128] put in evidence four types of string vessels, suggesting
different stages of string vessel formation, and an enhanced density of COLL4-positive string vessels
and ghost vessels that resembled remnants of string vessels [128]. Quite suggestive is the finding that
the higher quantities of string vessels that are described in work [128] have been found in the damaged
white matter parenchyma. However, in some cases, the Authors have found that after an endothelial
death, the empty basement membrane tubes could help the regrowth of new endothelial cells, which
can synthesize new basement membrane layers [137], which could give the reason of the multi-layered
vascular bags that are found in the original work [128].
3.3. Cholinergic Role in Small Vessel Disease
Moreover, small arteries undergo a systemic poorness of cholinergic network regulation.
Many hypotheses have been raised for a possible explanation, starting from an altered cholinergic
response to inflammation, which is a constant in chronic ischemic condition [138–141], up to a
disruption of the cholinergic networks, which subcortically approaches the basal forebrain, since this is
a preferential location of lacunar vascular infarcts and chronic hypoperfusion syndrome [142–145].
It has been widely demonstrated, either in animal models either in postmortem studies, that there
is a reduced level of acetylcholine (Ach) in patients with vascular dementia [146,147], both in the
cortical areas, in the hippocampus, and the cerebrospinal fluid [148–151]. A loss of cholinergic
neurons in 40% of demented vascular patients was reported, accompanied by reduced ACh activity
in the cortex, hippocampus, and striatum [152]. Post-mortem SVD studies revealed lower choline
acetyltransferase (ChAT) activity when compared with the controls [153], and SVD patients have
lower CSF concentrations of Ach [151,154–156]. In fact, in the experimental condition, the selective
muscarinic antagonism by atropine, for example, has dramatic consequences in the CA1 region, [157].
Other experiments demonstrated that the selective alpha-7-nicotinic AChR antagonism exacerbates
the hypoxic effects on the CA1 and CA3 cortical areas; on the contrary, non-selective nicotinic AChR
antagonists have a detrimental effect on the hippocampus, not in all the other cortical areas [158–161].
The chronic reduction of the cerebral blood flow can affect the control of the cholinergic networks, but
it happens that a proper cholinergic function is compulsory to well-regulation of the regional brain
blood flow [162,163]. Ach principally mediates the parasympathetic innervation of the Willis circle and
the pial vessels [164]. Ach stimulates in vitro the arterial relaxation, directly and via the promotion of
the synthesis of vasodilator endothelium agents [164], via the nitric oxide synthase [165] and the GABA
interneurons [166–168]. The stimulation of the Nucleus Basalis of Meynert results in increased blood
flow throughout the cerebral cortex in experimental animals [169]. Upon stimulation, perivascular
cortical afferents release Ach into endothelial M5 muscarinic receptors [148,170]. M5 receptors are
highly expressed in blood vessel walls [170]. Yamada et al. [148] prepared knockout mice (M5−/−) and
found that, as compared to wild-type mice, these animals lose the ability to dilate cerebral arteries, but
could still regulate extra-cerebral flow. Upon stimulation, perivascular cortical afferents release Ach
onto endothelial M5 muscarinic receptors [170,171]. Hamner et al. [172] demonstrated that cholinergic
control of the cerebral vasculature might be active at low frequencies, lower than 0.05 Hz when the
sympathetic nervous system appears to play a role in the cerebral auto-regulation, in a limited but
well-conducted study. At these low frequencies, the myogenic mechanisms appear to play any role; to
surprise, the correspondence between cholinergic and sympathetic cerebrovascular regulation above
0.05 Hz is striking, suggesting that the cerebral circulation engages different mechanisms to protect
itself [172]. There are many different reasons for the cholinergic impairment that was observed in
SVD. The cholinergic impairment for artery dysregulation that was observed in small vessel diseaseInt. J. Mol. Sci. 2020, 21, 1095 7 of 35
could derive from the deafferentation of the basal forebrain cholinergic bundle to the subcortical
structures, due to the most probable location of lacunar vascular events and the chronic hypoperfusion
consequences, aforementioned [36,38,173]. Bohnen et al. [36] demonstrated in vivo that the severity of
the periventricular white matter lesions is associated with lower AchE activity, in the middle-aged
