A genome scan of diversifying selection in Ophiocordyceps zombie ant fungi suggests a role for enterotoxins in co evolution and host specificity
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Received: 8 September 2017 | Accepted: 13 July 2018
DOI: 10.1111/mec.14813
ORIGINAL ARTICLE
A genome scan of diversifying selection in Ophiocordyceps
zombie‐ant fungi suggests a role for enterotoxins in
co‐evolution and host specificity
Noppol Kobmoo1,2 | Duangdao Wichadakul3,4 | Nuntanat Arnamnart2 |
Ricardo C. Rodríguez De La Vega1 | Janet J. Luangsa-ard2 | Tatiana Giraud1
1
Ecologie Systématique Evolution,
Université Paris-Sud, CNRS, AgroParisTech, Abstract
Université Paris-Saclay, Orsay, France Identification of the genes underlying adaptation sheds light on the biological func-
2
National Center for Genetic Engineering
tions targeted by natural selection. Searches for footprints of positive selection, in the
and Biotechnology (BIOTEC), National
Science and Development Agency (NSTDA), form of rapid amino acid substitutions, and the identification of species‐specific genes
Klhong Luang, Thailand
have proved to be powerful approaches to identifying the genes involved in host spe-
3
Chulalongkorn University Big Data
Analytics and IoT Center (CUBIC), cialization in plant‐pathogenic fungi. We used an evolutionary comparative genomic
Department of Computer Engineering, approach to identify genes underlying host adaptation in the ant‐infecting genus
Faculty of Engineering, Chulalongkorn
University, Bangkok, Thailand Ophiocordyceps, which manipulates ant behaviour. A comparison of the predicted
4
Center of Excellence in Systems Biology, genes in the genomes of species from three species complexes—O. unilateralis,
Faculty of Medicine, Chulalongkorn
O. australis and O. subramanianii—revealed an enrichment in pathogenesis‐associated
University, Bangkok, Thailand
functions, including heat‐labile enterotoxins, among species‐specific genes. Further-
Correspondence
more, these genes were overrepresented among those displaying significant footprints
Noppol Kobmoo, BIOTEC, NSTDA, Thailand
Science Park, Khlong Neung, Khlong Luang, of positive selection. Other categories of genes suspected to be important for viru-
12120 Pathum Thani, Thailand.
lence and pathogenicity in entomopathogenic fungi (e.g., chitinases, lipases, proteases,
Email: noppol.kob@biotec.or.th
core secondary metabolism genes) were much less represented, although a few candi-
Funding information
date genes were found to evolve under positive selection. An analysis including
H2020 Marie Skłodowska-Curie Actions,
Grant/Award Number: 655278; Thailand orthologs from other entomopathogenic fungi in a broader context showed that posi-
Research Fund, Grant/Award Number:
tive selection on enterotoxins was specific to the ant‐infecting genus Ophiocordyceps.
TRG5780162
Together with previous studies reporting the overexpression of an enterotoxin during
behavioural manipulation in diseased ants, our findings suggest that heat‐labile
enterotoxins are important effectors in host adaptation and co‐evolution in the Ophio-
cordyceps entomopathogenic fungi.
KEYWORDS
adaptation, enterotoxins, host specificity, Ophiocordyceps, positive selection
1 | INTRODUCTION of the copies (Ohno, 1970; Zhang, Zhang, & Rosenberg, 2002). Gene
losses can also be adaptive (Juárez‐Vázquez et al., 2017), particularly
The identification of genes underlying adaptation is a major goal in in pathogens, as the absence of a molecule recognized by the host
evolutionary biology, as it can shed light on the biological functions may enable the pathogen to colonize its host without triggering a
targeted by natural selection and the genetic mechanisms generating response from the host immune system (Albalat & Cañestro, 2016;
new, adaptive variants. Innovation may be generated during evolution Ghanbarnia et al., 2015; Rouxel & Balesdent, 2017). Gene duplications
by gene duplication followed by rapid amino acid substitutions in one and losses result in the presence of species‐specific genes, which are
Molecular Ecology. 2018;1–17. wileyonlinelibrary.com/journal/mec © 2018 John Wiley & Sons Ltd | 12 | KOBMOO ET AL.
often overrepresented among the genes involved in adaptation models in which to study the genomics of host specialization and
(Gladieux et al., 2014; Lespinet, Wolf, Koonin, & Aravind, 2002; Zhou co‐evolution. Specialist Metarhizium strains and species generally
et al., 2015). Adaptation may also occur through positive selection, have fewer genes, with notably fewer genes encoding host‐killing
with rapid amino acid substitutions, typically detected as higher rates toxins (Wang, Leclerque, Pava‐Ripoll, Fang, & St. Leger, 2009), but
of nonsynonymous substitutions (dN) than of synonymous substitu- more genes evolving under positive selection than generalists (Hu
tions (dS) among orthologous genes of closely related species (Ina, et al., 2014). However, it remains unclear how adaptation has
1996; Kimura, 1983). Comparisons of dN/dS ratios to neutral expecta- shaped the genomes of closely related fungal entomopathogens
tions therefore also constitute a powerful approach to identifying specializing on different hosts.
genes under recurrent positive selection (Yang & Nielsen, 1998; Yang, We therefore tried to identify genes involved in host speci-
Nielsen, Goldman, & Krabbe Pedersen, 2000; Yang, Wong, & Nielsen, ficity in three complexes of closely related species from the genus
2005). Ophiocordyceps (Hypocreales, Ascomycota): O. unilateralis sensu lato,
Pathogens are particularly interesting models for investigations O. subramanianii s.l. and O. australis s. l. One of the key features
of the genomic mechanisms of adaptation, as they are locked in an of these pathogens is their ability to manipulate their hosts to
arms race with their hosts, leading to continuous, rapid evolution promote their own dispersal. Infected ants, often described as
(Anderson et al., 2010; Kurtz, Schulenburg, & Reusch, 2016). Identifi- “zombie ants,” leave their nests and develop erratic behaviour,
cation of the genes underlying host‐specific adaptations in patho- wandering alone into vegetation and then biting into a leaf
gens improves our fundamental understanding of natural selection located at a precise height and orientation optimal for subsequent
and evolution, but it also has more applied implications, shedding fungal dispersal just before they die. Fungal spores produced from
light on major epidemics and disease emergence in plants and ani- the diseased ant are thus dispersed farther, from a height (de
mals (Möller & Stukenbrock, 2017). Bekker, Ohm, Evans, Brachmann, & Hughes, 2017; Hughes et al.,
Fungi are the principal pathogens of plants (Anderson et al., 2011, 2016; Pontoppidan, Himaman, Hywel‐Jones, Boomsma, &
2004), and they also represent threats to the health of many animals Hughes, 2009). Ophiocordyceps unilateralis s.l. is a highly diverse
(Fisher et al., 2012; Sexton & Howlett, 2006). Many studies have complex of pathogenic cryptic species specific to formicine ants. It
searched for genes under positive selection as a means of identifying is distributed worldwide, and many species occur together in sym-
genes and functions involved in the species‐specific adaptation of patry while displaying strong host specificity (Araújo, Evans,
fungal pathogens of plants (Aguileta, Refrégier, Yockteng, Fournier, Kepler, & Hughes, 2018; Evans, Elliot, & Hughes, 2011; Kobmoo,
& Giraud, 2009; Möller & Stukenbrock, 2017). For example, in the Mongkolsamrit, Tasanathai, Thanakitpipattana, & Luangsa‐Ard,
Microbotryum and Botrytis fungal plant pathogens, many such genes 2012; Kobmoo et al., 2015). Ants develop erratic behaviour only
have been identified through comparative transcriptomics studies as when infected with their specific pathogen species (de Bekker et
being under recurrent positive selection and they were involved in al., 2014; Sakolrak, Blatrix, Sangwanit, Arnamnart, & Kobmoo,
biological processes important for the recognition and cell signalling 2018). The taxonomy and phylogeny of the other ant‐manipulating
between the host and the pathogen (Aguileta et al., 2010, 2012). Ophiocordyceps species complexes have been studied in less detail,
More recent, next‐generation sequencing made it possible to per- but host specificity is also considered to be the rule for these
form genomewide scans in plant‐pathogenic fungi, resulting in the other taxa (Araújo et al., 2018).
