Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
JBC Papers in Press. Published on March 16, 2020 as Manuscript RA120.012697
The latest version is at https://www.jbc.org/cgi/doi/10.1074/jbc.RA120.012697
Structure-based discovery of a small-molecule inhibitor of methicillin-
resistant Staphylococcus aureus virulence
Jie Liu‡, Lina Kozhaya§, Victor J. Torres¶, Derya Unutmaz§, and Min Lu‡1
From the ‡Public Health Research Institute, Department of Microbiology, Biochemistry and
Molecular Genetics, New Jersey Medical School, Rutgers University, Newark, New Jersey
07103, the §Jackson Laboratory for Genomic Medicine, Farmington, Connecticut 06032, and the
¶
Department of Microbiology, New York University School of Medicine, New York, New York
10016
Running title: Discovery of a small-molecule inhibitor of MRSA virulence
1
To whom correspondence should be addressed: Public Health Research Institute, Department of
Microbiology, Biochemistry and Molecular Genetics, New Jersey Medical School, Newark, NJ
07103. Tel.: (973) 854-3260; E-mail: lum1@njms.rutgers.edu.
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
Keywords: Staphylococcus aureus (MRSA), pore-forming toxin, virulence factor, antivirulence
therapy, structure-based drug design, phosphatidylcholine, structural biology, Panton–Valentine
leukocidin (PVL), α-toxin, leukocidin ED (LukED)
protects primary human immune cells in vitro
ABSTRACT against cytolysis by PVL and α-toxin and hence
The rapid emergence and dissemination may serve as the basis for the development of
of methicillin-resistant Staphylococcus aureus an antivirulence agent for managing MRSA
(MRSA) strains poses a major threat to public infections.
health. MRSA possesses an arsenal of secreted
host-damaging virulence factors that mediate
pathogenicity and blunt immune defenses. Infection with Staphylococcus aureus
Panton–Valentine leukocidin (PVL) and α- can cause severe and devastating illness and is
toxin are exotoxins that create lytic pores in the one of the leading causes of death by any
host cell membrane. They are recognized as infectious agent in the United States (1, 2). S.
being important for the development of aureus is notorious for its ability to acquire
invasive MRSA infections and are thus genetic determinants of antibiotic resistance
potential targets for antivirulence therapies. and virulence that enhance fitness and
Here, we report the high-resolution X-ray pathogenicity (3, 4). Methicillin resistant S.
crystal structures of both PVL and α-toxin in aureus (MRSA) now accounts for >60% S.
their soluble, monomeric and oligomeric aureus isolates in US intensive care units,
membrane-inserted pore states in complex with severely restricting antibiotic treatment options
n-tetradecylphosphocholine (C14PC). The (2). MRSA also spreads rapidly among healthy
structures revealed two evolutionarily individuals in the community, causing
conserved phosphatidylcholine-binding predominantly skin and soft tissue infections
mechanisms and their roles in modulating host and life-threatening infections, including
cell attachment, oligomer assembly, and bacteremia, endocarditis, osteomyelitis and
membrane perforation. Moreover, we necrotizing pneumonia (2). Disturbingly,
demonstrate that the soluble C14PC compound MRSA can live in the biofilm state (5, 6), and
1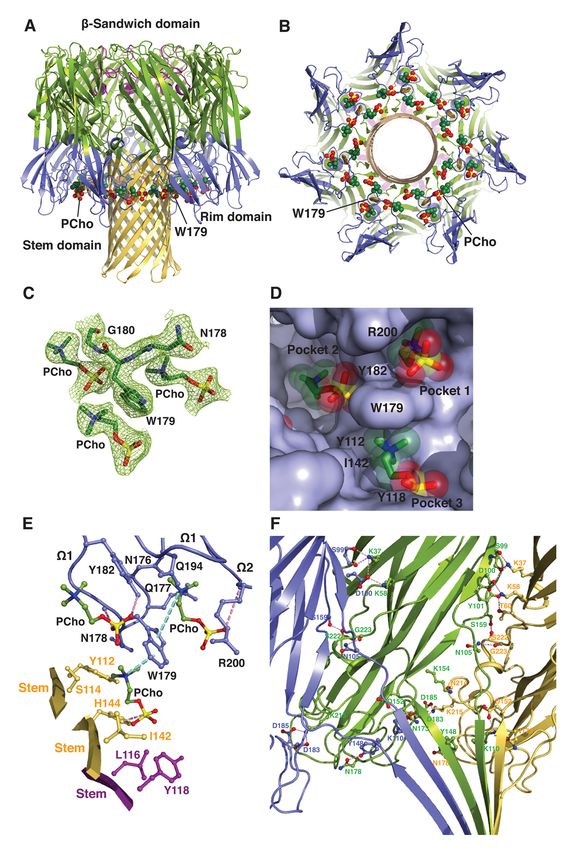
Discovery of a small-molecule inhibitor of MRSA virulence
it has long been recognized that biofilms prepore heterooctamer forms of HlgAB and
increase resistance to antimicrobial agents and HlgCB have been determined (27–30). These
the host immune response (7). MRSA is structures, and supporting biochemical and
currently treated with vancomycin, genetic data (26, 31–33) , suggest that members
clindamycin, linezolid and daptomycin (8) but of this subfamily share a common mechanism
resistance to these “last-resort” antibiotics has of cytolytic action (reviewed in Refs. 34 and
been reported (9–13). For these reasons, the 35). The cytolytic process begins with the
World Health Organization identifies MRSA binding of soluble toxin monomers to a cell
as one of six “high” priority pathogens that surface receptor (21, 36). The membrane-
pose an enormous threat to public health (14). bound monomers then associate to form a
Thus, new therapeutics with novel mechanisms nonlytic, oligomeric prepore. Finally, the
of action are desperately needed to combat this translocation of the prestem regions across the
high threat pathogen. membrane results in the bilayer-spanning b-
USA300 is the most prevalent strain of barrel pore structure and consequent membrane
MRSA in the US and represents a growing permeabilization and cell lysis.
threat in both community and healthcare MRSA strains that harbor the phage-
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
settings (15). Its heightened virulence and encoded PVL have been linked to highly
severity are related to the production of a virulent and severe community-acquired skin
cocktail of cytolytic pore-forming exotoxins infections (37), as well as necrotizing
that mediate virulence and impair host immune pneumonia and lethal necrotizing fasciitis (38).
defenses (3, 16). The pharmacological The role of PVL production in the pathogenesis
targeting of these cytotoxins has been of MRSA was demonstrated in both a rabbit
recognized as a promising new therapeutic model of necrotizing pneumonia and
approach to reducing morbidity and mortality humanized mouse models of skin infection and
associated with MRSA infection. Leukocidins pneumonia (39–41). PVL induces leukocyte
and a-toxin, secreted by S. aureus as water- destruction and tissue necrosis through
soluble, monomeric polypeptides, constitute interaction with the complement
the a-hemolysin subfamily of b-barrel pore- receptors C5aR and C5L2 (42–45). PVL, in
forming toxins (17). Five different bipartite conjunction with HlgAB, contributes to MRSA
leukocidins have been described, including biofilm-mediated killing of neutrophils (46).