and elderly subjects without dementia, as a result of cortical cholinergic deafferentation. In animal
SVD models, there is a concomitant reduction of vasopressin and histamine, which is interpreted as a
result of the interruption of the tracts that comes from the supra-optic and tuberomammillary nuclei
and ends in the basal forebrain [174]. The reduction of vasopressin and histamine seems to have a
redundant effect on hypoperfusion. Some clinical data confirm a reduction of the number of cholinergic
neurons in the Nucleus Basalis of Meynert in multi-infarct dementia, but not in SVD [175–177]. It has
been conveyed that a primary loss of the cholinergic neurons of Nucleus Basalis of Meynert does
not mediate cholinergic impairment [178,179], but is a consequence of the secondary cholinergic
deficits, due to the indirect, cholinergic endothelial effect, aforementioned. Though, the number of
muscarinic cholinergic receptors is markedly reduced in mixed dementia patients [179] and SVD
dementia. Cholinergic poorness promotes a less efficacious endothelium relaxation, even due to
an altered nitric oxide synthase and loss of efficacy of the GABA interneurons [165,166]. The two
mediators seem to be less efficient in influencing the small arteries contraction [180–183]. A final step
on this point has been written by a probabilistic tractography analysis [155]; this study tracked the
two primary white matter tracks which map to cholinergic pathways, identifying a significantly lower
fractional anisotropy in precocious form of SVD. Mediation analysis demonstrated that fractional
anisotropy in the tracked pathways could fully account for the executive dysfunction, and partly
mediate the memory and global cognition impairment. The recently published study [155] study
suggests that the fibers that are mapped into the cholinergic pathways, but not those of the Nucleus
Basalis of Meynert, are significantly damaged.
Finally, an alteration of the conceptualized “cholinergic anti-inflammation pathway” summarized
another possible cause for the cholinergic poorness [138,139]; these findings are based on the
knowledge that acetylcholine released from cholinergic axon terminals can interact with α7 nicotinic
Ach receptors on vicinal immune cells. The nicotinic receptors then translate the cholinergic signal into
the suppression of cytokine release, being involved in the inflammatory cascade [140,141]. A chronic
proinflammatory condition counterbalances the acetylcholine release and promotes its cascade effects
on the vasoregulation. The pathological cascade of events, which occurs as a consequence of all
the pathological alterations described, determines a decrease of the vascular tone, with a release of
the blood-brain barrier permeability, with a loss of the internal vascular remodeling and with major
vascular rarefactions. As a result, hypo-perfusion at rest occurs in the brain and it is associated with
impairment in the moment-to-moment control of CBF, with a decrease of adaptive vascular responses
and with a diminishment of the neurovascular coupling and auto-regulation system [145,180].
Insert Figure 2 approx here:Int.Int.
J. Mol. Sci.Sci.
J. Mol. 2020, 21,21,
2020, 1095
1095 8 of
8 35
of 35
Figure 2. Confirmed pathological processes underlying SVD.
Figure 2. Confirmed pathological processes underlying SVD.
4. Chronic Hypoxia and Brain Response
4. Chronic Hypoxia and Brain Response
The brain requires a disproportionate amount of the body’s energy. Up to 20% of cardiac output
Thetobrain
is devoted requires
meeting a disproportionate
the brain’s energy demands, amount of theaccounting
despite body’s energy.
for only Up2% to 20% of cardiac
of body output
mass [184].
is devoted to meeting the brain’s energy demands, despite accounting
The cerebral vasculature possesses well-developed mechanisms that enable cerebral blood flow (CBF) for only 2% of body mass [184].
The cerebral vasculature possesses well-developed mechanisms that
to remain constant during fluctuations in arterial pressure (autoregulation) and meet the increased enable cerebral blood flow
(CBF) demands
nutrient to remainwhen constant
localduring fluctuations
brain activity rises inin order
arterial pressurethese
to deliver (autoregulation) and meet
nutrients effectively andthe
increased nutrient demands when local brain activity rises in
protect the brain from hypoperfusion and ischemic damage [185,186]. Cerebral SVD significantlyorder to deliver these nutrients
andeffectively
chronicallyand protectthe
impairs theability
brain offrom
thehypoperfusion
cerebral vasculature and ischemic
to meet damage [185,186].