identification of an array of effectors under positive selection We conducted a comparative genomic study of ant‐infecting
(Badouin et al., 2017; Poppe, Dorsheimer, Happel, & Stukenbrock, Ophiocordyceps species, with the aim of identifying genes underlying
2015; Schirrmann et al., 2018; Stukenbrock et al., 2011; Wicker et host specificity by searching for species‐specific genes and genes
al., 2013) and species‐ or lineages‐specific genes underlying adapta- evolving under positive selection. We sequenced the genomes of
tions (Baroncelli et al., 2016; Hartmann, Rodríguez de la Vega, Bran- two closely related species of the O. unilateralis complex from Thai-
denburg, Carpentier, & Giraud, 2018). land: O. camponoti-leonardi and O. camponoti-saundersi, specific to
By contrast, far fewer such studies have been performed on the ants Colobopsis leonardi and C. saundersi, respectively. We also
entomopathogenic fungi (Wang & Wang, 2017), despite the impor- improved the available genome assembly of another species of this
tance of identifying genes underlying host‐specific adaptation for the complex, O. polyrhachis-furcata, specific to Polyrhachis furcata
use of these fungi as biological control agents against insect pests in (Wichadakul et al., 2015), and used the published genomes of other
agriculture (Wang & Feng, 2014). Furthermore, an understanding of ant‐infecting Ophiocordyceps species (de Bekker et al., 2017): one
host specificity and evolution in these insect pathogens is of funda- genome of each of two species of O. unilateralis s.l., O. kimflemingiae
mental interest in its own right, particularly for fungi able to manipu- from the United States infecting Camponotus castaneus (Araújo et al.,
late the behaviour of the insect host for their own benefit, as in the 2017; de Bekker et al., 2015) and O. camponoti-rufipedis from Brazil
“zombie‐ant” phenomenon. Most genomic studies on ento- specific to C. rufipes (Araújo et al., 2018; Evans et al., 2011); one
mopathogenic fungi have focused on species with agricultural appli- genome of O. subramanianii s.l. from a ponerine ant in Ghana; one
cations such as Beauveria bassiana and Metarhizium anisopliae (Gao genome of each of two strains of O. australis s.l. found on different
et al., 2011; Hu et al., 2014; Pattemore et al., 2014). However, these ponerine ant species, from Ghana and Brazil, probably belonging to
species have broad host ranges and may not, therefore, be the best different cryptic species (de Bekker et al., 2017).KOBMOO ET AL. | 3
The entomopathogenic fungi of the order Hypocreales are (Wichadakul et al., 2015); we aimed to improve the existing refer-
known to infect their host by penetrating the cuticle (Boomsma, Jen- ence genome, but the original strain BCC54312 could not be grown
sen, Meyling, & Eilenberg, 2014). This process requires an array of from the culture collection. We therefore collected three additional
proteinases, lipases and chitinases. The acquisition of nutrients from samples of this species (strains NK275ss‐12, NK142ss and NK294ss‐
the host requires proteases and glycoside hydrolases, including tre- 20), in 2013 and 2014, from the same site as the reference strain, in
halases in particular, as trehalose is a major carbon source present in Khao Yai National Park, Nakhon Ratchasima Province. The collected
the insect haemolymph (Thompson, 2003). Secondary metabolites, samples were isolated and grown as described by Wongsa, Tasana-
including toxins, help to combat the host immune system and even- tai, Watts, and Hywel‐Jones (2005). We complied with the Nagoya
tually kill the insect (Ortiz‐Urquiza, Riveiro‐Miranda, Santiago‐Álvarez, protocols on access and benefit‐sharing, by obtaining authorization
& Quesada‐Moraga, 2010; Schrank & Vainstein, 2010). Ophiocordy- from the Department of National Parks, Wildlife and Plant Conserva-
ceps polyrhachis-furcata has a more extensive family of genes encod- tion (DNP) at the Ministry of Natural Resources and Environment of
ing putative heat‐labile enterotoxins than other specialist Thailand for all strain collections. After two to three months of
entomopathogenic fungi (Wichadakul et al., 2015), and some of growth on Grace Insect Cell Medium (Sigma‐Aldrich), the mycelia
these genes are expressed during host‐specific behavioural manipula- and spores were harvested and DNA was extracted with the
tion. Heat‐labile enterotoxins may, therefore, act as neuromodulators NucleoSpin® Soil kit (Macherey‐Nagel). The long incubation period is
(de Bekker et al., 2015). We hypothesized that enterotoxin‐coding due to the fact that O. unilateralis species in Thailand are very fastid-
genes would be under recurrent positive selection in ant‐manipulat- ious to grow, requiring few steps of enlarging the culture scale to a
ing Ophiocordyceps fungi, as they have probably been involved in co‐ sufficient level for DNA extraction. Genomic libraries were con-
evolution with the host and in host‐specific adaptation. Small pro- structed (150‐bp paired‐end reads) for sequencing with an Illumina
teins secreted by fungal pathogens are often involved in interactions HiSeq3000 machine at the GenoToul platform (Toulouse, France).
with the host (Barrett & Heil, 2012; Rafiqi, Ellis, Ludowici, Hardham,
& Dodds, 2012). We therefore conducted genome scans for positive
2.2 | Read pretreatment, de novo assembly and
selection and focused on the heat‐labile enterotoxin gene family and
improvement of the reference genome
small secreted proteins. We conducted formal tests for positive
selection (statistical comparisons of models of evolution with and The raw reads were trimmed to remove adapters and low‐quality
without diversifying selection). As such tests detect only highly bases from their ends (q < 20). Duplicate reads were removed using
recurrent and rapid positive selection, we also investigated the 5% Picard Tools MarkDuplicate. The reference genomes for O. cam-
of genes with the highest dN/dS values. High dN/dS values, even if ponoti-leonardi and O. camponoti-saundersi were assembled de novo
below 1, may be indicative of positive selection at a few sites in the with SPAdes (Bankevich et al., 2012), which progressively integrates
protein, although they may also result from relaxed selection. In sev- k‐mers of increasing size. The k‐mer sizes used were 21, 33, 55, 77,
eral classes of genes thought to be important for virulence and 99, 119 and 127 for NK405ss‐6, and 21, 33, 55, 77, 99 and 115 for
pathogenicity in entomopathogenic fungi (e.g., chitinases, lipases, NK511ss‐8. The appropriate maximum k‐mer sizes were estimated
proteases, small secreted proteins), only a few genes showed signs with Kmergenie (Chikhi & Medvedev, 2014).
of selection or species specificity. By contrast, we found that heat‐ The reads obtained for the new O. polyrhachis-furcata samples
labile enterotoxins were overrepresented among both the species‐ were used to fill gaps in the existing reference genome of this spe-
specific genes and the genes with significant footprints of positive cies with GapFiller (Boetzer et al., 2012), which mapped the reads
selection. An analysis including enterotoxin‐encoding genes from onto the reference sequence over the regions flanking the gaps and
other entomopathogenic fungi (Hypocreales), that do not manipulate identified a consensus between reads overlapping the gaps. In total,
host behaviour, showed that positive selection was specific to the 175 of 3,915 gaps were closed (identifying around 1.6 Mb from a
ant‐infecting genus Ophiocordyceps. These findings suggest that total gap length of 2.4 Mb in the reference genome).
heat‐labile enterotoxins are important effectors involved in host
adaptation and co‐evolution in entomopathogenic Ophiocordyceps
2.3 | Gene prediction and functional annotation
fungi.
Gene prediction was based exclusively on scaffolds of more than
1 kb in length and involved a two‐round approach based on MAKER
2 | MATERIALS AND METHODS
(Cantarel et al., 2008). Gene sets were initially predicted with
CEGMA (Parra, Bradnam, & Korf, 2007) and GeneMark‐ES (Lom-
2.1 | Sampling and sequencing
sadze, Ter‐Hovhannisyan, Chernoff, & Borodovsky, 2005) and were
In 2015, we collected a sample of O. camponoti-leonardi (strain then used as inputs into MAKER for the first round of prediction.
