Ercc1 Deficiency Promotes Tumorigenesis and Increases Cisplatin Sensitivity in a Tp53 Context-Specific Manner
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Oncogenes and Tumor Suppressors Molecular
Cancer
Research
Ercc1 Deficiency Promotes Tumorigenesis and
Increases Cisplatin Sensitivity in a Tp53
Context-Specific Manner
Mladen Jokic 1,2, Ignacija Vlasic
1,2, Miriam Rinneburger1,2, Niklas Klu € mper3,
4 3 3
Judith Spiro , Wenzel Vogel , Anne Offermann , Christiane Ku € mpers3,
Christian Fritz , Anna Schmitt , Arina Riabinska , Maike Wittersheim5,
1,2 1,2 1,2
Sebastian Michels6, Luka Ozretic 5, Alexandra Florin5, Daniela Welcker1,2,7,
Mehmet Deniz Akyuz , Michael Nowak9, Martin Erkel10, Ju
8
€ rgen Wolf6,
Reinhard Bu€ ttner , Bjo
5
€ rn Schumacher , Ju 8
€ rgen Thomale , Thorsten Persigehl4,
10
David Maintz , Sven Perner , and Hans Christian Reinhardt1,2
4 3
Abstract
KRAS-mutant lung adenocarcinoma is among the most com- tumors that relapsed after cisplatin treatment in our model devel-
mon cancer entities and, in advanced stages, typically displays op a robust etoposide sensitivity that is independent of the Ercc1
poor prognosis due to acquired resistance against chemotherapy, status and depends solely on previous cisplatin exposure. Our
which is still largely based on cisplatin-containing combination results provide a solid rationale for further investigation of the
regimens. Mechanisms of cisplatin resistance have been extensive- possibility of preselection of lung adenocarcinoma patients
ly investigated, and ERCC1 has emerged as a key player due to its according to the functional ERCC1- and mutational TP53 status,
central role in the repair of cisplatin-induced DNA lesions. How- where functionally ERCC1-incompetent patients might benefit
ever, clinical data have not unequivocally confirmed ERCC1 status from sequential cisplatin and etoposide chemotherapy.
as a predictor of the response to cisplatin treatment. Therefore, we
employed an autochthonous mouse model of Kras-driven lung Implications: This study provides a solid rationale for the
adenocarcinoma resembling human lung adenocarcinoma to stratification of lung adenocarcinoma patients according to
investigate the role of Ercc1 in the response to cisplatin treatment. the functional ERCC1- and mutational TP53 status, where
Our data show that Ercc1 deficiency in Tp53-deficient murine lung functionally ERCC1-incompetent patients could benefit from
adenocarcinoma induces a more aggressive tumor phenotype that sequential cisplatin and etoposide chemotherapy. Mol Cancer Res;
displays enhanced sensitivity to cisplatin treatment. Furthermore, 14(11); 1110–23. 2016 AACR.
1
Department I of Internal Medicine, University Hospital of Cologne, Introduction
Weyertal 115B, 50931, Cologne, Germany. 2Cologne Excellence Cluster
on Cellular Stress Response in Aging-Associated Diseases, University Nucleotide excision repair (NER) is a complex DNA repair
of Cologne, Weyertal 115B, 50931, Cologne, Germany. 3Pathology of system that has evolved to cope with helix-distorting lesions,
the University Medical Center Schleswig-Holstein, Campus Luebeck
and the Research Center Borstel, Leibniz Center for Medicine and
such as those inflicted by nitrosamines, benzo(a)pyrenes
Biosciences, 23538 Luebeck and 23845 Borstel, Germany. 4Depart- (from cigarette smoke), cyclobutane pyrimide dimers, and
ment of Radiology, University Hospital of Cologne, Kerpener Str. 62, pyrimidine (6-4) pyrimidone photoproducts (induced by UV
50937, Cologne, Germany. 5Institute of Pathology, University Hospital
of Cologne, Kerpener Str. 62, 50937, Cologne, Germany. 6Department I
light), as well as crosslinks between guanine bases (induced
of Internal Medicine, University Hospital of Cologne, Kerpener Str. 62, by the chemotherapeutic agents cisplatin, carboplatin, and
50937, Cologne, Germany. 7Department II of Internal Medicine, Uni- oxaliplatin; refs. 1–4). The NER mechanism consists of four
versity Hospital Cologne, Kerpener Str. 62, 50937, Cologne, Germany. distinct steps, namely (i) recognition of the DNA lesion; (ii)
8
Institute for genome stability in ageing and disease, CECAD Research
Center, Joseph-Stelzmann-Str. 26, 50931, Cologne, Germany. 9Insti- DNA unwinding, (iii) endonuclease-mediated incisions 30
tute of Pathology, University Hospital Bonn, Sigmund-Freud-Str. 25, and 50 of the lesion; and (iv) DNA resynthesis and ligation
53127, Bonn, Germany. 10Institute for Cell Biology, University Hospital (1–4). Lesion recognition occurs through one of two mechan-
Essen, Hufelandstraße 55, 45122, Essen, Germany.