Panton-Valentine leukocidin (PVL), On the other hand, the chromosomally encoded
leukocidin ED (LukED), two g-hemolysins a-toxin lyses epithelial and endothelial cells,
(HlgAB and HlgCB) and leukocidin AB red blood cells, lymphocytes and monocytes by
(LukAB; also known as LukGH), each of targeting its receptor, the metalloprotease
which consists of two distinct polypeptides ADAM10 (36, 47). The elevated expression of
referred to as the S and F subunits (reviewed in a-toxin in the USA300 clone and in historic
Ref. 18). Their cellular tropism and species human epidemic strains correlates with
specificity are determined by the S subunits increased pathogenicity in mouse models of
LukS-PV, LukE, HlgA, HlgC and LukA (19– skin and soft tissue infection, pneumonia and
21). The S and F subunits and single- sepsis (48, 49). a-Toxin also plays a role in
component a-toxin share a unique modular biofilm formation by clinical MRSA isolates
structure consisting of the amino latch and (50). Moreover, LukED relies on the
prestem regions and the b-sandwich and rim chemokine receptor CCR5 to kill T
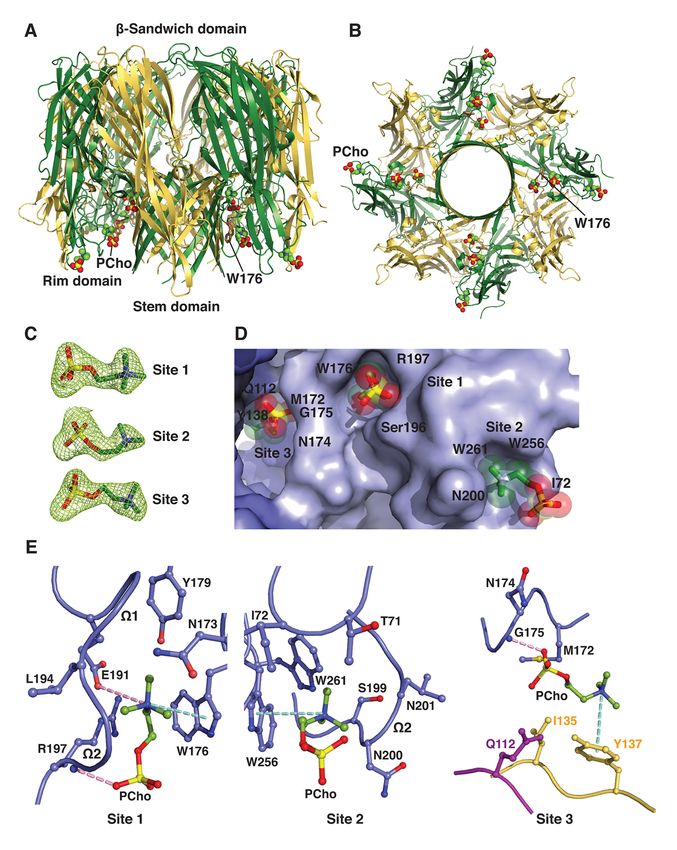
domains (see Fig. 1A) (22–26). The X-ray lymphocytes, macrophages and dendritic cells,
crystal structures of the membrane-inserted as well as CXCR1 and CXCR2 to kill
pore oligomer forms of a-toxin, HlgAB and leukocytes (19, 51). Inhibition of the
LukGH and of the membrane surface-bound interaction between LukED and CCR5 has
2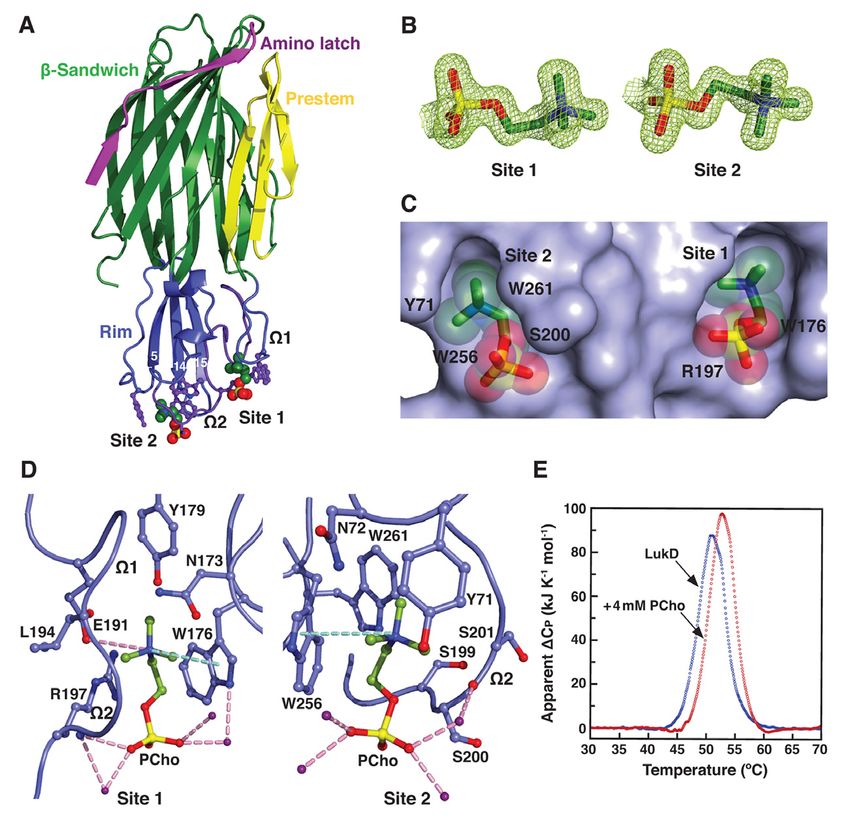
Discovery of a small-molecule inhibitor of MRSA virulence
been shown to block cytotoxicity and attenuate C14PC binds to the rim domain of LukD at two
S. aureus infection in mice (19). Together, adjacent but distinct sites
these cytotoxins can modulate phagocytic cell To better understand the molecular basis
functions via their specific receptors and for the recognition of PCho by the leukocidin F
contribute to MRSA immune evasion and subunits, we determined the crystal structures
disease pathogenesis. As such, the discovery of LukD with and without C14PC at 1.5 Å and
and development of new antivirulence agents 1.75 Å resolution, respectively (Table 1).
that protect from the combined immune C14PC was selected in the present study as a PC
cytolytic activities of this subfamily of pore- mimic for its high micellization efficiency due
forming toxins is of utmost importance. to low critical micelle concentration. The two
There is considerable evidence pointing protein structures are closely similar, with a
to the role of phosphatidylcholine (PC) in the r.m.s.d. for Ca atoms of 0.69 Å. The rim
mechanism of pore formation by these toxins. domain forms an antiparallel, three-stranded
PC is an absolute requirement for pore open-face b-sandwich toppled by two surface-
formation by a-toxin, HlgAB and HlgCB and exposed consecutive W loops (residues 180–
has been shown to inhibit their cytolytic effects 194, W1 and 195–202, W2) (Fig. 1A). Two
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
(52–56). Particularly, crystallographic studies PCho moieties that bind to opposite sides of the
revealed the presence of single, highly W2 loop were unexpectedly discovered upon
conserved phosphocholine (PCho) binding examination of the difference electron density
sites on the rim domains of the monomeric F map in the C14PC-bound structure (Fig. 1B).
subunit HlgB and the a-toxin protomer in the Average B-factor for these two moieties is 22
heptameric pore complex (22, 57). These Å2 and for surrounding solvent molecules and
binding sites have been shown by mutational protein atoms 21 Å2. The two binding sites are
analysis to be required for membrane targeting approximately 16 Å apart (Fig. 1C). The PCho
and cytolytic function of the two toxins (32, 58). moiety at the first binding site (site 1) is lodged
It is generally accepted that a-toxin and the F into a concave pocket similar to one in HlgB
subunits LukD, LukF-PV, LukB and HlgB also (PDB code 3LKF). This pocket is formed by
function in cell attachment through the two extended segments (residues 171–173 and
engagement of their rim domains with the PC 176–179, respectively) and the W1–W2
head group in the plasma membrane of target junction (191–197) (Fig. 1, A and D). The
cells (52, 54, 57). In this report, we demonstrate quaternary ammonium group of the PCho
that the soluble, monomeric and oligomeric moiety engages in a cation–p interaction with
pore forms of both PVL and a-toxin deploy Trp176 while forming a salt bridge to Glu191
two distinct modes to recognize and bind the (3.79 Å) (Fig. 1D). Its N-methyl and methylene
PC-containing membrane and suggest a novel groups are in van der Waal contacts (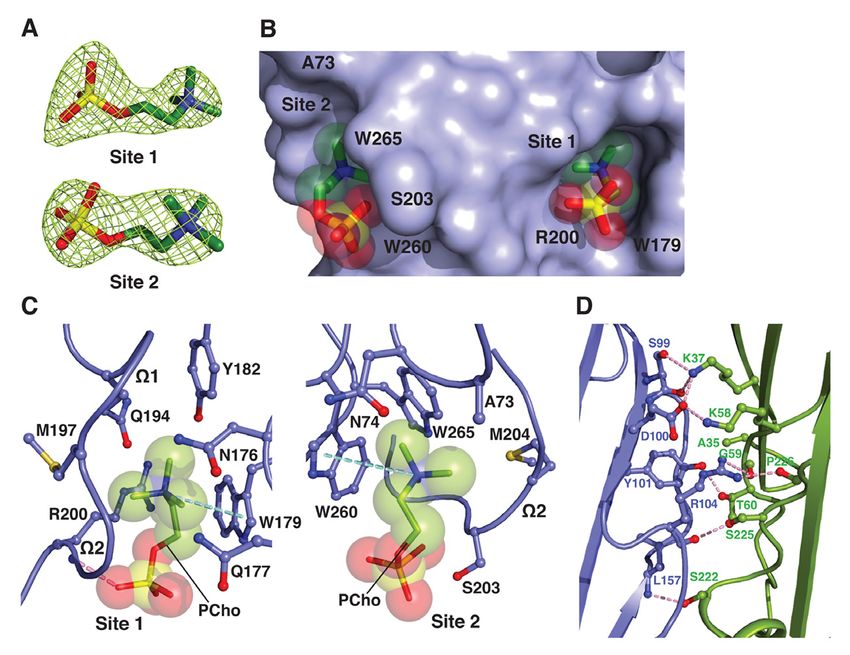
Discovery of a small-molecule inhibitor of MRSA virulence
Immediately adjacent to site 1 is a novel formation (Fig. 1E). Thus, our results suggest a
second binding site (site 2), where the PCho revised mode of PC recognition and membrane
moiety occupies a shallow surface pocket that targeting by the rim domain loops.