NK511ss‐8) from Kalayaniwattana district, in Chiang Mai province in The predicted proteins and transcripts identified in previous studies
Thailand, and a sample of O. camponoti-saundersi (strain NK405ss‐6) on O. polyrhachis-furcata (Wichadakul et al., 2015) were also used as
from the Phu Kiew National Park, in Chaiyaphum province. We used a training set for MAKER. The predicted gene set from this first
the reference genome of O. polyrhachis-furcata (strain BCC54312) round was then fed into SNAP (Korf, 2004) and Augustus (Keller,4 | KOBMOO ET AL.
Kollmar, Stanke, & Waack, 2011). The output of these two tools was 2016). Fisher's exact tests were used to compare gene counts
then fed back into MAKER for a second round of prediction. between paralogous species‐specific or complex‐specific groups and
The predicted proteins were annotated with InterProScan 5 the whole gene set for the species or complex, respectively.
(Jones et al., 2014), which also associated the protein domains Sequences within all orthologous groups were aligned with MACSE
detected with sequences in the Pfam (Finn et al., 2016) and KEGG (Ranwez, Harispe, Delsuc, & Douzery, 2011) for further analyses. A
(Kanehisa, Sato, Kawashima, Furumichi, & Tanabe, 2016; Ogata et phylogenetic tree with bootstrap support was constructed according
al., 1999) databases and with Gene Ontology (GO) terms. Small to the GTRCAT model under RAXML-HPC v8.1.5 (Stamatakis, 2014),
secreted proteins (SSPs) were identified as proteins of 10) ratios, were discarded. The functions overrepresented among
and on gene positions on scaffolds. SMGCs were predicted with the the 5% of genes with the highest dN/dS ratios were inferred by an
fungal version of antiSMASH (Weber et al., 2015). SMGC homology analysis of enrichment in GO terms. A mean dN/dS>1 for a given
across species was inferred with BiG‐SCAPE (Navarros‐Munõz J., gene indicates positive selection, whereas high dN/dS values below
https://git.wageningenur.nl/medema-group/BiG-SCAPE/wikis/home), 1 can be due to positive selection on a small number of sites within
which classified SMGCs into families based on Jaccard similarity the protein or to relaxed selection.
indices between clusters. RepeatMasker was used to predict repeti- We also formally tested for positive selection by performing site‐
tive elements for the three species from Thailand. model likelihood ratio tests (LRTs) with the CODEML program imple-
mented in PAML v.4.8a (Yang, 2007), excluding gaps and ambiguous
sites and using trees inferred under GTRCAT model from respective
2.4 | Orthology and phylogenomics
orthologous groups. CODEML estimates the parameter omega (ω = dN/
In addition to the predicted proteins from the de novo assembled dS) by maximum‐likelihood methods, allowing variation between
and improved genomes of O. unilateralis species from Thailand, we sites. While the pairwise measures above only approximate synony-
also included in our analyses the predicted proteins of other ant‐ mous and nonsynonymous rates, likelihood ratio tests (LRTs) statisti-
infecting Ophiocordyceps fungi specific to different ant species and cally compare two models of evolution, one in which ω < 1 (null
originating from different geographic areas (de Bekker et al., 2017). model) at all sites and another in which ω > 1 at some sites (alterna-
We used the available genomes from two additional O. unilateralis tive hypothesis of positive selection); LRTs thus indicate whether a
s.l. species (O. kimflemingiae from the United States and O. cam- model with positive selection is more likely than a model without
ponoti-rufipedis from Brazil), from two cryptic species of O. australis positive selection. We compared the M7 (beta distribution of ω) and
s.l., from Ghana and Brazil (de Bekker et al., 2017), and from O. sub- M8 (beta distribution of ω with a proportion of sites with ω > 1;
ramanianii s.l., also from Ghana. The predicted proteins correspond- Nielsen & Yang, 1998; Yang et al., 2000) models, and the M8a (simi-
ing to all these genomes were subjected to Blast comparisons with lar to M8 but with a category of sites evolving with ω = 1) and M8
−5
each other, with a significance threshold e‐value of 1e . The Blast (Swanson, Nielsen, & Yang, 2003) models in LRTs. Only genes with a
results were used as input for orthAgogue (Ekseth, Kuiper, & Miro- p‐value below 0.05 after false‐discovery rate (FDR) correction were
nov, 2014), a tool for the rapid inference of orthologous groups with considered significant. The M7 vs. M8 test is known to lack robust-
the Markov clustering algorithm (MCL, Dongen, 2000). This algo- ness when the probability mass is located around ω = 1, in which
rithm recovers species‐specific paralogous groups, with genes from a case this test gives a high proportion of false positives; under these
given species considered to be more closely related to each other conditions, the M8a vs. M8 test is preferred (Swanson et al., 2003).
than to any other gene in any other species. The functional annota- We ensured the robustness of our results by considering only genes
tions obtained for O. polyrhachis-furcata were transferred to the in which significant evolution under positive selection was detected
other species for gene copies in the same orthologous group. Spe- in both tests. We checked for enrichment in particular GO terms
cies‐specific paralogous genes were annotated as described above. among the genes evolving under positive selection.
We analysed GO term enrichment among species‐ or complex‐speci- We also investigated whether genes encoding heat‐labile entero-
fic paralogs, with the TOPGO package in R (Alexa & Rahnenfuhrer, toxins evolved under positive selection specifically in ant‐infectingKOBMOO ET AL. | 5
Ophiocordyceps and not in other Hypocrealean fungi. We therefore mate‐pair libraries (Wichadakul et al., 2015) (Table 1). These gen-
downloaded predicted gene sequences from other Hypocrealean omes are markedly larger than those reported for O. kimflemingiae
fungi that were annotated as putative heat‐labile enterotoxins from (OKi: 23.91 Mb), O. camponoti-rufipedis (OCR: 21.91 Mb), O. australis
the Ensembl Genome database (Herrero et al., 2016). Putative heat‐ s.l. from Brazil (OAB: 23.32 Mb) and from Ghana (OAG: 22.19 Mb),
labile enterotoxin genes were retrieved for 14 entomopathogenic and O. subramanianii s.l. (OSS: 32.31 Mb), but all these previously
fungi (one strain per species) (Supporting Information Table S1): published genomes were more fragmented than our assemblies
Metarhizium anisopliae ARSEF23 (24 genes), M. acridum CQMa 102 (Table 1).
(three genes) (Pattemore et al., 2014); M. album ARSEF1941 (12 Despite the differences in genome size, the numbers of predicted
genes), M. brunneum ARSEF3297 (32 genes), M. guizhouense genes were of a same order of magnitude across species (Table 1),
ARSEF977 (32 genes), M. majus ARSEF297 (32 genes) (Hu et al., although the number of predicted genes was nevertheless largest for
2014); M. rileyi RCEF4871 (three genes), Isaria fumosorosea OSS. For the three species from Thailand, OPF had the largest num-
ARSEF2679 (five genes), Aschersonia aleyrodis RCEF2490 (14 genes), ber of predicted genes, probably because the protein and transcript
Cordyceps confragosa RCEF1005 (six genes), C. brongniartii training set used for prediction came from this species. The number
RCEF3172 (30 genes) (Shang et al., 2016), Cordyceps militaris CM01 of SSPs was similar between the three Thai species. The number of
(one gene, Zheng et al., 2011), Beauveria bassiana ARSEF2860 (six genes with assigned Pfam domains or InterPro classification and the
genes, Xiao et al., 2012); and Ophiocordyceps sinensis Co18 (13 complete predicted gene sets obtained by core eukaryotic genes
genes, Xia et al., 2017). We also included putative heat‐labile entero- mapping (CEGMA) were also very similar in the three species (~95%:
toxin sequences from two nematode‐killing fungi: Purpureocillium Table 1), but smaller than those for species from the New World
lilacinum PLBJ‐1 (two genes, Wang et al., 2016) and Pochonia (~99%).