these demands dueCerebral
to severalSVD
significantly and chronically impairs the ability of the cerebral vasculature
structural and functional changes, which ultimately result in brain injury, cognitive decline, and to meet these demands
due to several
dementia. Cerebral structural and functional
vasoconstrictor changes, responses
and vasodilator which ultimately result in
are important brain injury,
mechanisms by cognitive
which
decline,
brain and isdementia.
blood flow maintained, Cerebral vasoconstrictor
as aforementioned. In SVD, and vasodilator
chronic hypoperfusion responses
leads toarea decrease
important
mechanisms
in cerebral bloodby flow, which
hypoxia,brain blood stress,
oxidative flow and is maintained, as aforementioned.
triggers inflammatory responses, which In SVD,leadschronic
to a
hypoperfusion leads to a decrease in cerebral blood flow, hypoxia,
potentiated hypoperfusion condition. The induced lesions are mostly expressed in the white matter oxidative stress, and triggers
inflammatory
(WM) and especiallyresponses, which leads to aWM,
in the periventricular potentiated hypoperfusion
basal ganglia, condition. Hypoxia-induced
and hippocampus. The induced lesions
oxidative stress leads then to mitochondrial dysfunction, neuronal damage, and apoptosisWM,
are mostly expressed in the white matter (WM) and especially in the periventricular basal
via the
ganglia,
nitric oxide and hippocampus.
synthase (NOS) pathway Hypoxia-induced
[187–190]. Chronic oxidative stress
hypoxia leads then
profoundly to mitochondrial
influences vascular
control, altering both vasoconstrictors as well as vasodilator responses in isolated cerebral [187–190].
dysfunction, neuronal damage, and apoptosis via the nitric oxide synthase (NOS) pathway vessels;
Chronic
indeed, hypoxia
chronic hypoxiaprofoundly
alters theinfluences
contractile vascular
response control, altering cerebral
of the isolated both vasoconstrictors as well as
vessels [191]. Chronic
vasodilator
hypoxia is known responses in isolated
to influence cerebral
Nitric Oxide (NO)vessels; indeed,ofchronic
modulation contractilehypoxia altersInthe
response. onecontractile
animal
response of the isolated cerebral vessels [191]. Chronic hypoxia is known
study, the authors showed that chronic hypoxia augmented contractile sensitivity to the thromboxane to influence Nitric Oxide
(NO) modulation of contractile response. In one animal study, the authors
mimetic U-46619 in isolated cerebral vessels as the result of reduced nitric oxide (NO) production and showed that chronic
hypoxia
activity. augmented
A decrease in NO contractile
productionsensitivity to theand
of L-arginine thromboxane mimetic
oxygen increased NO U-46619 in isolated
degradation cerebral
or reduced
vessels as the result of reduced nitric oxide (NO) production
cyclic guanosine-3-5-monophosphate (cGMP) production (involved in smooth muscle relaxation). and activity. A decrease in NO
In production
this case, the of administration
L-arginine and of oxygen increased NO
the nonspecific NO degradation
synthase (NOS) or reduced
inhibitor cyclic guanosine-3-5-
nitro-L-arginine
monophosphate (cGMP) production (involved in smooth muscle
(NLA) eliminated the difference in contractile sensitivity between the vessels from the normoxic relaxation). In this case,andthe
administration of the nonspecific NO synthase (NOS) inhibitor nitro-L-arginine
chronically hypoxic animals, which suggests that a reduction in NO production and activity was (NLA) eliminated
the difference
responsible for the in increased
contractilecontractile
sensitivitysensitivity
between that the vessels from the
was observed normoxic
[192]. and chronically
Such effects may be
hypoxic animals, which suggests that a reduction in NO production and activity was responsible for
the increased contractile sensitivity that was observed [192]. Such effects may be significant in theInt. J. Mol. Sci. 2020, 21, 1095 9 of 35
significant in the adult in whom disorders involving cerebral circulation occur under conditions of
acute and chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a prevalent pathophysiological
state in patients with Alzheimer’s disease (AD) and vascular dementia (VaD). CCH has been identified
as one of the initial conditions that are critical in the development of cognitive dysfunction [193].