is framed by the C-terminal half of the W2 loop
(residues 198–202) and the b14–b15 loop Binding mode of C14PC to the rim domain of
(257–260) and flanked by the side chains of LukF-PV
Tyr71, Asn72, Trp256 and Trp261 (Fig. 1, A To validate this binding mode, we co-
and D). The quaternary ammonium group is crystallized LukF-PV with C14PC and solved
sandwiched between the aromatic rings of its structure at 1.78 Å resolution (Table 1). In
Tyr71 and Trp256 through cation–p effect, PCho moieties engage the
interactions, and the two indole rings of the aforementioned two adjacent binding pockets
latter residue and Trp261 interact with each on the rim domain surface (Fig. 2, A and B). At
other in an edge-to-face fashion to engage the site 1, the quaternary ammonium group of the
N-methyl and methylene groups, which also PCho moiety forms both a cation–p interaction
make contacts with the main chain atoms of with Trp176 and a salt bridge to Glu191 (3.84
Ser199, Ser200 and Ser201 and with the side Å); its N-methyl and methylene groups interact
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
chain of Asn72 (Fig. 1D). The phosphate group with both the main chain atoms of Leu194 and
is secured by a water-mediated hydrogen Gly195 and the side chains of Asn173, Trp176,
bonding interaction with the main chain Tyr179, Glu191 and Arg197; and the
carbonyl of Ser200 (O2–H2O = 2.53 Å and phosphate group is held in place by a hydrogen
H2O–O = 2.76 Å), whose Ca and Cb atoms pack bond between its O2 oxygen and the main
against the O1, O2 and O4 oxygens (Fig. 1D). chain amide of Arg197 (2.72 Å), along with the
The highly complementary interactions side chain of this residue lying against the O2
between the two adjacent binding sites and the and O3 oxygens (Fig. 2B). At site 2, the
PCho moieties are ostensibly important for quaternary ammonium group participates in a
specific recognition and binding. The buried cation–p interaction with Trp256 (Fig. 2B).
solvent accessible surface area of PCho is 262 Further contacts are made between the N-
Å2 at site 1 and 231 Å2 at site 2, which methyl and methylene groups and both the
correspond to approximately 77% and 69% of main chain atoms of Ser199, Asn200 and
the unbound PCho surface area, respectively. Leu201 and the side chains of Asn200, Trp256
The side chains of the conserved Trp176– and Trp261. Polar interactions are also
Arg197 and Ser200–Trp256–Trp261 residues, observed between the phosphate and both the
seen below, that define site 1 and site 2, main chain atom of Asn200 and the side chain
respectively, become more ordered upon of Asn202 (Fig. 2B).
binding to C14PC. This side chain flexibility The solvent accessible surface area of
could allow these two adjacent, largely PCho buried by the LukF-PV interaction
preformed pockets to efficiently accommodate comprises 264 Å2 (79%) at site 1 and 214 Å2
the PCho moieties that have distinct binding (63%) at site 2. DSC measurements reveal that
poses and residue interactions (Fig. 1, C and D). the Tm of LukF-PV increased from 50.3 °C to
Consistent with this argument, in differential 52.3 °C when it was bound to PCho. We note
scanning calorimetry (DSC) experiments, that the PCho moiety at site 2 has considerably
LukD (10 µM) was found to unfold in a single higher average B-factor and poorer electron
cooperative transition, with a midpoint melting density than that at site 1 (70 Å2 as compared
temperature (Tm) of 51.0 °C, while this Tm with 31 Å2), suggesting that the former moiety
value was shifted to 52.8 °C in the presence of is less tightly bound and exhibits greater spatial
PCho (4 mM), representing the enhanced or temporal disorder. In LukD, the aromatic
thermal stability that accompanies complex side chain of Tyr71 contributes to the cation–p
4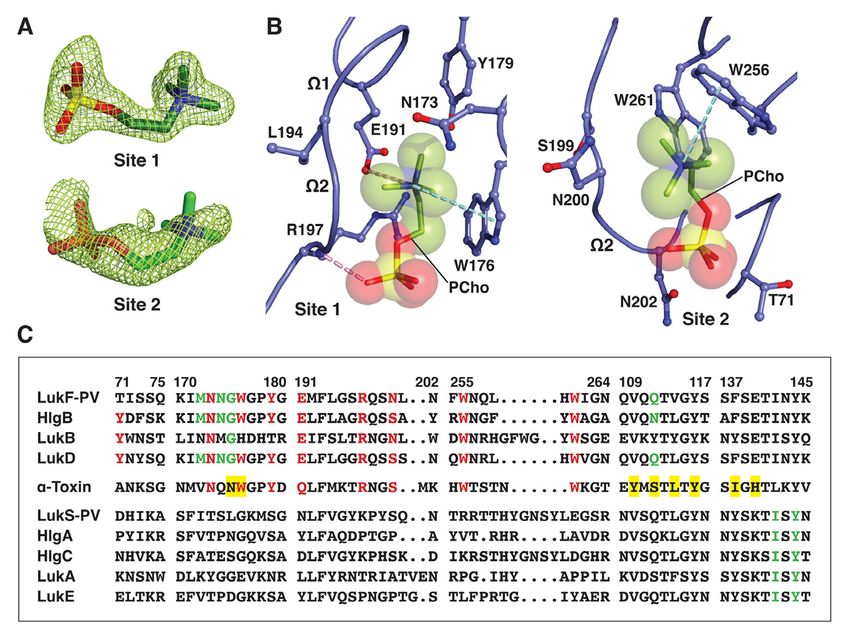
Discovery of a small-molecule inhibitor of MRSA virulence
binding interaction to site 2 (see Fig. 1D), Asn74 and Trp265 (Fig. 3B). The phosphate
whereas the corresponding residue in LukF-PV group is clearly visible in the electron density
(Thr71) cannot make this interaction (Fig. 2C), map, although the fine detail of the oxygens is
likely accounting for the lower affinity binding not clear. There are contacts of 3.19 Å between
site. The critical functional role of this affinity the phosphate and Ser203 and of 3.62 Å
difference is highlighted by the observation between the phosphate and Trp260 (Fig. 3C).
that replacement of Thr71 with a tyrosine Upon binding to a-toxinH35A, PCho buries 268
endows LukF-PV with the ability to bind Å2 (79%) and 203 Å2 (61%) of its solvent
human erythrocytes and acquire hemolytic accessible surface area at site 1 and site 2,
activity when combined with the S subunit of respectively. DSC analysis shows that the
HlgAB (33). Therefore, the elaborate structural addition of PCho increased the Tm of a-
features of the two distinct, adjacent PCho toxinH35A from 50.8 °C to 52.4 °C. We also
binding sites on the leukocidin F subunits may observed that the average B-factor for the PCho
be explained by a selective pressure for moiety at site 2 is significantly higher than that
membrane PC itself acting as their cell surface at site 1 (112 Å2 as compared with 75 Å2). As
receptor. discussed in the preceding section, the
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
decreased affinity of site 2 for PCho may arise
C14PC binding by monomeric a-toxinH35A from the presence of an alanine at position 73
To discern the mechanism in the (corresponding to LukD Tyr71) (Fig. 2C).
attachment of a-hemolysin subfamily Closer examination of the positions and
members to host cells, we determined the 2.80 conformations of the two PCho moieties in the
Å crystal structure of C14PC in complex with superimposed cocrystal structures of C14PC
the oligomerization-defective His35→Ala with a-toxinH35A, LukD and LukF-PV revealed
mutant of a-toxin (a-toxinH35A) (59) (Table 1). remarkable similarities. There are few
The asymmetric unit contains two nearly differences in the positions of the five key
identical protein monomers (rmsd for Ca atoms binding site amino acid side chains (Trp179,
of 0.44 Å), each bound to two PCho moieties Arg200, Ser203, Trp260 and Trp265 in a-toxin;
(Fig. 3A). These moieties occupy the two equivalent to Trp176, Arg197, Ser/Asn200,
adjacent binding pockets described above (Fig. Trp256 and Trp261 in LukD and LukF-PV) in
3B). At site 1, which is similar to that on the a- these structures. The three Trp side chains
toxin protomer in the heptameric pore complex provide two important anchor points for
(57), the quaternary ammonium group of the locating the PCho moieties in the two adjacent
PCho moiety makes a cation–p interaction with binding sites, and the Arg and Ser/Asn residues
Trp179 (Fig. 3C). Its N-methyl and methylene are critical determinants in the binding of the
groups are surrounded by the main chain atoms two phosphate groups. Evidently, PC
of Met197 and Lys198 and by the side chains recognition specificity is achieved by a
of Asn176, Gln177, Trp179, Tyr182, Gln194, combination of stacking and hydrogen bonding
Met197 and Arg200. Importantly, the O2 interactions, and van der Waals contacts. Our
oxygen of the phosphate group establishes a study shows that membrane PC serves as the
strong hydrogen bond to the main chain amide common receptor for a-toxin and the
of Arg200 (2.64 Å) that also makes side chain leukocidin F subunits, in agreement with
contacts with the O2 and O4 oxygens (Fig. 3C). previous observations (52, 54, 57). The
At site 2, the quaternary ammonium group presence of the two adjacent PC binding sites
forms a cation–p interaction with Trp260, and on the toxin monomer is consistent with the
the N-methyl and methylene groups interact estimated cross-sectional areas of the PC-
with the main chain atoms of Gly202, Ser203 bound rim domain (~150 Å2) and one PC
and Met204 and with the side chains of Ala73, molecule (~70 Å2) (60).