chlamydosporia 170 (four genes). Orthologs between these
sequences and the putative enterotoxins of O. unilatealis species
3.2 | Orthology and phylogenomics
studied here were identified. The occurrence of clade‐specific posi-
tive selection in O. unilateralis was assessed with branch‐model LRTs The genomes used in this study were sequenced from individuals
in PAML (Yang, 1998; Yang & Nielsen, 1998) and with the BUSTED belonging to one of the three species complexes: O. unilateralis s.l.,
test, an alignment‐wide test of episodic positive selection (Murrell et O. australis s.l. and O. subramanianii s.l. Most of the genes were com-
al., 2015). Both these tests are log‐likelihood ratio tests comparing a mon to all three complexes (Figure 1a): 8,554 orthologous groups
model in which positive selection is allowed in the foreground were retrieved, 5,718 of which were common to all complexes. For
branches (i.e., the clade of interest) to the null model in which posi- orthologous groups present in only one of the three complexes (Sup-
tive selection is not allowed. The branch model (Yang & Nielsen, porting Information Table S2), pathogenesis (GO:0009405) was the
1998), as implemented in PALM, detects positive selection by allow- function displaying the most significant enrichment in all complexes
ing a candidate clade to have a dN/dS ratio higher than those of the (Bonferroni‐corrected p‐values: 2e−10 for O. unilateralis s.l., 0.016 for
other branches (background branches) without taking into account O. australis s.l., 3.4e−5 for O. subramanianii s.l.), mostly due to the
variation between sites or allowing variation between branches of presence of genes encoding putative heat‐labile enterotoxins in
the same category. By contrast, BUSTED is a stochastic test using these complex‐specific genes. Complex‐specific genes were also
information from all sites and branches; it is therefore considered to found to be enriched in interspecies interactions and multi‐organism
have greater statistical power (Murrell et al., 2015). process functions.
Within each species complex, most of the genes were common
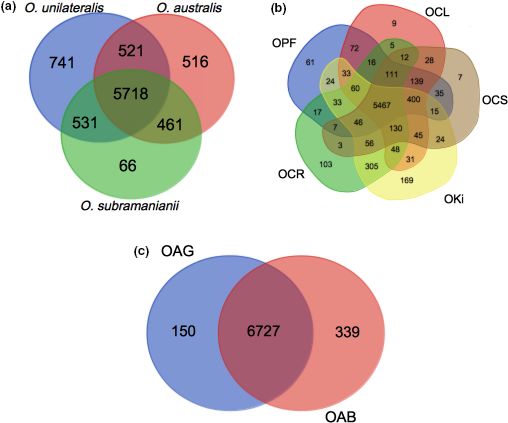
to several species (Figure 1b,c). The function pathogenesis was
3 | RESULTS
found to be overrepresented among species‐specific genes (Support-
ing Information Tables S3 and S4), due to the presence of genes
3.1 | General genome features
encoding heat‐labile enterotoxins, and SSPs (Tables 2 and 3). In par-
The three reference genomes of closely related species sequenced ticular, we detected an overrepresentation of SSPs among the genes
here differed considerably in size, O. camponoti-saundersi (OCS) unique to O. kimflemingiae (p‐value = 0.003) relative to O. unilateralis
being the largest (49.26 Mb), followed by O. polyrhachis-furcata s.l. complex, and among the genes unique to O. australis from Brazil
(OPF) (43.25 Mb) and O. camponoti-leonardi (OCL) (37.91 Mb). These relative to O. australis s.l. complex (p‐value = 0.001). None of these
differences probably partly reflect methodological differences as the species‐specific SSPs had a predicted function, suggesting an expan-
OPF genome is an improved version of a genome sequenced with a sion of rapidly evolving families of effectors (Kim et al., 2016).
different technology (454 pyrosequencing combined with Illumina There were 4,651 orthologous groups common to all eight gen-
mate‐pair sequencing, Wichadakul et al., 2015). OCL and OCS were omes. We used a subset of 4,014 single‐copy orthologous groups
sequenced and assembled with the same methodology, so the common to all species to construct a phylogenetic tree (Figure 2). This
observed differences probably reflect genuine differences in genome tree recovered the expected relationships between the sibling species
size. OCS also had more scaffolds (1700) than OCL (531). OPF had from Thailand, with O. polyrhachis-furcata being the most closely
fewer scaffolds and larger contigs, due to the use of variable‐size related to O. camponoti-leonardi (Kobmoo et al., 2012, 2015); the6 | KOBMOO ET AL.
T A B L E 1 Genome summary statistics for the ant‐infecting Ophiocordyceps species used in this study
OPF OCL OCS OKi OCR OAB OAG OSS
Species (sample name) (BCC54312)a (NK511ss‐8) (NK405ss‐6) (SC16a)b (Map‐16)b (Map‐64)b (1348a)b (1346)b
Genome size in Mb (scaffolds >1 kb) 43.25 37.91 49.26 23.91 21.90 23.32 22.19 32.30
Number of scaffolds (>1 kb) 68 531 1,700 1,64 2,204 595 2,296 3,395
Largest scaffold (kb) 5,272.94 574.15 755.06 167.40 146.68 427.81 117.86 138.81
N50 (kb) 2,974.013 139.47 102.43 26.91 23.06 111.99 17.42 17.59
GC content (%) 45.03 45.88 40.13 55.92 56.1 53.13 53.48 60.35
Number of Ns per 100 kb 5,426.84 11.32 15.22 739.17 13.02 403.43 554.75 376.08
Number of protein‐coding genes 8,988 7,059 6,970 8,629 7,621 8,174 7,995 11,275
Number of exons per gene 3.57 3.00 2.98 3.00 2.00 2.00 2.00 2.00
Exon length (median) 146 303 303 220 273 268 290 266
Core eukaryotic gene mapping 95.56 95.16 95.97 99.13 98.69 99.13 98.25 98.47
(CEGMA) completeness (%)
Repetitive content (% of the genome) 5.23 5.41 5.65 6.83 6.59 2.87 2.45 4.06
Number of genes with SignalP 716 811 761 914 840 802 681 1,064
Number of small secreted proteins 270 252 239 373 802 776 648 1,027
(SSPs)
Notes. OAB: Ophiocordyceps australis from Brazil; OAG: Ophiocordyceps australis from Ghana; OCL: Ophiocordyceps camponoti-leonardi; OCR: Ophiocordy-
ceps camponoti‐rufipedis; OCS: Ophiocordyceps camponoti-saundersi; OKi: Ophiocordyceps kimflemingiae; OPF: Ophiocordyceps polyrhachis-furcata; OSS:
Ophiocordyceps subramanianii. aImproved from Wichadakul et al. (2015). bTaken from de Bekker et al. (2017).
F I G U R E 1 Inference of orthologous
groups: Venn diagram showing the number
of orthologous groups common to and
specific to species complexes and species
a. between the three ant‐infecting
Ophiocordyceps species complexes used in
this study; (b) between the species in the
O. unilateralis complex
(OPF = O. polyrhachis-furcata,
OCL = O. camponoti-leonardi,
OCS = O. camponoti-saundersi,
OKi = O. kimflemingiae,
OCR = O. camponoti-rufipedis), (c) between
the species in the O. australis complex
(OAG = O. australis from Ghana,
OAB = O. australis from Brazil)
species from the Americas, O. kimflemingiae and O. camponoti-rufipe-
3.3 | Variation of dN/dS across genomes and
dis, clustered together but were separate from those from Thailand,
putative functions
corresponding to the separation between the Old and New Worlds
observed in a previous study (Evans, Araújo, Halfeld, & Hughes, 2018). The median pairwise dN/dS ratio was 0.081, indicating that most sin-
The two O. australis s.l. species were grouped together and formed, gle‐copy orthologs evolved under strong purifying selection (Fig-
with O. subramanianii, an outgroup to the O. unilateralis complex. ure 3a). No orthologous group had dN/dS > 1 (SupportingKOBMOO ET AL. | 7
T A B L E 2 Characteristics of orthologous groups specific to different species among the Ophiocordyceps unilateralis sensu lato complex
Number of
predicted genes
Number of (number of genes Species‐specific SSPs/genes,
specific with Pfam whole‐genome SSPs/genes
orthologous domains/InterPro (p‐value for enrichment analysis
Species groups classification) Enriched functions (GO term; FDR p‐value) of SSPs)
O. polyrhachis-furcata 61 519 (25) Pathogenesis (GO:0009405;0.0054) 18/519, 270/7678 (0.60)
Interspecies interaction between organisms
(GO:0044419; 0.0021)
Multiorganism process (GO:0051704; 0.0021)
O. camponoti-leonardi 9 9 (8) ‐ ‐
O. camponoti-saundersi 7 9 (6) ‐ ‐
O. kimflemingiae 169 185 (83) ‐ 17/185,373/7457(0.003)
O. camponoti-rufipedis 103 122 (49) Pathogenesis (GO:0009405;0.023) 17/122,802/6868(0.396)
Interspecies interaction between organisms
(GO:0044419; 0.023)
Multiorganism process (GO:0051704; 0.023)
Note. SSPs: small secreted proteins.