isms, either global genome repair (GG-NER), which constant-
Note: Supplementary data for this article are available at Molecular Cancer ly scans noncoding and nontranscribed regions of the
Research Online (http://mcr.aacrjournals.org/).
genome, or transcription-coupled repair (TC-NER), which
M. Jokic, I. Vlasic and M. Rinneburger contributed equally to this article as first is activated upon detection of stalled RNA polymerase II
authors. (1–6). The ERCC1/XPF endonuclease complex constitutes a
Corresponding Author: Mladen Jokic, Department I of Internal Medicine, critical component of the NER mechanism, which mediates
University Hospital of Cologne, Weyertal 115B, Cologne 50931, Germany. the DNA incision 50 of the lesion (7, 8). In addition to its
Phone: 4922-1478-96702; Fax: 4922-1478-96719; E-mail: contribution to NER, excision repair cross-complementation
mladen.jokic@uk-koeln.de
group 1 (ERCC1) is involved in recombination-mediated
doi: 10.1158/1541-7786.MCR-16-0094 DNA repair, as well as the repair of interstrand crosslinks
2016 American Association for Cancer Research. (9, 10). In humans, four distinct ERCC1 splice variants exist,
1110 Mol Cancer Res; 14(11) November 2016
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.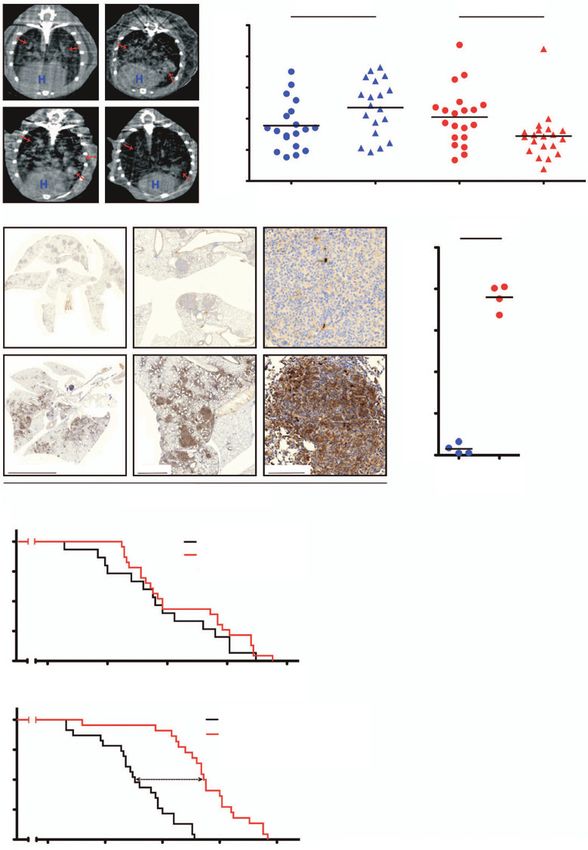
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
of which only one (ERCC1-202) encodes a protein product adenocarcinomas (22). In addition, we combined a conditional
that is capable of mediating repair of cisplatin-induced DNA Ercc1 allele (Ercc1flox) (23) with the K and KP models to obtain
damage (11). KrasLSL.G12D/wt;Ercc1flox/flox (KE) and KrasLSL.G12D/wt;Tp53flox/flox;
Given its prominent role in NER-mediated repair of cisplatin- Ercc1flox/flox (KPE) mice, respectively. Mice were kept on a mixed
induced DNA lesions, ERCC1 expression levels have been eval- C57Bl6/FVB background. To induce lung tumors, 8-week-old
uated as a potential biomarker to predict the response of cancer KP mice were anesthetized with ketamine (100 mg/kg) and
patients to cisplatin-based chemotherapy regimens. Although xylazine (20 mg/kg) by intraperitoneal injection, prior to intra-
numerous studies indicate that low levels of ERCC1 mRNA or tracheal application of a replication-deficient adenovirus expres-
protein expression predict enhanced overall survival in response sing Cre [Ad-CMV-Cre, 2.5 107 plaque-forming unit (PFU)],
to platinum-based chemotherapy in human non–small cell lung as described previously (22). Five weeks after Ad-CMV-Cre
cancer (NSCLC; refs. 12–14), several groups have reported con- inhalation, lungs of KP and KPE mice were scanned by mCT
tradictory results (11, 15–18). Thus, there is conflicting data on imaging (Aloka, Latheta LCT-100) under ketamine/xylazine anes-
the role of ERCC1 expression as a predictor for cisplatin sensitivity thesia (as described above) to confirm tumor formation. K and
in human lung adenocarcinomas. Of note, the method of ERCC1 KE mice were imaged 12 weeks after tumor induction in the
detection varied between the studies, where some used RT-PCR same manner. After mCT-confirmed tumor formation, mice of all
(14, 15, 18) and others used IHC (11–13, 16, 17) to determine four genotypes (K, KE, KP, and KPE) were treated with 7.5 mg/kg
ERCC1 status. Particularly different batches of the commonly cisplatin (Sigma-Aldrich, P4394) once a week for 3 weeks. One
used ERCC1 antibody 8F1 were found to deliver non-reproduc- week following administration of the last dose, mice were reim-