5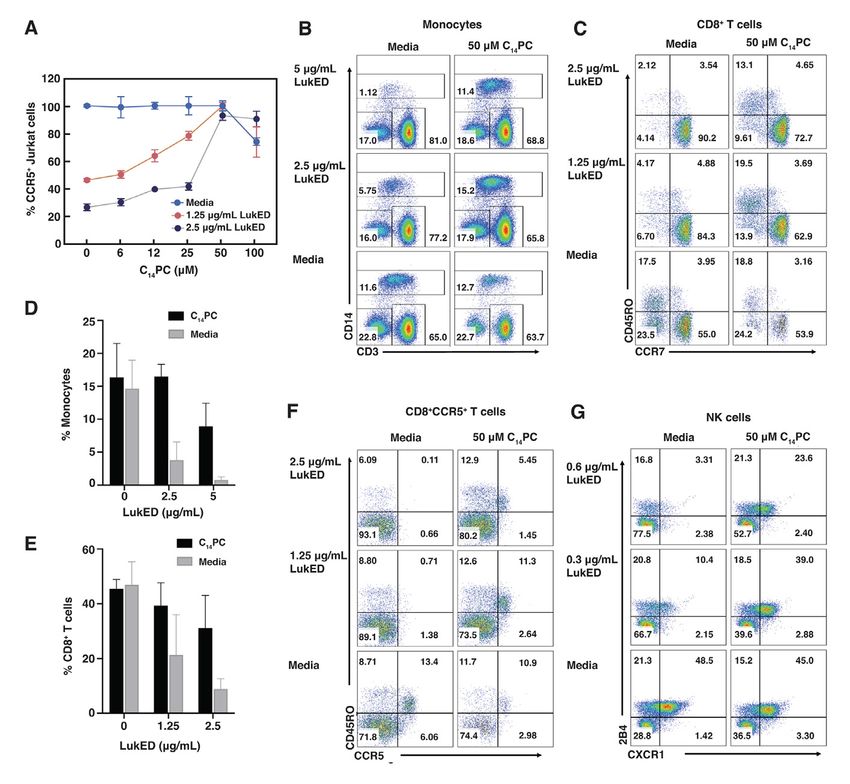
Discovery of a small-molecule inhibitor of MRSA virulence
Intermolecular contacts between the heterodimerization with its S subunit
above two a-toxinH35A monomers comprising counterpart. Likewise, membrane binding by
the crystal asymmetric unit are formed by a-toxin, mediated by PC and/or ADAM10,
residues in the b-sandwich domain (Fig. 3D). irrevocably commits the monomers to
Comparison of the conformation of these dimerization. The remarkable high degree of
contact residues with their interprotomeric conservation of the two adjacent PC binding
equivalents in the unliganded and C14PC- sites among a-toxin and the F subunits reflects
bound heptamers of wild-type a-toxin (PDB a strong selective pressure on the ability of
code 7AHL; see Fig. 5) reveals no local these two sites to help anchor toxin monomers
conformational changes involving the main- to the cell surface and to form intermolecular
chain or side-chain atoms. Superposition of the contacts that prime the ensuing formation of
a-toxinH35A dimer onto two adjacent promoters the oligomeric, membrane-inserted pore
in the above two wild-type toxin heptamers complex.
yields overall Ca r.m.s.d. values of 0.99 and In summary, the bivalent rim domain
0.95 Å, respectively, indicating their structural interaction with PC provides a mechanism by
similarity. Dimer interfaces have similar buried which soluble toxin monomers can recognize
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
surface area values, from 2,061 to 2,171 Å2. It and target the PC-containing membrane,
is also important to note that the crystal thereby promoting dimer-nucleated pore
structure of unliganded a-toxinH35A (PDB code assembly. The relatively low affinity of PC-
4YHD) lacks the aforementioned mediated binding may facilitate subsequent
intermolecular contacts between six establishment of the final geometry of the
independent monomers in the asymmetric unit. oligomeric pore complex, which we discuss
In this structure, both the amino latch and below. a-Toxin and the leukocidin S subunits
prestem regions have well-defined density with also bind their cognate proteinaceous receptors
the exception of the six-residue prestem loop (19–21, 47), and these interactions likely work
and pack against the b-sandwich core of the in concert with the PC targeting mechanism to
protein. By contrast, these two regions are modulate toxin binding, pore formation and
apparently disordered in the C14PC-bound cytotoxicity. Finally, and most importantly,
structure. Our results suggest that a-toxinH35A structural elucidation of the two conserved,
may be trapped in a PC-bound dimeric state, adjacent PC binding pockets on a-toxin and the
which may represent an on-pathway leukocidin F subunits will guide the rational
intermediate in the assembly of the heptameric development of PC analogs as decoy receptors
pore complex. that prevent the cytotoxin from binding to
Given their expected importance in susceptible cells.
membrane targeting, the five key PC binding
site residues are highly conserved or invariant Structure of the C14PC-bound PVL
in both a-toxin and the leukocidin F subunits heterooctamer
but are absent in the S subunits, with the In light of previous studies suggesting
exception of a histidine at position 176 in LukB that PC plays a crucial role in the assembly and
(Fig. 2C). Of particular importance, LukB function of the a-toxin heptamer (54, 57), we
exists as a soluble heterodimeric complex with co-crystallized the LukS-PV and LukF-PV
LukA (61). This finding is consistent with the proteins with C14PC in the presence of n-octyl-
central role of the conserved Trp176 of the b-glucoside. The structure of the complex was
three other F subunits in their binding to the PC solved at 2.04 Å resolution by molecular
bilayer (22, 52, 54; this study). We therefore replacement (Table 1). The asymmetric unit
propose that the binding of the F subunit to the contains one LukF-PV/LukS-PV heterodimer
PC-rich membrane is allosterically coupled to and a single LukS-PV molecule. The
6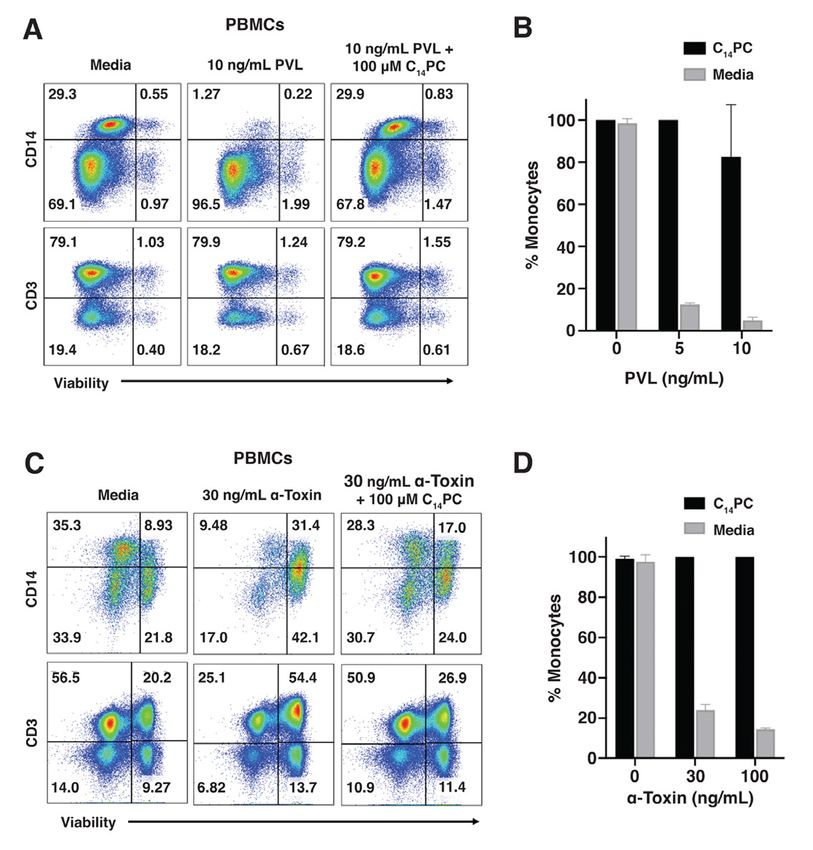
Discovery of a small-molecule inhibitor of MRSA virulence
heterodimer interacts with three 4D; see Fig. 2B), differing only in the presence
crystallographic 4-fold symmetry-related of more stabilizing molecular contacts at site 2
copies of itself to generate a heterooctamer (Fig. on the heterooctamer. Specifically, the
4, A and B). In this b-barrel pore complex, four quaternary ammonium group of the PCho
LukF-PV protomers (denoted A, C, E and G) moiety makes a cation–p interaction with
and four LukS-PV protomers (B, D, F and H) Trp256, and the indole ring of this residue
are arranged in an alternating fashion around establishes an edge-to-face interaction with the
the central axis of pore, in which the stem indole ring of Trp261 to pack against the N-
domain folds into an antiparallel b-barrel methyl and methylene groups, which are also
composed of 16 b-strands. We could not in contact with the main chain atoms of Thr71,
discern electron density corresponding to the Ser199, Asn200 and Leu201 and with the side
bottom third of the stem domain in our chain of Ile72 (Fig. 4E). At site 3, the aromatic
structure. Two distinct interfaces between ring of Tyr137 of protomer H forms a cation–p
neighboring protomers involve residues that interaction with the quaternary ammonium
are distributed among the amino latch region group and stacks against the N-methyl and
and the b-sandwich and stem domains, and methylene groups that are also lined with the
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
bury 2,644 Å2 and 1,902 Å2 of solvent side chain of Ile135 of protomer H (Fig. 4E).