T A B L E 3 Characteristics of orthologous groups specific to different species among the Ophiocordyceps australis sensu lato complex
Number of
Number of predicted genes Species‐specific SSPs/genes,
specific (number of genes whole‐genome SSPs/genes
orthologous with Pfam domains/ (p‐value for enrichment analysis
Species groups InterPro classification) Enriched functions (GO term; FDR p‐value) of SSPs)
O. australis Ghana 150 173 (88) ‐ 14/173,648/7414 (0.892)
O. australis Brazil 339 356 (201) Pathogenesis (GO:0009405; 1.07e−5) 66/356,776/7558 (0.001)
Interspecies interaction between organisms
(GO:0044419; 1.07e−5)
Multiorganism process (GO:0051704; 1.07e−5)
Note. SSPs: small secreted proteins.
F I G U R E 2 The best maximum‐
likelihood tree based on 4,014 single‐copy
orthologous groups with bootstrap
supports. The horizontal scale bar
represents the branch length based on
substitution rates 0.098 | KOBMOO ET AL.
(a) (b)
F I G U R E 3 Distribution of pairwise nonsynonymous‐to‐synonymous substitution ratios (dN/dS) for the genes in all single‐copy orthologous
groups with at least four species of ant‐infecting Ophiocordyceps represented. (a) Whole‐genome dN/dS distributions, (b) Boxplots of pairwise
dN/dS values for the whole genome (small secreted protein‐coding genes or SSPs, in blue, vs. non‐SSPs, in red) and between different
categories of genes suspected a priori to be involved in pathogenesis and virulence, that is, with the putative functions of enterotoxins, core
proteins of secondary metabolism (SM), lipases, proteases (including subtilisin‐like, trypsin and aspartyl proteases) and trehalases. The dotted
line represents the mean dN/dS value for the whole genome (0.145)
Information Table S5). We investigated the putative functions of the higher than that of other genes (t test: Bonferroni‐corrected
5% of genes with the highest dN/dS values (297 genes) (Supporting p‐value = 0.885) (Figure 3b). The genes encoding putative SSPs had
Information Table S5), even if these ratios were below 1, as this a significantly higher dN/dS ratio than non‐SSP genes (t test:
could be indicative of positive selection at a small number of sites in p‐value = 2.2e‐16) (Figure 3b). This suggests that a higher proportion
the protein, although relaxed selection cannot be excluded for dN/dS of genes may evolve under positive selection among SSP‐encodingKOBMOO ET AL. | 9
T A B L E 4 Results of the gene ontology (GO) term enrichment analyses for the genes with significant likelihood ratio test (LRT) results for
positive selection in both the M7 vs. M8 (Nielsen & Yang, 1998) and M8a vs. M8 (Swanson et al., 2003) comparisons
GO Category GO.ID Term p‐value
Biological process GO:0009405 Pathogenesis 0.033
GO:0044419 Interspecies interaction between organisms 0.033
GO:0051704 Multiorganism process 0.033
Molecular function GO:0090729 Toxin activity 5.7e‐4
GO:0005524 ATP Binding 0.013
GO:0032559 Adenyl ribonucleotide binding 0.013
GO:0030554 Adenyl nucleotide binding 0.013
GO:0016301 Kinase activity 0.013
GO:0000166 Nucleotide binding 0.013
GO:1901265 Nucleoside phosphate binding 0.013
GO:0016772 Transferase activity, transferring phosphorus‐containing group 0.013
GO:0036094 Small molecule binding 0.013
GO:0016773 Phosphotransferase activity, alcohol group as an acceptor 0.013
Cellular compartment GO:0005615 Extracellular space 3.7e‐4
GO:0044421 Extracellular region part 5.3e‐4
GO:0005576 Extracellular region 5.3e‐4
F I G U R E 4 Percentages of genes in
various functional categories for which
likelihood ratio tests (LRTs) for positive
selection (M7 vs. M8 and M8a vs. M8)
yielded significant results (false‐discovery
rate‐corrected p‐value < 0.05).
Proteases = subtilisin, trypsin and aspartyl
proteases, SM = core genes of secondary
metabolites. The total number of genes in
each category is indicated above the bars
high‐energy molecule (ATP) to a substrate and are involved in vari- essential for pathogen growth and survival and, thus, for pathogene-
ous cellular processes. The proportion of kinases evolving under pos- sis and virulence (Lee et al., 2016).
itive selection was lower than that of heat‐labile enterotoxins The functions relating to hydrolytic enzymes important for
(Figure 4), but the numbers of kinases and heat‐labile enterotoxins pathogenesis (glycoside hydrolases, lipases, proteases) were not
evolving under positive selection were similar, and these two func- overrepresented among the genes evolving under positive selection.
tions were overrepresented among the genes evolving under positive Indeed, the proportions of genes in these families found to be under
selection. Most of these kinases were annotated as protein kinases, positive selection were markedly smaller than those for heat‐labile
histidine kinases and phosphatidylinositol 3 and 4‐kinases. These enterotoxins (Figure 4). Nevertheless, several of the genes from
families of kinases are well known to be involved in cell signalling, these gene families were found to evolve under positive selection in10 | KOBMOO ET AL.
model tests (Supporting Information Table S7) and can be considered cytochrome b5‐like haem/steroid‐binding domain. The first of these
good candidates for involvement in co‐evolution and host specificity. genes was shown to be associated with iron uptake in yeast (Roman,
One to three of the 11 chitinases (GH18) presented significant foot- Dancis, Anderson, & Klausner, 1993), whereas the product of the
prints of positive selection depending on the evolution model con- second mediates iron‐free electron transfer. The third of these genes
sidered, and significant p‐values were obtained in all tests for one of may encode a nitrate reductase or sulphite oxidase, both of which
these enzymes. Chitinases are involved in the degradation of the are involved in nitrogen assimilation. These putative neuromodula-
insect cuticle, a major component of the insect exoskeleton, and in tors thus seem to be involved in host resource utilization. The neu-
the degradation and remodelling of fungal cell walls (Adams, 2004; rological disorder displayed by zombie ants infected with
Langner & Göhre, 2016). Other GH families converging to various Ophiocordyceps may result from the pathogen outcompeting the host
functions, such as cellulase, glucanase, glucosidase and galactosidase for iron and nitrogen.
(e.g., GH5, GH16, GH47, GH76), also included a few genes display-
ing significant tests of positive selection (Supporting Information
3.5 | Positive selection of heat‐labile enterotoxin
Table S7). Neither of the two trehalases (GH37), which are thought
genes specific to the ant‐manipulating O. unilateralis
to play important roles in nutrient acquisition within the host body,
species complex
displayed significant signs of positive selection.