In several studies, deranged energy metabolism, glial activation, apoptosis, oxidative stress, neuronal
damage, and white matter lesions that are caused by cerebral hypoperfusion have been found to
contribute to the pathophysiological mechanisms that lead to cognitive impairment [194,195]. Animal
models of CCH showed that such compensatory actions induce abnormal activation of the frontal
cortex and the hippocampus. Hypoxemia, in addition to hypoperfusion, exacerbates ischemic brain
damage and it is associated with more severe white matter lesions. Abnormal cerebral hypoxia induces
compensatory and adaptive mechanisms to prevent hypoperfusion injury and preserve recovery of
brain function [194,196]. Part of those adaptive mechanisms involves increased capillary diameter,
neovascularization, and enhanced expression of vascular endothelial growth factor (VEGF). In the
condition of CCH, hypoxia-inducible factor 1 (HIF-1) is one of the most important transcription factors
that are involved in the endogenous adaptive response. HIF-1α then leads to the expression of a large
number of genes. It regulates more than 2% of the genes in human vascular endothelial cells [197] and is
recognized today as a regulator of the vast majority of hypoxia-inducible genes that are responsible for
the cell adaptation to hypoxia, including angiogenesis, anaerobic metabolism, mitochondrial biogenesis,
erythropoiesis, vasomotor control, and cell proliferation, such as vascular endothelial growth factor
(VEGF), glucose transporter-1 (GLUT-1), and erythropoietin (EPO), all factors that lead to survival
under hypoxic conditions [198,199]. HIF-1a is also involved in hypoxia-dependent inflammation,
apoptosis, and cellular stress. Animal models showed that the neuron-specific knockdown of HIF-1a
aggravates brain damage after a 30 min. middle cerebral artery occlusion (MCAO) and reduces the
survival rate of those mice, and an impairment of learning and memory after four weeks of CCH has
been reported. Cerebral angiogenesis is reduced, while oxidative damage is also promoted with the
proliferation of astrocytes and microglia in the cortex and some sub-regions of the hippocampus [200].
In other studies it is reported that the lowering of oxygen induces hypoxia-inducible factor-1α (that is
involved in neuroinflammatory response), which has the direct consequence of the hyper-production of
free radicals and proteases, BBB disruption, vasogenic edema, and myelin damage; all these effects may
lead to white matter (WM) damage and vascular cognitive impairment. Moreover, hypoxia-induced
MMP-9 expression leads to vascular leakage, which MMP inhibition could reduce. Pharmacological
blockage of MMP-9 or MMP-9 gene deletion confers neuroprotection in traumatic brain injury and
stroke [201].
Protective mechanisms that are triggered by hypoxia are characterized by decreasing the O2
demand, increasing the O2 supply, or a combination of both. Some animals can reduce the O2
demand through a condition called hypometabolism, but, in the human brain, this condition is poorly
expressed. Hypoxia is always associated to early signs of failure that are represented by marked
falls in pH and tissue creatine phosphate levels, followed by a dysfunction of Na+/K+ ATPase and
lethal ion imbalance [202,203]. In the human brain, pro-survival pathways and improving brain
oxygenation actions are activated. During cerebral hypoxia, in brain, HIF-2, also known as EPAS-1
(endothelial PAS domain protein 1), is expressed, principally in endothelial cells, including brain
capillary endothelial cells [204]. HIF-2 is active during prolonged mild hypoxia and it might be involved
in brain microvascular response. In one paper, the authors provided evidence that HIF-mediated
pro-survival responses are dominant in rats with CCH. The activation of HIF-1 is part of a homeostatic
response that is aimed at coping with the deleterious effects of CCH [200]. While considering these
premises, a large number of clinical trials tried to identify protective strategies against cerebral
impairment after hypoxia through the identification of endogenous neuroprotective pathways. Based
on animal work, it has been shown that spontaneously hypertensive/stroke-prone rats (SHR/SP) with
unilateral carotid artery occlusion had white-matter damage while being treated with a permissive
Japanese diet. One week after, white matter showed a significant increase in hypoxia-inducibleInt. J. Mol. Sci. 2020, 21, 1095 10 of 35
factor-1α
Int. J. Mol. Sci.(HIF-1α), which
2020, 21, 1095increased further by three weeks. The BBB disruption was supposed to 35
10 of be
secondary to hypoxia and related to a matrix metalloproteinase-9 (MMP-9)-mediated infiltration of
mediated
leukocytes.infiltration of leukocytes.