accessible surface area, respectively. The Furthermore, the O3 oxygen of the phosphate
electron density map revealed clearly the group hydrogen bonds to the main chain amide
presence of PCho moieties at three distinct of Gly175 of protomer A (2.65 Å), and the O1
binding sites on each of the four protomeric and O3 oxygens engage both the main chain
units of the PVL heterooctamer (Fig. 4, C and atoms of Asn174 and Gly175 of protomer A
D). The two adjacent binding sites are and the side chains of Met172 of protomer A
essentially the same as those on the above- and Gln112 of protomer G (Fig. 4E). The
described toxin monomer, whereas the other, solvent accessible surface area of PCho buried
novel site lies at the interface between the rim upon complex formation is 258 Å2 (77%) at site
domain of a LukF-PV protomer (e.g. protomer 1, 189 Å2 (57%) at site 2 and 224 Å2 (65%) at
A) and the proximal stem domain regions of site 3.
protomers G and H. The average B-factor for Our results suggest that multivalent
the three PCho moieties is significantly higher binding of the PVL heterooctamer to PC on the
than that for the surrounding residues (60 Å2 as membrane surface leads to localized alterations
compared with 31 Å2), possibly due to greater in the lipid bilayer and thus promotes the
disorder and/or subunitary occupancy. insertion of amphipathic b-hairpins to produce
Superposition of the PVL hetereooctamer the b-barrel piercing the bilayer. Critical
bound to C14PC onto the unliganded HlgAB residues Tyr137 of LukS-PV and Gly175 of
(PDB code 3B07) and LukGH (PDB code LukF-PV at site 3 are invariant in the
4TW1) heterooctamers yields Ca rmsds of 0.67 leukocidin S and F subunits, respectively (Fig.
and 1.14 Å, respectively, suggesting that the 2C), underscoring their functional importance.
PVL pore does not undergo large Furthermore, three similar PC binding pockets
conformational changes upon binding to C14PC. also exist in protomers of the C14PC-bound a-
The three PCho binding sites on a single toxin heptamer described below.
protomeric unit are contained within a water-
accessible crevice between the inner surface of Binding mode of C14PC to the a-toxin
the rim domain and the upper portion of the heptamer
stem domain (Fig. 4, A and B). As noted above, To evaluate the binding of the a-toxin
the two adjacent sites correspond to those on heptamer to the PC head group in a membrane-
the rim domain of monomeric LukF-PV (Fig. mimicking environment, we determined the
7Discovery of a small-molecule inhibitor of MRSA virulence
crystal structure of its complex with C14PC at its O2 oxygen with the main chain amide of
2.35 Å resolution (Table 1). In this structure, Gly180 (2.84 Å) while in the cis rotamer.
three PCho moieties are bound to each of the The third pocket is located at the
seven protomeric units in the water-accessible interface between the rim domain of protomer
crevice between the rim and stem domains (Fig. A and the proximal stem domain regions of
5, A and B). The indole ring of Trp179 mediates protomers E and F (Fig. 5E), in contrast to the
three-way interactions with these three other pockets that are constituted solely by
moieties (Fig. 5C). Their conformations are residues from the rim domain. The third pocket
clearly defined in three partially overlapping is formed by residues Asn178 and Trp179 from
but distinct binding pockets of the crevice (Fig. the rim domain of protomer A, by Leu116 and
5D). One pocket corresponds to site 1 on the Tyr118 from the stem domain of protomer E
toxin monomer described above, while the and by Tyr112, Ser114, Ile142, Gly143 and
other two are novel heptamer-specific binding His144 from the stem domain of protomer F
sites (see below). The average B-factor for the (Fig. 5E). The indole ring of Trp179 is situated
three PCho moieties is 60 Å2 and for to produce a cation–p interaction with the
surrounding protein atoms 33 Å2. The structure quaternary ammonium group of the PCho
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
of the C14PC-bound heptamer is very similar to moiety (Fig. 5E). The N-methyl and methylene
that of the unliganded heptamer (PDB code groups participate in extensive contacts with
7AHL; rmsd for Ca atoms of 0.48 Å), with only the main chain atoms of Gly143 and Asn178
minor changes in the positions of side chains and with the side chains of Tyr112, Ser114 and
involved in direct contact with C14PC. The Ile142. The PCho moiety is further stabilized
pairwise rmsds between protomers A–G in the by a hydrogen bond between the O3 oxygen of
heptamer span a range from 0.13 to 0.17 Å for the phosphate group and the ND1 atom of
Ca atoms. The PCho moieties at each of the His144 (2.78 Å) and by contacts between the
three binding sites have essentially identical O1, O2 and O3 oxygens and the side chains of
conformation and orientation in each of the Leu116, Tyr118 and His144 (Fig. 5E). The
seven protomeric units, with average rmsds of solvent accessible surface areas buried upon
0.34 Å for the first pocket, 0.32 Å for the binding of the PCho moieties to the first,
second pocket and 0.41 Å for the third pocket. second and third pockets are 260 A2 (76%), 207
For this reason, the following structural A2 (60%), 285 A2 (83%), respectively.
analysis of these binding pockets applies to all These results strengthen the hypothesis
of the protomeric units. that multivalent binding of the PC bilayer by
The first pocket, defined by Trp179 and the a-toxin heptamer may help overcome the
Arg200, is the same as that on monomeric a- energetic barrier to deformation of the
toxinH35A (see Fig. 3), albeit the hydrogen bond membrane during assembly of the b-barrel pore
between the phosphate group of the PCho lining, thereby driving the conversion of the
moiety and the main chain amide of Arg200 is prepore to the transmembrane pore complex.
considerably longer and weaker in the latter Indeed, replacement of Trp179 and Arg200
(Fig. 5E). The second pocket lined by all four with alanines in a-toxin is known to lead to an
residues on strand b12 of the rim domain arrested prepore state in which only the top half
snugly accommodates the PCho moiety (Fig. 5, of the cytolytic b-barrel pore has formed (26).