Zero to four of the nine subtilisin‐like (MEROPS family S08 and The above results and those of previous studies (de Bekker et al.,
S53) and trypsinlike proteases (MEROPS family S01), which are con- 2015; Wichadakul et al., 2015) suggest that heat‐labile enterotoxin
sidered to act as cuticle‐degrading proteases, presented significant genes are candidate genes for host‐specific adaptation. We there-
footprints of positive selection, depending on the evolution model fore investigated whether the positive selection detected above
considered. Zero to one of 16 putative aspartyl proteases (MEROPS was specific to the ant‐infecting Ophiocordyceps species or general
family A01) was found to evolve under positive selection following to Hypocrealean entomopathogenic and nematode‐killing fungi.
different models. However, none of these proteases yielded signifi- Thirty‐six orthologous groups of heat‐labile enterotoxin genes were
cant results in both tests (Figure 4). Two to six of the 39 putative inferred for a group of 16 Hypocrealean entomopathogenic and
lipases yielded significant p‐values in positive selection tests, and nematode‐killing fungi in addition to our eight focal species (Sup-
only one yielded significant p‐values in both tests (Figure 4; Support- porting Information Table S1); 22 of these orthologous groups
ing Information Table S7). included at least one sequence from the ant‐infecting Ophiocordy-
One to four of the seven core genes of secondary metabolites ceps, and 10 (42%) of these groups included only sequences from
displayed significant signatures of positive selection, depending on the ant‐infecting Ophiocordyceps species. We further analysed the
the evolution model considered (Supporting Information Table S7). only group (ORTHAgEnt13) common to at least four of the ant‐
The only gene to yield significant p‐values in both tests (orthologous infecting Ophiocordyceps species considered and sequences recov-
group ORTHAg2248, Supporting Information Table S7) encoded a ered from other species from Hypocreales, for which both site‐
polyketide synthase (PKS)‐like protein with a beta‐ketoacyl synthase model LRTs for positive selection were significant. This group
domain. Beta‐ketoacyl synthase is involved in fatty acid biosynthesis included five sequences each from an O. unilateralis species. In a
and has been shown to be involved in the production of polyketide maximum‐likelihood tree, all the O. unilateralis sequences were
antibiotics in fungi (Beck, Ripka, Siegner, Schiltz, & Schweizer, 1990). located on the same branch (Figure 6). The PAML branch‐model
The gene encoding this enzyme is part of a secondary metabolic LRTs indicated that this gene was evolving under positive selec-
gene cluster that is highly syntenic across the species of the O. uni- tion specifically in the O. unilateralis clade (p‐values < 0.001). The
lateralis complex, but located in different clusters in O. australis and branch at the base and the internal branches of the O. unilateralis
in O. subramanianii (Figure 5). clade therefore had significantly higher dN/dS ratios than the
We also investigated whether the genes previously identified as other branches (Figure 6). The BUSTED test, which is similar to
encoding possible “neuromodulators” (de Bekker et al., 2017), based PAML branch tests but considered more powerful, also gave a sig-
on their overexpression during the manipulation of ant behaviour, nificant result (p‐value = 6.16e‐14).
showed signs of positive selection. In total, 12 to 41 of these genes
yielded significant results in tests for positive selection (Supporting
4 | DISCUSSION
Information Table S8). Five genes yielded significant results in both
tests. These genes encoded a short‐chain dehydrogenase, a DNA
4.1 | Enterotoxin genes as major candidate genes
mismatch repair protein (MutC), a DNA replication factor, an ATPase
underlying host adaptation
and a protein with no functional annotation. Seven other genes
yielded results only in the M8a vs. M8 test, which is more robust Comparative genomic studies of closely related species of fungal
than the M7 vs. M8 test. These seven genes included oxidoreduc- pathogens have shown that, in general, genes involved in adaptation,
tases clearly involved in metabolic reactions: a protein with a ferric‐ particularly those involved in virulence and pathogenicity, are spe-
reductase transmembrane‐like domain, a flavodoxin oxidoreductase cies‐specific, highly divergent and/or under diversifying selection, as
and an oxidoreductase binding to a molybdopterin cofactor with a a result of the arms race between host and pathogen orKOBMOO ET AL. | 11 F I G U R E 5 Homology of putative secondary metabolic gene clusters (SMGCs) with the core gene under positive selection according to log‐ likelihood ratio tests (M7 vs. M8 models and M8a vs. M8 models). The dashed lines indicate orthology between the putative polyketide synthase (PKS)‐like core gene. The phylogenetic tree was inferred from Jaccard similarity indices between alignments of common gene domains within families. OCS = Ophiocordyceps camponoti-saundersi, OCL = O. camponoti-leonardi, OPF = O. polyrhachis-furcata, OKi = O. kimflemingiae, OCR = O. camponoti-rufipedis, OAG = O. australis from Ghana, OAB = O. australis from Brazil, OSS = O. subramanianii specialization on new hosts (Ghanbarnia et al., 2015; Huang, Si, differences between species, suggesting the occurrence of diversi- Deng, Li, & Yang, 2014; Plissonneau et al., 2017; Stukenbrock et al., fying selection, which was confirmed by formal tests comparing 2011). We therefore used an evolutionary comparative genomic models with and without positive selection. Furthermore, in the approach for identifying genes underlying host adaptation in ant‐ cases in which orthologs of enterotoxin genes were found in infecting Ophiocordyceps from three species complexes (O. unilater- other entomopathogenic fungi, we inferred that positive selection alis s.l., O. australis s.l. and O. subramanianii s.l.). Genome comparisons was specific to the ant‐infecting Ophiocordyceps clade. These find- showed that species complex‐specific genes were enriched in genes ings support the view that heat‐labile enterotoxins are effectors associated with the function pathogenesis which included genes involved in host adaptation, as previously suggested based on encoding heat‐labile enterotoxins. The species‐specific genes were observations of enterotoxin overexpression during manipulation of also enriched in this function. However, most species‐specific genes the behaviour of the diseased ants (de Bekker et al., 2015) and of lacked functional annotation, perhaps due to their rapid evolution as the species‐specific nature of behavioural manipulation (de Bekker part of the arms race between pathogen and host, resulting in et al., 2014; Sakolrak et al., 2018). The proximal mechanisms via homology no longer being detectable. Most of the small secreted which enterotoxins act during infection and the manipulation of proteins (SSPs), in particular, lacked predicted functions, but these host behaviour remain unclear, but it has been suggested that proteins were particularly abundant among the species‐specific these molecules interfere with the chemical communication of genes. SSPs may act as effectors in pathogenicity, but the validation social insects; bacterial enterotoxins have been shown to affect of their function requires further studies. pheromone production in boll weevils (Wiygul & Sikorowski, 1986, Heat‐labile enterotoxin genes were also overrepresented in the 1991). Alterations to chemical communication may contribute to orthologous groups with the highest rates of amino acid the modification of behaviour in infected ant hosts.
12 | KOBMOO ET AL.
F I G U R E 6 The best RAxML tree based on the GTRCAT model for the orthologous group ORTHAgEnt13 of putative heat‐labile
enterotoxins in entomopathogenic and nematode‐killing fungi of the order Hypocreales. The numbers above the nodes are bootstrap supports.