ible results (11, 13). Further uncertainty was spurred by the recent aged by mCT to assess tumor response. The percentage of tumor
discovery that 8F1 does not only engage ERCC1, but also cross- versus normal lung volume before and after cisplatin treatment
reacts with another otherwise unrelated protein due to a common was analyzed in each animal, and the results were confirmed
epitope (19). These conflicting results, together with the lack of a by a specialized radiologist, using the semiautomatic segmenta-
reliable and clinically applicable method of ERCC1 detection, led tion software Imalytics (Philips). Tumor tissue volume (mm3)
to considerable confusion in the field and a certain hesitance to and lung volume (mm3) were measured by regions of interest
pursue ERCC1 as a potential biomarker. using a 3D Hounsfield unit threshold technique, and the per-
Despite the development of novel, molecularly targeted ther- centage (%) of tumor and normal lung volumes was calculated.
apeutics for the treatment of many different cancer entities, In addition, acute tumor response was assessed 24 hours fol-
cisplatin remains a cornerstone in the frontline regimens of most lowing administration of a single dose of cisplatin (7.5 mg/kg),
solid tumors, including NSCLC, which is the most common using immunohistochemical staining for cleaved caspase-3 and
malignancy after non-melanocytic skin cancer, with deaths from Pt-(GpG) adducts. Second line of chemotherapy based on etopo-
lung cancer exceeding those from any other malignancy (20). side (Sigma-Aldrich, E1383, 20 mg/kg, 2 consecutive days per
Cisplatin-based combination regimens are particularly important week, for 2 weeks, i.p.) treatment was applied on a cohort of KP
for the treatment of KRAS-mutant lung adenocarcinoma, which (n ¼ 7) and KPE (n ¼ 10) mice that had previously undergone
represents the largest subentity of lung adenocarcinoma and cisplatin treatment as described above. Relapse of the tumors
for which no targeted therapeutic approach is currently available resistant to cisplatin was verified by mCT. Mice were treated with
(20, 21). It is thus interesting to identify genetically defined etoposide and monitored for survival. Mice used for the syngeneic
scenarios in which KRAS-mutant tumors respond to cisplatin. allograft experiment (n ¼ 5) were anesthetized (2.5% isoflurane)
Here, we employed genetically engineered mouse models of and injected with 106KPE (n ¼ 1) control cells at the upper part of
Kras-driven lung adenocarcinoma to address the role of Ercc1 in the back and with 106KPE post-cisplatin cells (n ¼ 5) in the
NSCLC tumorigenesis and cisplatin response. Conditional dele- lower part of the back. After tumor formation was verified by
tion of Ercc1 in Kras-driven NSCLC models with concomitant palpation and mCT, mice were subjected to etoposide treatment
Tp53 deficiency leads to more aggressive tumors that result in (20 mg/kg, 3 consecutive days per week, for 2 weeks, i.p.), and
significantly reduced overall survival than in Ercc1-proficient tumor progression was monitored by mCT reimaging one week
disease. In addition, we show that Ercc1-deficient tumors display after the last etoposide treatment. Tumor volume was assessed by
a massively enhanced cisplatin response. Finally, we demonstrate OsiriX, free and open-source DICOM viewer (OsiriX v.5.8.2,
that in vivo cisplatin treatment of mice bearing conditionally Pixmeo). All animal procedures were approved by the local
Ercc1-deficient lung adenocarcinomas leads to the selection of animal protection committee and the local authorities.