D and E). It mediates a network of van der Together with analysis of intermediate stages
Waals contacts involving both the main chain of the a-toxin assembly process with
atoms of Gly180 and Pro181 and the aromatic engineered disulfide bonds (34), our study also
rings of Trp179 and Tyr182, forming hydrogen suggests that the interaction between the a-
bonds via its hydroxyl group towards the O3 toxin prepore and the PC head group may
oxygen of the phosphate group (2.69 Å) and via induce a large conformational change in the
8Discovery of a small-molecule inhibitor of MRSA virulence
prestem region, which is essential for pore assembly pathway, involving the initial
formation. membrane binding of toxin monomers and
membrane-dependent dimerization and
Structure of the a-toxinH35A heptamer in oligomerization, followed by the prepore-to-
complex with C14PC pore transition and membrane perforation. It
In the a-toxin pore structure, His35 is should be stressed that our crystallographic
located in the crucial interprotomeric contact results demonstrate that the interactions
region (27), and nonconservative replacements between PCho and the oligomeric pore forms
at this position (including H35A) have been of a-toxin and PVL differ considerably.
shown to abolish heptamer formation and thus Importantly, atomic-level insight of the toxin
cytolytic activity and lethal toxicity (62–64). In oligomer–PC interactions obtained here will
light of our findings that the PC bilayer binding facilitate the development of PC analogs that
might promote both the oligomerization of a- inhibit pore formation and thus block the
toxin monomers and the structural immune cytolytic effects of this subfamily of
rearrangements that accompany the prepore-to- proteins.
pore conversion, we hypothesized that a high
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
concentration of C14PC could facilitate the Inhibition of the cytotoxicity of LukED, PVL
assembly of the a-toxinH35A pore complex. To and a-toxin by C14PC
directly test this hypothesis, we have The presence of the conserved PC
determined the structure of the a-toxinH35A binding sites in the leukocidins and a-toxin
heptamer crystallized in the presence of 25 mM (see above) suggests that PC mimetic
C14PC at 2.5 Å resolution (Table 1; Fig. 5F); it compounds may interfere with toxin-mediated
is worth noting the use of 5 mM C14PC for the killing of primary human immune cells.
crystallization of the a-toxinH35A monomer (see Therefore, flow cytometry experiments were
Fig. 3 and Experimental Procedures). In this conducted to first evaluate the ability of C14PC
mutant pore complex, PCho moieties bind in to diminish the cytolytic activity of LukED in
the first and second pockets described above on Jurkat cells expressing CCR5. This Jurkat cell
the rim domain of each protomer. In essence, line has been shown to be susceptible to the
the C14PC-bound structures of the a-toxinH35A toxin (19). LukED at a concentration of 2.5
and wild-type heptamers are nearly identical, µg/mL resulted in ~80% lysis of Jurkat cells
with rmsds of 0.04–1.42 Å over 2,051 Ca atoms. within 1 h at 37 °C (Fig. 6A). We found that
The positions and conformations of the two C14PC inhibited the lysis in a concentration-
PCho moieties are also similar. However, dependent manner, with an IC50 value between
C14PC does not bind to the aforementioned 15–25 µM (Fig. 6A). In sharp contrast, PCho
interprotomer pocket on the a-toxinH35A pore, did not show appreciable inhibitory activity up
while B-factors for this mutant pore are to 0.5 mM. We conclude that C14PC produces
considerably higher than those for the wild- effective toxin inhibition by presenting
type one (24–201 Å2 as compared with 13–73 multiple copies of the PC head group on its
Å2), consistent with the pronounced effect of micellar surface, in accordance with previous
the H35A mutation on cytotoxicity (59). These observations (54).
results support our hypothesis that the PC-rich To investigate the protective effects of
membrane acts as a critical effector of C14PC on LukED-induced lysis of primary
oligomerization and pore formation by a-toxin. human leukocytes expressing CCR5 and
In summary, despite their different CXCR1 in vitro, LukED at concentrations of
subunit composition and stoichiometry, a- 2.5 and 5 µg/ml was first preincubated with 50
toxin and the leukocidins likely follow an µM C14PC at 4 °C and was subsequently added
evolutionarily conserved PC-dependent pore to PBMCs labelled with specific cell surface
9Discovery of a small-molecule inhibitor of MRSA virulence
markers. After 1–1.5 h at 37 °C, the cells were of MRSA to form biofilms on necrotic tissues
stained with fixable viability dye eFluor 506 and medical devices is also an important
and analyzed by flow cytometry. Inhibition of virulence mechanism that complicates
LukED by C14PC was assessed by determining infections (5, 6). As antibiotic resistance
the relative abundance of viable cells after continues to emerge, disarming the major
challenge with the toxin or media. As expected, virulence mechanisms of MRSA strains has
CD14+ monocytes were significantly absent by potential to become an alternative therapeutic
2.5 and 5 µg/mL of LukED (Fig. 6D), while approach aimed at limiting host tissue damage
pretreatment with 50 µM C14PC produced a while aiding immune clearance. The a-
70–90% protective effect against monocyte hemolysin subfamily of cytotoxins represents a
lysis (Fig. 6, B and D). Likewise, 50 µM C14PC prime target for antivirulence drug
blocked the lysis of CD8+ effector memory T development, owing to their critical roles in
cells by 50–75% (Fig. 6, C and E) and of inactivating host immune defenses, destroying
CD8+CCR5+ T cells by 50–95% (Fig. 6F). tissue barriers and modulating inflammatory
Moreover, 50 µM C14PC also rescued 50–85% responses (3, 16). Monoclonal antibodies
of NK cells (Fig. 6G), which are highly (mAbs) targeting a-toxin have been shown to
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
susceptible to LukED due to their surface prevent human lung cell injury in vitro and
expression of CXCR1 (19). These results protect experimental animals against lethal S.
demonstrate that C14PC confers target cell aureus pneumonia (65). Several such mAbs are
protection by blocking the interaction between currently in clinical trials, including mAbs
LukED and membrane PC. MEDI4893 and KBSA301 (Refs. 66–69).
We next sought to assess the ability of Given the variability of MRSA immune
C14PC to abrogate the cytolytic activities of evasion determinants, such single-target drugs
PVL and a-toxin using the in vitro cell viability are most likely to be inadequate to achieve a
assay described above. Addition of 2 and 10 therapeutic effect (68). Our structural
ng/mL of PVL led to 85–95% lysis of elucidation of the two conserved, adjacent
monocytes after 1.5 h of incubation at 37 °C PCho binding pockets on the rim domains of a-
(Fig. 7, A and B). Pretreatment with 100 µM toxin and the leukocidin F subunits will guide
C14PC suppressed the lysis by 90% (Fig. 7, A the rational development of PC analogs that
and B). Similarly, a-toxin at concentrations of prevent cytotoxin assembly and pore formation
30 and 100 ng/mL caused 75–90% lysis of in the susceptible cell membrane, thereby
monocytes and ~50% lysis of CD3+ T cells blocking the cytolytic effects of this subfamily
after incubation at 37 °C for 24 h, while 100 of proteins. Using a combined structural
µM C14PC caused 75–90% reduction of the biology and pharmacological approach, we
lytic activity (Fig. 7, C and D). We conclude have been able to demonstrate that C14PC is a
that C14PC is a broad-spectrum small-molecule novel broad-spectrum inhibitor of PVL,
inhibitor of LukED, PVL and a-toxin and that LukED and a-toxin in vitro. In light of the
membrane PC contributes to the mechanism of safety of miltefosine (hexadecylophosphocline,
their cytolytic action. C16PC), an oral drug used for the treatment of
leishmaniasis (70), we expect that C14PC will
Implication for MRSA drug discovery likewise be well tolerated in humans.
The high prevalence of highly pathogenic Considering its conserved mechanism of action
MRSA is creating a crisis in modern healthcare and low production costs, C14PC may provide
due to the limited therapeutic options available, the basis for the development of prophylactic
the toll of severe disease and mortality it and therapeutic agents that reduce the virulence
inflicts, and the enormous cost of inpatient care of MRSA infection.
to which it contributes (3, 4). The ability
10Discovery of a small-molecule inhibitor of MRSA virulence
Experimental Procedures chromatography on a GE Superdex 200 10/300
Chemicals GL equilibrated with 50 mM sodium acetate,
All chemicals used were of analytical pH 5.4, 100 mM NaCl. Fractions containing the
grade. Unless otherwise indicated, chemicals toxin were pooled, concentrated to ~20 mg/mL
were purchased from Sigma-Aldrich. and stored at –80 °C until use. The
Detergents were from Anatrace. concentration of the toxin in purified
preparations was determined through UV
Cloning and protein purification absorbance measurements.