The numbers below the branches are the ratios of nonsynonymous‐to‐synonymous substitution rates (dN/dS)
(Boomsma et al., 2014; Ortiz‐Urquiza & Keyhani, 2013; Wang, Fang,
4.2 | Minor role of the cuticle in exerting selective
Wang, & St. Leger, 2011; Wang & St. Leger, 2005). Nevertheless, as
pressure leading to diversifying selection
the fungi in the three ant‐infecting complexes considered here are
Hypocrealean entomopathogenic fungi are known to infect their all pathogens of formicine and ponerine ants, our findings do not
insect hosts by penetrating the cuticle from the outside (Boomsma rule out diversifying selection occurring across larger phylogenetic
et al., 2014). An array of hydrolytic enzymes, including chitinases, scales. These enzymes may be highly conserved among pathogens of
lipases and proteases, is required to break through the insect cuticle formicine and ponerine ants, providing a common arsenal for attack-
(Ortiz‐Urquiza & Keyhani, 2013). Chitins are major constituents not ing taxonomically related ants. There may also be constraints in the
only of insect cuticles, but also of fungal cell walls (Langner & Göhre, host or the fungus preventing rapid co‐evolution through changes to
2016) while lipids are a major component of the epicuticle waxy these molecules.
layer (Jarrold, Moore, Potter, & Charnley, 2007; Pedrini, Ortiz‐
Urquiza, Huarte‐Bonnet, Zhang, & Keyhani, 2013). Proteases are
4.3 | Utilization of host resources
important for the penetration of the cuticle by fungi and have been
shown to be virulence factors for the infection of insect hosts (Shah, Once inside the host, the pathogen requires other hydrolases for
Wang, & Butt, 2005). Subtilisin proteases have been shown to play a carbon assimilation. Efficient nutrient uptake from the host allows
particularly important role in regulating insect host specificity optimal proliferation of the fungus within its host and ultimately
through the differential expressions of specific isoforms (Bye & leads to insect death (Luo, Qin, Pei, & Keyhani, 2014). It has, there-
Charnley, 2008; Mondal, Baksi, Koris, & Vatai, 2016). We therefore fore, been suggested that host resource utilization is crucial for host
hypothesized that the genes encoding chitinases, proteases and specificity (Gillespie, Bailey, Cobb, & Vilcinskas, 2000). Trehalases, in
lipases might have evolved under diversifying selection. However, particular, probably play an important role in this respect. Indeed,
we found footprints of positive selection for only a few putative the fly pathogen Entomophthora muscae (Entomophthorales) carries
genes encoding these enzymes in the ant‐infecting Ophiocordyceps more trehalase‐encoding genes in its genome than its close relative,
species. This challenges the widely accepted view that the insect the generalist Conidiobolus coronatus, which is a nonobligate patho-
cuticle, as a major barrier to infections, exerts a strong selective gen (De Fine Licht, Jensen, & Eilenberg, 2017). We identified two
pressure on entomopathogenic fungi, leading to different host ranges trehalases with no positive selection signature as conserved acrossKOBMOO ET AL. | 13
all species. Other glycoside hydrolases and lipases may be involved Ferrara, & Perrone, 2013). We detected significant footprints of pos-
in breaking down primary carbon sources (Ortiz‐Urquiza & Keyhani, itive selection in some of the core genes of secondary metabolites.
2013; Schrank & Vainstein, 2010). However, the evidence for posi- The most notable case concerned a PKS‐like function involved in
tive selection is less robust for these enzymes. Thus, diversifying lipid biosynthesis. Lipids have been shown to be involved in patho-
selection in ant‐pathogenic Ophiocordyceps fungi probably acts less physiological processes in pathogenic fungi, but the role of the lipid
strongly on the function of carbon assimilation than on enterotoxins. signalling network in host‐specific pathogenicity remains to be deter-
Again, there may be constraints preventing the rapid evolution of mined (Singh & Poeta, 2011). Kinases are also known to participate
host cuticle or fungal hydrolase and lipase functions. in lipid signalling pathways, and the kinases with significant foot-
Nitrogen also plays a key role in the proliferation of ento- prints of positive selection identified included phosphatidylinositol 3
mopathogenic fungi (Luo et al., 2014). However, our results suggest and 4‐kinases. The phosphorylated form of phosphatidylinositol plays
that initial nutrient acquisition via proteinases is not under strong an important role in lipid and cell signalling (Funaki, Katagiri, Inukai,
diversifying selection. Genes evolving under positive selection were Kikuchi, & Asano, 2000). Lipid metabolism thus seemed to be subject
not enriched in functions related to the assimilation of nitrogen or to diversifying selection, although to a much lesser extent than heat‐
amino acid synthesis. labile enterotoxins.
In addition to carbon and nitrogen, iron uptake is also crucial for
pathogen success (Bairwa, Hee Jung, & Kronstad, 2017; Haas, 2012;
Sutak, Lesuisse, Tacherzy, & Richardson, 2008). The candidate neu- 5 | CONCLUSIONS
romodulator genes found to be under positive selection included
iron‐related oxidoreductases. In particular, one of the proteins identi- We focused on three ant‐infecting species complexes from the
fied had a ferric‐reductase transmembrane domain, and another was genus Ophiocordyceps, including closely related species. Complex‐
a flavodoxin oxidoreductase. Proteins with ferric‐reductase trans- and species‐specific genes were found to be enriched in genes for
membrane domains have been shown to be crucial for ferric iron heat‐labile enterotoxins, and this gene family was found to be evolv-
uptake in yeast (Roman et al., 1993), whereas flavodoxin is an iron‐ ing under positive selection to a greater extent than other candidate
free electron‐transfer protein facilitating a range of metabolic reac- gene families. Our results thus suggest that the specific adaptation
tions in the absence of iron. Specialist entomopathogens kill their and co‐evolution of specialist species in the ant‐infecting Ophiocordy-
hosts more slowly than generalists (Boomsma et al., 2014). In such a ceps fungi to their hosts is dependent on selection occurring within
context, ant‐specific Ophiocordyceps might be expected to have the body of the host rather than during cuticle penetration. By con-
developed strategies for hijacking resources from the host. The effi- trast, we detected little positive selection on lipases, proteases or
cient acquisition of iron and an ability to divert its use may be the chitinases, although we did identify a few interesting candidate
key to outcompeting the host during infection. genes from these groups. Comparative genomic studies of ento-
mopathogenic fungi remain scarce, and the few studies that have
been performed have focused exclusively on species of agricultural
4.4 | The role of kinases and signal transduction
or medical interest. The findings of this study improve our under-
Kinase enzymes are widely recognized as participating in various cel- standing of the mechanisms of fungal adaptation to insect hosts, and
lular processes, crucial to growth and survival (Lee et al., 2016). The future studies on fungal pathogens associated with other groups of
genes under positive selection in the ant‐infecting Ophiocordyceps insects should provide more general insight into the adaptation of
were enriched in kinase‐related functions. Most were clearly related entomopathogenic fungi and a more documented comparison with
to signal transduction, which plays a crucial role in interactions the mechanisms of adaptation in fungal pathogens of plants. The
between hosts and pathogens (Bahia, Satoskar, & Dussurget, 2018). insect innate immune response seems to be much more specific than
Pathogens sense and respond to environmental stimuli, including the that in plants, suggesting a certain level of acquired immune
expression of virulence factor regulatory systems, in the hostile con- response (Cooper & Eleftherianos, 2017). Fungal pathogens of
ditions of the host immune system. As extremely specialized patho- insects would be expected to display extensive expansions and con-
gens, ant‐infecting Ophiocordyceps fungi must fine‐tune their tractions of gene families, as observed in plant pathogens, but the
responses in the host body. target functions may be different. Additional insight gleaned from
entomopathogenic fungi would help to improve our general under-
standing of the mechanisms of adaptive evolution in eukaryotes.
4.5 | Importance of lipid metabolism
Many entomopathogenic fungi are also thought to deploy a plethora
ACKNOWLEDGEMENTS
of metabolites and toxins within the bodies of their hosts (Schrank &
Vainstein, 2010; Singh, Son, & Lee, 2016). The nature of these mole- This work was supported by the Marie Sklodowska Curie Action No
cules probably differs between groups of insect‐pathogenic fungi 655278 and Thailand Research Fund (TRF) Young Scientist Grant
and remains to be precisely determined, but the principal molecules (TRG5780162) to NK. We would like to thank Alodie Snirc for
include polyketides (PKs) and nonribosomal peptides (NRPs) (Gallo, advice concerning DNA extraction, Antoine Branca for suggestions14 | KOBMOO ET AL.
about bioinformatic protocols, Rayan Chikhi for training in genome Badouin, H., Gladieux, P., Gouzy, J., Siguenza, S., Aguileta, G., Snirc, A.,
assembly, Jérome Collemare and Jorge C. Navarro‐Muñoz for their … Giraud, T. (2017). Widespread selective sweeps throughout the
genome of model plant pathogenic fungi and identification of effec-
guidance on using antiSMASH and BiG‐SCAPE, and Suchada
tor candidates. Molecular Ecology, 26, 2041–2062. https://doi.org/10.