In those animals, In those
treatment animals, treatment
with minocycline with reduced
significantly minocycline significantly
the lesion size and
reduced
improvedthecerebral
lesion size andflow.
blood improved cerebralprolonged
Minocycline blood flow.survival
Minocycline
[205].prolonged survival
The results are far [205].
from toThebe
results
appliedare
in thefarhuman
from chronic
to be hypoperfusion
applied in thecondition
human for
chronic hypoperfusion
the aforementioned condition
cascades for the
of events that
aforementioned cascades ofinevents
appear to be determinant humanthat
SVD. appear to be determinant in human SVD.
Insert
InsertFigure
Figure33approx.
approx.here:
here:
Figure 3. The pathological circuit of chronic hypoxia damage in the brain.
Figure 3. The pathological circuit of chronic hypoxia damage in the brain.
5. Endothelium and SVD
5. Endothelium and SVD
The brain endothelium, even in severe SVD (presenting an almost complete loss of myocytes
andThe
other mural cells) remains
brain endothelium, even intact, even ifSVD
in severe the endothelium
(presenting an is one
almostof the main targets
complete loss ofofmyocytes
the redox
altered process and inflammation (and both these processes are highly activated
and other mural cells) remains intact, even if the endothelium is one of the main targets of the redox in SVD) [134,206–209].
This paradoxical
altered process and survival of the brain
inflammation (andendothelium is also evident
both these processes in patients
are highly with CADASIL
activated [206,207].
in SVD) [134,206–
On the contrary, systemic endothelium activation is quite different in SVD.
209]. This paradoxical survival of the brain endothelium is also evident in patients with CADASIL
Thus,
[206,207]. Onindirectly, brain
the contrary, endothelium
systemic suffersactivation
endothelium in SVD conditions. Mitochondrial
is quite different in SVD. senescence of
the Thus,
endothelium walls
indirectly, hasendothelium
brain a catastrophic effectinon
suffers SVD cerebral endothelial
conditions. cells [210];
Mitochondrial this alteration,
senescence of the
which is over-expressed in SVD [211], is generally related to an impaired
endothelium walls has a catastrophic effect on cerebral endothelial cells [210]; this alteration, response to the threewhich
major
isendothelium-derived
over-expressed in SVD nitric oxide-vasodilators
[211], is generally related[212], toprostacyclin
an impaired [213], and endothelium-derived
response to the three major
hyperpolarizing factors (EDHF) [214]. The reduction of NO production is
endothelium-derived nitric oxide-vasodilators [212], prostacyclin [213], and endothelium-derived derived from an impairment
of the mitochondrial functions, being caused by a hyperproduction
hyperpolarizing factors (EDHF) [214]. The reduction of NO production is derived of the anti-oxidative defensefromsystem,
an
and an increased
impairment of theO2 anions reaction
mitochondrial with NO,
functions, producing
being causedperoxynitrite [215]. The activity
by a hyperproduction of endothelial
of the anti-oxidative
NO synthase
defense system, (eNOS),
and an which catalyzes
increased O2 the production
anions reactionofwithNO, declines with aging
NO, producing [216], but is[215].
peroxynitrite even more
The
activity of endothelial NO synthase (eNOS), which catalyzes the production of NO, declineskinase
impaired in SVD, where an important downstream target of Rho is the Rho-associated protein with
(ROCK)
aging [217].