The full-length LukD (residues 1–301),
LukE (1–283), LukF-PV (1–301), LukS-PV Crystallization
(1–284) and a-toxin (1–293) constructs, All crystallization experiments were
excluding their signal peptides, were subcloned performed at room temperature using the
individually into a modified pET3a vector hanging drop-vapor diffusion method by
(Novagen). Site-directed mutagenesis was mixing 1 µL of protein solution with an equal
carried out using the Kunkel method. All volume of precipitant solution. Crystals of
constructs were verified by DNA sequencing. LukD were grown from protein at 12 mg/mL in
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
E. coli BL21(DE3)pLysS cells transformed 10 mM sodium acetate, pH 5.4, and precipitant
with each plasmid were grown at 37 °C in LB solution (20% PEG MME 2000, 10 mM NiCl2,
medium until the A600 was between 0.6 and 0.8. and 0.1 M Tris-HCl, pH 8.5). For data
IPTG was then added to a final concentration collection, the crystals were cryoprotected with
of 0.5 mM, and incubation was continued for 15% glycerol in the mother liquor and then
24 h at 16 °C. Cells were harvested by flash-cooled in liquid nitrogen. The C14PC–
centrifugation, suspended in 50 mM sodium LukD complex was crystallized from protein at
acetate buffer, pH 5.4, 25% sucrose, 5 mM 10 mg/mL in 10 mM sodium acetate, pH 5.4,
EDTA, and 5 mM DTT and lyzed at 4 °C using 10 mM C14PC, and 30 mM n-octyl-b-D-
an Avestin Emulsiflex C3 homogenizer. glucoside (bOG), and precipitant solution
Inclusion bodies were isolated by (28% PEG 400, 0.2 M MgCl2, and 0.1 M
centrifugation, washed twice with the same HEPES, pH 7.5). The crystals were flash-
buffer and subsequently incubated overnight at cooled by plunging directly into liquid
4 °C in 50 mM sodium acetate buffer, pH 5.4, nitrogen. Crystals of LukF-PV complexed with
5 mM DTT, and 6 M guanidine-HCl or 8 M C14PC were grown from protein at 10 mg/mL
urea. Insoluble material was removed by in 10 mM sodium acetate, pH 5.4, 10 mM
centrifugation, and the protein solution was C14PC, and 30 mM bOG, and precipitant
then dialyzed for 2 days at 4 °C against three solution (2.6 M ammonium sulfate, 5% PEG
changes of buffer A (50 mM sodium acetate, 400, and 0.1 M HEPES, pH 8.5). The crystals
pH 5.4, and 1 mM EDTA). After removal of the were flash-cooled in liquid nitrogen. The
insoluble material by centrifugation, the C14PC–a-toxinH35A complex was crystallized
refolded recombinant toxin was loaded onto a from protein at 10 mg/mL in 10 mM sodium
CM-Sepharose CL-6B column equilibrated acetate, pH 5.4, 5 mM C14PC, 40 mM bOG,
with buffer A and eluted using a linear gradient and 0.4 mM Deoxy-Big CHAP, and precipitant
from 0 to 1 M NaCl. Fractions containing the solution (1.5 M ammonium sulfate, 0.25 M
recombinant toxin were pooled, dialyzed potassium sodium tartrate, and 0.1 M sodium
against buffer A, concentrated and loaded onto citrate, pH 6.0). The crystals were transferred
a GE Mono S 5/50 GL equilibrated with buffer into stabilizing solution (2.25 M ammonium
A, and the toxin was eluted using a linear sulfate, 5% glycerol, 20 mM C14PC, and 0.1 M
gradient from 0 to 0.5 M NaCl. The toxin was sodium citrate, pH 6.0) and then allowed to
further purified using size exclusion equilibrate against 3 M ammonium sulfate for
11Discovery of a small-molecule inhibitor of MRSA virulence
1 h at room temperature prior to flash freezing refinement was performed in Refmac5 (76).
in liquid nitrogen. The PVL heterooctamer in Crystallographic data and refinement statistics
complex with C14PC was crystallized from are summarized in Table 1.
LukF-PV at 6.7 mg/mL and LukS-PV at 6.3
mg/mL in 10 mM sodium acetate, pH 5.4, 15 Structural analyses
mM C14PC, and 40 mM bOG, and precipitant Model quality was judged using the
solution (0.16 M magnesium formate). The programs Rampage, Procheck and Sfcheck
crystals were transferred into dehydrating (77–79). Protein-ligand contacts for the toxin–
solution (2.7 M ammonium sulfate, and 20 mM C14PC complex structures were analyzed using
C14PC) and then allowed to equilibrate against the program COOT (80). The r.m.s.d. values
3 M ammonium sulfate for 3 h at room were calculated using the program SuperPose
temperature prior to flash freezing in liquid (81). Molecular and solvent-accessible
nitrogen. Crystals of the a-toxin heptamer– surfaces were calculated with the AREAIMOL
C14PC complex were grown from protein at 8 program (82) from the CCP4 suite (83).
mg/mL in 10 mM sodium acetate, pH 5.4, 15 PyMOL (DeLano Scientific) was used to
mM C14PC, and 30 mM bOG, and precipitant render structure figures.
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
solution (2 M ammonium sulfate, 0.2 M
potassium sodium tartrate, and 0.1 M sodium Differential scanning calorimetry
citrate, pH 6.0). The crystals were flash-frozen Protein thermal stability was determined
in liquid nitrogen. The a-toxinH35A heptamer in by differential scanning calorimetry (DSC)
complex with C14PC was crystallized from using a Nano-DSC model 602000 calorimeter
protein at 10 mg/mL in 10 mM sodium acetate, (TA instruments). Protein solutions in buffer A
pH 5.4, 25 mM C14PC, and 40 mM bOG, and (20 mM sodium acetate, pH 5.8, and 50 mM
precipitant solution (1.9 M ammonium sulfate, NaCl) in the presence and absence of 4 mM
0.25 M potassium sodium tartrate, and 0.1 M PCho were subjected to a temperature increase
sodium citrate, pH 5.2). The crystals were of 1 °C/min from 0 °C to 100 °C under a
flash-cooled in liquid nitrogen. pressure of 3 atm, and the evolution of heat was
recorded as a differential power between
Data collection and structure determination reference (buffer A) and sample (10 µM
Diffraction data were collected at 100 K protein in buffer A) cells. The resulting
at beamline X4C at the National Synchrotron thermograms (after buffer subtraction) were
Light Source at Brookhaven National used to derive thermal transition midpoints
Laboratory, at the Cornell High Energy (Tm’s). Fitting to the two-state scaled model
Synchrotron Source (CHESS) beamline F1, provided in NanoAnalyze software was used to
and at the Stanford Synchrotron Radiation obtain a Tm value. The experiments were
Lightsource (SSRL) beamline 9-2. The repeated two times with consistent results.
diffraction data were processed with HKL-
2000 (71). Initial phases were determined by Isolation of human peripheral blood
molecular replacement using Phaser (72) with mononuclear cells
respective models of HlgB (PDB code 1LKF), Blood samples were obtained from
LukF-PV (1PVL), a-toxinH35A (4YHD), the healthy, consenting donors as Buffy coats
HlgAB heterooctamer (3B07) and the a-toxin (New York Blood Center) and leukopaks
heptamer (7AHL). Refinement was carried out (AllCells, Alameda, CA). Human peripheral
in Refmac5 (73), alternating with manual blood mononuclear cells (PBMCs) were
rebuilding and adjustment in COOT (74). isolated from peripheral blood by density
Coordinates for C14PC were generated using gradient centrifugation using Ficoll-Paque Plus
LibCheck (75). TLS (GE life sciences).
12Discovery of a small-molecule inhibitor of MRSA virulence
Cytolysis inhibition assay Data availability: All data are contained
Flow cytometry was used to assay within the manuscript. The atomic coordinates
permeabilization of the plasma membrane and structure factors (codes 6U33, 6U2S,
(pore formation) by LukED, PVL and a-toxin 6U3F, 6U3T, 6U3Y, 6U49 and 6U4P) have
in Jurkat cells and primary human immune been deposited in the Protein Data Bank
cells as described previously (84). Briefly, (http://www.rcsb.org/).
C14PC (6 µM to 100 µM) was preincubated
individually with different concentrations of Acknowledgments: We thank the beamline
the LukD and LukF-PV F subunits and a-toxin personnel at the Cornell High Energy
in V-bottom 96 well-plate for 30 min at 4 °C. Synchrotron Source and the Stanford
These mixtures were then added to prestained Synchrotron Radiation Lightsource for data
PBMCs and incubated with the cognate LukE collection, J. Cai for her participation and
and LukS-PV S subunits for 1–1.5 h and with assistance in the early stage of the project, M.
a-toxin for 24 h in a humidified 5% CO2 Zhang and Q. Li for technical assistance, and J.
incubator at 37 °C. The cytotoxin-treated cells Nunberg, N. Kallenbach and J. Lu for
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
were stained with a viability dye and analyzed comments on the manuscript.
by FACS. 50% inhibitory concentration (IC50)
values are calculated using GraphPad Prism by Conflict of interest: The authors declare that
fitting data to single-slope dose-response they have no conflicts of interest with the
curves constrained to 0% and 100% values. contents of this article. The content is solely the
responsibility of the authors and does not
Staining and FACS analysis necessarily represent the official views of the
PBMCs were differentially stained with National Institutes of Health.
specific cell surface markers prior to
intoxication in order to identify distinct cell Author contributions: JL and ML performed
populations. Antibodies used for flow the biochemical and biophysical experiments
cytometric staining included CD3-Alexa 532 and the co-structure determinations. LK and
(clone UCHT1) (eBioscience, San Diego, CA), DU performed the toxin activity and inhibition
CD4-Brilliant Violet 570, CD8-Pacific Blue, measurements. ML wrote the paper with DU.
CD45RO-APCCy7, CD14-Alexa 700, CD27- ML, DU and VT initiated the project.