Mongkholsamrit and Kanoksri Tasanathai for the organization of 1111/mec.13976
sampling trips. We also would like to sincerely thank Clarissa de Bahia, D., Satoskar, A., & Dussurget, O. (2018). Cell signalling in host‐
Bekker and David P. Hughes for kindly sharing their data on the pathogen interactions: The host point of view. Frontiers in Immunol-
ogy, 9, 1–4. https://doi.org/10.3389/fimmu.2018.00221
candidate neuromodulators.
Bairwa, G., Hee Jung, W., & Kronstad, J. (2017). Iron acquisition in fungal
pathogens of human. Metallomics: Integrated Biometal Science, 9(3),
215–227. https://doi.org/10.1039/c6mt00301j
AUTHOR CONTRIBUTION
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov,
N.K., J.J.L. and T.G. designed the study. N.K. and N.A. conducted A. S., … Pevzner, P. A. (2012). SPAdes: A new genome assembly
algorithm and its applications to single‐cell sequencing. Journal of
sampling and DNA extraction. N.K., D.W. and RCRSLV analysed
Computational Biology, 19(5), 455–477. https://doi.org/10.1089/cmb.
sequencing and comparative genomic data. N.K. and T.G. wrote the 2012.0021
manuscript, with contributions from all the authors. Baroncelli, R., Amby, D. B., Zapparata, A., Sarrocco, S., Vannacci, G., Le
Floch, G., … Thon, M. R. (2016). Gene family expansions and con-
tractions are associated with host range in plant pathogens of the
DATA ACCESSIBILITY genus Colletotrichum. BMC Genomics, 17(555), 1–17. https://doi.org/
10.1186/s12864-016-2917-6
The de novo assemblies of Ophiocordyceps camponoti-leonardi (NCBI Barrett, L. G., & Heil, M. (2012). Unifying concepts and mechanisms in
Biosample SAMN07662903) and O. camponoti-saundersi (NCBI the specificity of plant‐enemy interactions. Trends in Plant Science, 17
Biosample SAMN07662932) have been deposited with the NCBI as (5), 282–292. https://doi.org/10.1016/j.tplants.2012.02.009
Beck, J., Ripka, S., Siegner, A., Schiltz, E., & Schweizer, E. (1990). The
whole‐genome shotgun (WGS) projects with Accession nos.
multifunctional 6‐methylsalicylic acid synthase gene of Penicillium pat-
PDHP00000000 and PDHQ00000000, respectively. The O. polyrha- ulum. Its gene structure relative to that of other polyketide synthases.
chis-furcata genome was updated based on the improved assembly European Journal of Biochemistry, 192(2), 487–498. https://doi.org/10.
from this study (LKCN00000000.2). 1111/j.1432-1033.1990.tb19252.x
de Bekker, C., Ohm, R. A., Evans, H. C., Brachmann, A., & Hughes, D. P.
(2017). Ant‐infecting Ophiocordyceps genomes reveal a high diversity
REFERENCES
of potential behavioral manipulation genes and a possible major role
Adams, D. J. (2004). Fungal cell wall chitinases and glucanases. Microbiol- for enterotoxins. Scientific Reports, 7, 12508. https://doi.org/10.
ogy, 150(7), 2029–2035. https://doi.org/10.1099/mic.0.26980-0 1038/s41598-017-12863-w
Aguileta, G., Lengelle, J., Chiapello, H., Giraud, T., Viaud, M., Fournier, E., de Bekker, C., Ohm, R. A., Loreto, R. G., Sebastian, A., Albert, I., Merrow,
… Gout, L. (2012). Genes under positive selection in a model plant M., … Hughes, D. P. (2015). Gene expression during zombie ant bit-
pathogenic fungus, Botrytis. Infection, Genetics and Evolution, 12(5), ing behavior reflects the complexity underlying fungal parasitic
987–996. https://doi.org/10.1016/j.meegid.2012.02.012 behavioral manipulation. BMC Genomics, 16(620), 1–23. https://doi.
Aguileta, G., Lengelle, J., Marthey, S., Chiapello, H., Rodolphe, F., Gen- org/10.1186/s12864-015-1812-x
drault, A., … Giraud, T. (2010). Finding candidate genes under posi- de Bekker, C., Quevillon, L. E., Smith, P. B., Fleming, K. R., Ghosh, D., Pat-
tive selection in non‐model species: Examples of genes involved in terson, A. D., & Hughes, D. P. (2014). Species‐specific ant brain
host specialization in pathogens. Molecular Ecology, 19(2), 292–306. manipulation by a specialized fungal parasite. BMC Evolutionary Biol-
https://doi.org/10.1111/j.1365-294X.2009.04454.x ogy, 14(166), 1–12. https://doi.org/10.1186/s12862-014-0166-3
Aguileta, G., Refrégier, G., Yockteng, R., Fournier, E., & Giraud, T. (2009). Boetzer, M., Pirovano, W., Zerbino, D., Birney, E., Simpson, J., & Wong,
Rapidly evolving genes in pathogens: Methods for detecting positive K. (2012). Toward almost closed genomes with GapFiller. Genome
selection and examples among fungi, bacteria, viruses and protists. Biology, 13(6), R56. https://doi.org/10.1186/gb-2012-13-6-r56
Infection, Genetics and Evolution, 9(4), 656–670. https://doi.org/10. Boomsma, J. J., Jensen, A. B., Meyling, N. V., & Eilenberg, J. (2014). Evo-
1016/j.meegid.2009.03.010 lutionary interaction networks of insect pathogenic fungi. Annual
Albalat, R., & Cañestro, C. (2016). Evolution by gene loss. Nature Reviews Review of Entomology, 59, 467–485. https://doi.org/10.1146/annure
Genetics, 17(7), 379–391. https://doi.org/10.1038/nrg.2016.39 v-ento-011613-162054
Alexa, A., & Rahnenfuhrer, J. (2016). topGO: Enrichment Analysis for Gene Bye, N. J., & Charnley, A. K. (2008). Regulation of cuticle‐degrading sub-
Ontology. R package version 2.28.0. tilisin proteases from the entomopathogenic fungi, Lecanicillium spp:
Anderson, P. K., Cunningham, A. A., Patel, N. G., Morales, F. J., Epstein, Implications for host specificity. Archives of Microbiology, 189(1), 81–
P. R., & Daszak, P. (2004). Emerging infectious diseases of plants: 92. https://doi.org/10.1007/s00203-007-0296-8
Pathogen pollution, climate change and agrotechnology drivers. Cantarel, B. L., Korf, I., Robb, S. M. C., Parra, G., Ross, E., Moore, B., …
Trends in Ecology and Evolution, 19(10), 535–544. https://doi.org/10. Yandell, M. (2008). MAKER: An easy‐to‐use annotation pipeline
1016/j.tree.2004.07.021 designed for emerging model organism genomes. Genome Research,
Anderson, J. P., Gleason, C. A., Foley, R. C., Thrall, P. H., Burdon, J. B., & 18, 188–196. https://doi.org/10.1101/gr.6743907
Singh, K. B. (2010). Plants versus pathogens: An evolutionary arms Chikhi, R., & Medvedev, P. (2014). Informed and automated k‐mer size
race. Functional Plant Biology, 37(6), 499–512. https://doi.org/10. selection for genome assembly. Bioinformatics, 30(1), 31–37.
1071/FP09304 https://doi.org/10.1093/bioinformatics/btt310
Araújo, J. P. M., Evans, H. C., Kepler, R., & Hughes, D. P. (2018). Zombie‐ Cooper, D., & Eleftherianos, I. (2017). Memory and specificity in the
ant fungi across continents: 15 new species and new combinations insect immune system: Current perspectives and future challenges.
with Ophiocordyceps. I. Myrmecophilous hirsutelloid species. Studies in Frontiers in Immunology, 8(539), 1–6. https://doi.org/10.3389/fimmu.
Mycology, 90, 119–160. 2017.00539You can also read

















