[216], butThese
is evenubiquitously
more impaired expressed
in SVD, serine/threonine
where an important protein kinases aretarget
downstream involved in diverse
of Rho is the
Rho-associated protein kinase (ROCK) [217]. These ubiquitously expressed serine/threonine proteinof
cellular activities, including apoptosis, smooth muscle contraction, cell adhesion, and remodeling
the extracellular
kinases are involved matrix [218]. In
in diverse the regulation
cellular activities,ofincluding
endothelial cell, migration
apoptosis, smoothROCK
muscleinteracts with
contraction,
cell adhesion, and remodeling of the extracellular matrix [218]. In the regulation of endothelial cell,
migration ROCK interacts with ezrin, radixin, and moesin (also known as the ERM proteins) that
function as cross-linkers between the plasma membrane and actin filaments [217] and are
indispensable for the leukocyte adhesion molecules coordination, being essential for barrier functionInt. J. Mol. Sci. 2020, 21, 1095 11 of 35
ezrin, radixin, and moesin (also known as the ERM proteins) that function as cross-linkers between
the plasma membrane and actin filaments [217] and are indispensable for the leukocyte adhesion
molecules coordination, being essential for barrier function [219]. Moreover, the ROCK/RhoA complex
regulates the eNOS, as previously exposed [217]. NO-induced vasodilation occurs via the activation
of myosin light chain phosphatase (MLCP) in a cGMP dependent manner. RhoA/ROCK counteracts
this through MLCP inactivation and calcium desensitization [217,220]. ROCK/Rho decreases eNOS
expression and affects the availability of NO [221]; it has also been proven in brain small vessels, even
if these effects have been largely studied in major vessel disease (coronary) [82]. Three potentially
functional eNOS polymorphisms (T-786C, intron 4ab, G894T) located toward the 50 flanking end of the
gene are known to be considered as being present in SVD and also in isolated lacunar infarction and
ischemic leukoaraiosis [222]. RhoA inhibition overwhelms VEGF-enhanced endothelial cell migration
in response to vascular injury, without, or better said, with a minimal effect, on basal endothelial cell
migration [223,224]. The maintenance of the endothelial barrier is a prior role of the endothelium
cells, mainly through the operative system of RhoA [225], also being mediated through the regulation
of Vascular endothelium cadehrins (VE-cadherins) [226]. In diabetes (one of the main risk factors
associated to SVD), advanced glycation end products (AGEs) accumulate in the vasculature, triggering
a series of purposeful and morphologic changes of endothelial cells, such as the increase of the
activation of the RhoA/ROCK pathway; the significant consequence is an increased endothelial cell
permeability [227]. It can also act as a VEGF inducer, which indirectly causes microvascular endothelial
hyper-permeability [228].
Therefore, it should be argued that the endothelium seems to be functionally impaired in SVD,
even if morphologically and structurally undamaged [229].
The endothelial NO downregulation in SVD is a marker of decreased endothelial regulatory
capacity, in response to external stimuli, such as hypercapnia [230,231]. Living studies have
demonstrated a significant baseline CBF reduction in SVD–affected subjects, together with an impaired
CBF autoregulation [232–234]. Endothelial activation refers to the change in the expression of
many different surface markers [235–238]. These circulating markers of endothelial activation include
intercellular adhesion molecule-1 (ICAM-1), which has been considered as a generic expression of white
matter progression [239], soluble thrombomodulin (sTM), interleukin-6 (IL-6), plasminogen activator
inhibitor-1 (PAI-1), von Willebrand factor, and others [207,240–242]. Moreover, an upregulation of
hypoxia-endothelial-related markers has been proven, such as HIF 1 alpha, VEGFR2, and neuroglobin,
when white matter lesions appear to be confluent [243]. The matter is even more impressive when
it appears evident that endothelium in overall activated, as described above, but, according to some
authors, not specifically in the human gray matter [209,241,244,245]. Though, the brain endothelium
NO dysregulation implies not only a direct inhibition of the vessel tone, but indirectly, more critically,
a decrease of the dynamic neurovascular control mechanism [246,247].
Moreover, the permanent status of oxidative stress-induced should be taken into account, which
causes a superimposed macroscopic alteration of the cerebral endothelium.