PeCy7, CD244 (2B4)-Percp Cy5.5, CXCR1-
APC (Biolegend, San Diego, CA), CCR5-PE
(BD Biosciences, San Diego, CA) and CCR7-
FITC (R&D systems, Minneapolis, MN). After
intoxication, cells were collected, washed with
phosphate buffered saline and stained with
Fixable viability dye eFluor 506 (eBioscience,
San Diego, CA). Data were acquired on BD
LSRFortessa X-20 instrument (BD Biosciences,
CA) using FACSDiva software, iQue Screener
PLUS (Intellicyt, MI) using ForeCyt Software
or SP6800 Spectral Analyzer (Sony
Biotechnology, CA). Data analysis was
performed using FlowJo software (TreeStar Inc,
Ashland, OR). Statistical analysis was
performed using GraphPad Prism 8 software.
13References
1. Lowy, F. D. (1998) Staphylococcus aureus infections. N Engl J Med 339, 520-532
2. Klevens, R. M., Morrison, M. A., Nadle, J., Petit, S., Gershman, K., Ray, S., Harrison, L.
H., Lynfield, R., Dumyati, G., Townes, J. M., Craig, A. S., Zell, E. R., Fosheim, G. E.,
McDougal, L. K., Carey, R. B., Fridkin, S. K., and for the Active Bacterial Core
surveillance MRSA Investigators. (2007) Invasive methicillin-resistant Staphylococcus
aureus infections in the United States. JAMA 298, 1763-1771
3. Otto, M. (2010) Basis of virulence in community-associated methicillin-resistant
Staphylococcus aureus. Annu Rev Microbiol 64, 143-162
4. Chambers, H. F., and Deleo, F. R. (2009) Waves of resistance: Staphylococcus aureus in
the antibiotic era. Nat Rev Microbiol 7, 629-641
5. Jones, S. M., Morgan, M., Humphrey, T. J., and Lappin-Scott, H. (2001) Effect of
vancomycin and rifampicin on meticillin-resistant Staphylococcus aureus biofilms. Lancet
357, 40-41
6. Otto, M. (2008) Staphylococcal biofilms. Curr Top Microbiol Immunol 322, 207-228
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
7. Bowler, P. G. (2018) Antibiotic resistance and biofilm tolerance: a combined threat in the
treatment of chronic infections. J Wound Care 27, 273-277
8. Liu, C., Bayer, A., Cosgrove, S. E., Daum, R. S., Fridkin, S. K., Gorwitz, R. J., Kaplan, S.
L., Karchmer, A. W., Levine, D. P., Murray, B. E., M, J. R., Talan, D. A., and Chambers,
H. F. (2011) Clinical practice guidelines by the infectious diseases society of america for
the treatment of methicillin-resistant Staphylococcus aureus infections in adults and
children: executive summary. Clin Infect Dis 52, 285-292
9. Liu, C., Bayer, A., Cosgrove, S. E., Daum, R. S., Fridkin, S. K., Gorwitz, R. J., Kaplan, S.
L., Karchmer, A. W., Levine, D. P., Murray, B. E., M, J. R., Talan, D. A., Chambers, H.
F., and Infectious Diseases Society of America (2011) Clinical practice guidelines by the
infectious diseases society of america for the treatment of methicillin-resistant
Staphylococcus aureus infections in adults and children. Clin Infect Dis 52, e18-55
10. Han, L. L., McDougal, L. K., Gorwitz, R. J., Mayer, K. H., Patel, J. B., Sennott, J. M., and
Fontana, J. L. (2007) High frequencies of clindamycin and tetracycline resistance in
methicillin-resistant Staphylococcus aureus pulsed-field type USA300 isolates collected at
a Boston ambulatory health center. J Clin Microbiol 45, 1350-1352
11. Mendes, R. E., Sader, H. S., Deshpande, L. M., Diep, B. A., Chambers, H. F., and Jones,
R. N. (2010) Characterization of baseline methicillin-resistant Staphylococcus aureus
isolates recovered from phase IV clinical trial for linezolid. J Clin Microbiol 48, 568-574
12. Mangili, A., Bica, I., Snydman, D. R., and Hamer, D. H. (2005) Daptomycin-resistant,
methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis 40, 1058-1060
13. Wilson, P., Andrews, J. A., Charlesworth, R., Walesby, R., Singer, M., Farrell, D. J., and
Robbins, M. (2003) Linezolid resistance in clinical isolates of Staphylococcus aureus. J
Antimicrob Chemother 51, 186-188
14. Willyard, C. (2017) The drug-resistant bacteria that pose the greatest health threats. Nature
543, 15
15. Diekema, D. J., Richter, S. S., Heilmann, K. P., Dohrn, C. L., Riahi, F., Tendolkar, S.,
McDanel, J. S., and Doern, G. V. (2014) Continued emergence of USA300 methicillin-
resistant Staphylococcus aureus in the United States: results from a nationwide surveillance
study. Infect Control Hosp Epidemiol 35, 285-29216. Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., Lin, F.,
Lin, J., Carleton, H. A., Mongodin, E. F., Sensabaugh, G. F., and Perdreau-Remington, F.
(2006) Complete genome sequence of USA300, an epidemic clone of community-acquired
meticillin-resistant Staphylococcus aureus. Lancet 367, 731-739
17. Gouaux, E., Hobaugh, M., and Song, L. (1997) alpha-Hemolysin, gamma-hemolysin, and
leukocidin from Staphylococcus aureus: distant in sequence but similar in structure.
Protein Sci 6, 2631-2635
18. Alonzo, F., 3rd, and Torres, V. J. (2014) The bicomponent pore-forming leucocidins of
Staphylococcus aureus. MMBR 78, 199-230
19. Alonzo, F., 3rd, Kozhaya, L., Rawlings, S. A., Reyes-Robles, T., DuMont, A. L., Myszka,
D. G., Landau, N. R., Unutmaz, D., and Torres, V. J. (2013) CCR5 is a receptor for
Staphylococcus aureus leukotoxin ED. Nature 493, 51-55
20. Gauduchon, V., Werner, S., Prevost, G., Monteil, H., and Colin, D. A. (2001) Flow
cytometric determination of Panton-Valentine leucocidin S component binding. Infect
Immun 69, 2390-2395
21. Spaan, A. N., van Strijp, J. A. G., and Torres, V. J. (2017) Leukocidins: staphylococcal bi-
Downloaded from http://www.jbc.org/ by guest on October 27, 2020
component pore-forming toxins find their receptors. Nat Rev Microbiol 15, 435-447
22. Olson, R., Nariya, H., Yokota, K., Kamio, Y., and Gouaux, E. (1999) Crystal structure of
staphylococcal LukF delineates conformational changes accompanying formation of a
transmembrane channel. Nat Struct Biol 6, 134-140
23. Pedelacq, J. D., Maveyraud, L., Prevost, G., Baba-Moussa, L., Gonzalez, A., Courcelle, E.,
Shepard, W., Monteil, H., Samama, J. P., and Mourey, L. (1999) The structure of a
Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-
soluble species of a family of transmembrane pore-forming toxins. Structure 7, 277-287
24. Guillet, V., Roblin, P., Werner, S., Coraiola, M., Menestrina, G., Monteil, H., Prevost, G.,
and Mourey, L. (2004) Crystal structure of leucotoxin S component: new insight into the
Staphylococcal beta-barrel pore-forming toxins. J Biol Chem 279, 41028-41037
25. Nocadello, S., Minasov, G., Shuvalova, L., Dubrovska, I., Sabini, E., Bagnoli, F., Grandi,
G., and Anderson, W. F. (2016) Crystal structures of the components of the Staphylococcus
aureus leukotoxin ED. Acta Crystallogr D Struct Biol 72, 113-120
26. Sugawara, T., Yamashita, D., Kato, K., Peng, Z., Ueda, J., Kaneko, J., Kamio, Y., Tanaka,
Y., and Yao, M. (2015) Structural basis for pore-forming mechanism of staphylococcal
alpha-hemolysin. Toxicon 108, 226-231
27. Song, L., Hobaugh, M. R., Shustak, C., Cheley, S., Bayley, H., and Gouaux, J. E. (1996)
Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science
274, 1859-1866
28. Badarau, A., Rouha, H., Malafa, S., Logan, D. T., Hakansson, M., Stulik, L., Dolezilkova,
I., Teubenbacher, A., Gross, K., Maierhofer, B., Weber, S., Jagerhofer, M., Hoffman, D.,
and Nagy, E. (2015) Structure-function analysis of heterodimer formation, oligomerization,
and receptor binding of the Staphylococcus aureus bi-component toxin LukGH. J Biol
Chem 290, 142-156
29. Yamashita, K., Kawai, Y., Tanaka, Y., Hirano, N., Kaneko, J., Tomita, N., Ohta, M.,
Kamio, Y., Yao, M., and Tanaka, I. (2011) Crystal structure of the octameric pore of
staphylococcal gamma-hemolysin reveals the beta-barrel pore formation mechanism by
two components. Proc Natl Acad Sci U S A 108, 17314-17319
15You can also read

















