The immediate consequence of the endothelial dysfunction has two significant consequences, the
reduction of the resting flow in the marginally perfused white matter and macroscopic alterations
of the BBB permeability [247]; these two aspects lead to additional oxidative stress, by inducing
tissue hypoxia and extravasation of the plasma proteins [247], and both of them potentiate the
inflammation pathway, through the Nuclear Factor Kappa-Light-Chain-Enhancer of activated B cells
(NFkBeta) dependent transcription. The modern view gives the endothelium the control role of
the propagation of vasomotor signals [248], even if the question is still unresolved. In systemic
vessels, the endothelium is well known to participate in the retrograde propagation of vascular
signals [81,249], but in the brain the mechanisms by which endothelium interacts with the spread of
the vascular signal is still debated. It has been proven that a highly localized lesion of the endothelium
failed to propagate beyond the lesion site, and altered the amplitude and temporal dynamics of the
go-ahead vascular sign, with weaker temporal coordination [250]. It has been demonstrated that brainInt. J. Mol. Sci. 2020, 21, 1095 12 of 35
endothelium is enriched with KIR channels, and not by KCa channels; these channels are sensitive to
high K flow, being derived from neural activity, and are transmitted by the synapses or by astrocytic
end-feet [249,251]. It has been recognized that K+ is recognized in the endothelium, and the upstream
penetrating arteriole is the effector of the vasodilatation [251], and its rapid propagation is probably
conducted by ionic currents traveling through the endothelium via gap junctions and then through
the myoendothelial junctions [249]. Therefore, KIR suppression avoids the increase of CBF that is
produced by cortical activation [251]. The most intriguing aspect of the endothelial conductance is
the fact that the conducted vasomotor responses, either being a dilatation, either vasoconstriction,
can be generated by different neuromodulators, i.e., Acetylcholine, ATP, prostaglandin F-2alpha, and
NO, but their effects on neurovascular coupling has never been determined [252]. The evidence is
increasing on pial arterioles: signals that are generated by the neuronal activity, deep in the brain,
should be conveyed to upstream arterioles, remote from the area of activation, to increase flow
efficiently [249]. Vascular mapping and fMRI demonstrated that vascular responses are first seen in
the deep cortical lamina during somatosensory activation, and then, more superficially, suggesting a
retrograde propagation of the vascular response [253]. A possible scenario for the transmission and
coordination of the vascular response is described [249], as follows: activation-induced increase in
extracellular potassium triggers the hyperpolarization of capillary endothelial cells and pericytes [254].
The hyperpolarization propagates upstream and reaches smooth muscle cells in penetrating arterioles,
producing relaxation [249,251]. At the same time, metabolic modifications (reduced viscosity, increased
deformability of blood cells) on the endothelium of feeding arterioles increment the smooth muscle
cell relaxation (the so-called flow-mediated vasodilation). In upstream pial arterioles, remote from the
site of activation, there is vasodilation, by propagation from arteriole downstream and acting as a local
flow-mediated and myogenic response. For all of the conditions mentioned above, SVD is defective in
neurovascular coupling, even for endothelial and pericytes failure.
6. Astrocytes and SVD
Neurons, astrocytes, oligodendrocytes, as well as vascular and perivascular cells, are intimately
related to metabolic control and they act as trophic determinants in brain development, function,
and reaction to injury. Specifically, astrocytes play integral roles in the formation, maintenance, and
elimination of synapses in development and disease [255]. In addition to their well-established
interactions with neurons, astrocytes are also needed for the development and maintenance of BBB
characteristics in endothelial cells [256], and for the reorganization of vascular networks after brain
injury [257]. In turn, the endothelial cells regulate glycolytic metabolism in astrocytes through the
production of NO [258]. The release of vasoactive substances, such as prostanoids from astrocytes,
can couple cerebral blood flow to neuronal energy demand, and astrocytes supply neurons with vital
metabolites, such as lactate in response to neuronal activity [259]. Additional homeostatic functions
of astrocytes include water, ion, and glutamate buffering, as well as tissue repair after insult or
injury [260,261]. The reactive astrocytes can release a wide variety of extracellular molecules, including
inflammatory modulators, chemokines and cytokines, and various neurotrophic factors. These factors
can be either neuroprotective (e.g., cytokines, such as interleukin-6 [IL-6], and transforming growth
factor-b [TGF-b]) or neurotoxic (such as IL-1b and tumor necrosis factor-a [TNF-a]) [262]. The process
of glial scar formation exemplifies the interplay between the neuroprotective and neurotoxic effects of
reactive gliosis. The glial scar serves to isolate the damaged area and it prevents the damage extension
by restricting the infiltration of inflammatory cells. However, molecules that are secreted by reactive
scar-forming astrocytes can also be refractory to neurite growth [262,263]. An example of what the above
written can be found in AD, where reactive astrocytes are found in the postmortem brain of affected
patients [264–266]. It has been demonstrated that astrocytes can internalize Amyloid-beta plaques,
exerting a scavenger-like function [267,268]. Nonetheless, it has been proved that those astrocytes
with amyloid-beta are irreversibly compromised, likely showing altered calcium homeostasis [269].
Moreover, it is believed that astrocytes in AD are seriously compromised through the altered expressionsYou can also read

















