rare tumor cells that retain one functional allele of Ercc1. These
cells then constitute relapsing tumors, which display an increased Mouse lung adenocarcinoma grading system
sensitivity to etoposide. Together, our data suggest that KRAS- Formalin-fixed paraffin-embedded (FFPE) mice lung sam-
and TP53-mutant NSCLC patients could be stratified on the basis ples were cut into 4-mm thick sections and mounted on slides.
of functional ERCC1 expression levels, as low functional ERCC1 After staining with hematoxylin and eosin (H&E), the tumors
expression might be associated with favorable cisplatin response were assessed by a pathologist to evaluate the tumor burden.
and sensitivity toward etoposide in the second line. A tumor grading system with three tumor grades (1, 2, and 3)
based on the relative amount of diffuse or nodular tumor
growth was applied. Grade 1 ("diffuse") tumors were defined
Materials and Methods by a predominantly diffuse tumor architecture (>90% diffuse).
Autochthonous and allograft murine lung adenocarcinoma Grade 3 ("nodular") tumors predominantly show a nodular
model growth pattern (>90% nodular), and in between, grade 2
KrasLSL.G12D/wt (K) and KrasLSL.G12D/wt;Tp53flox/flox (KP) mice ("mixed") contains tumors with a mixed architecture (>10%
were used as autochthonous models of KRAS-mutant lung diffuse and >10% nodular).
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1111
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.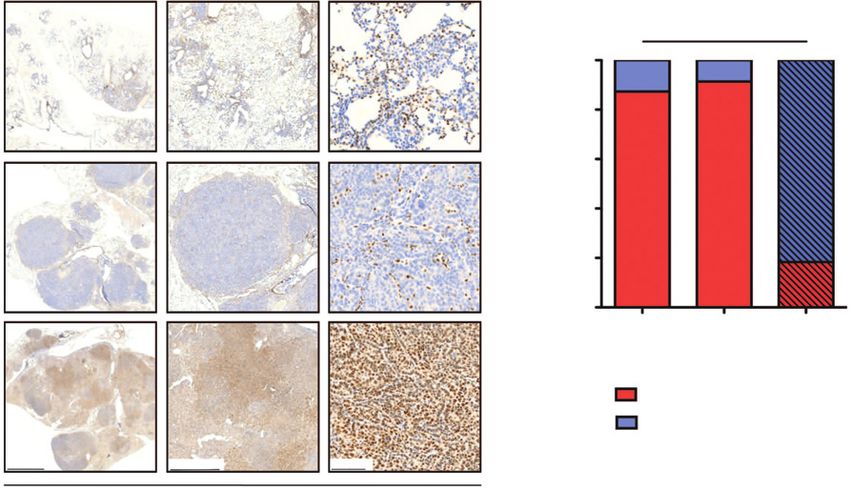
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
A
100 KrasLSL.G12D/wt (K) 100 KrasLSL.G12D/wt; Trp53fl/fl(KP)
KrasLSL.G12D/wt; Ercc1fl/fl(KE)
KrasLSL.G12D/wt; Trp53fl/fl; Ercc1fl/fl(KPE)
P = 0.571
75 75 ***P < 0.0001
Survival (%)
Survival (%)
32d
50 50
25 25
0 0
0 60 90 120 150 180 210 240 0 60 90 120 150
Time (days) Time (days)
B C
100 12 w after Ad-Cre
Overall lung volume (%)
Early-stage tumors
90 Normal tissue
80 Tumor tissue
70
60
50
40
30
20
10
Late-stage tumors
0
K KE KP KPE
(n = 7) (n = 3) (n = 3) (n = 9)
K KE KP KPE
5,000 µm 1,000 µm 100 µm
G1 G2 G3
D P = 0.7878 P = 0.4512
100
Ercc1-positive index (%)
90
80
K
70
60
50
40
30
20
10
0
KE
4 wKE 12 wKE 12 w K
(n = 4) (n = 3) (n = 3)
Ercc1-negative cells
Ercc1-positive cells
2,000 µm 1,000 µm 100 µm
P = 0.7854 *P = 0.0178
100
Ercc1-positive index (%)
90
80
KP
70
60
50
40
30
20
10
0
KPE
4 wKPE 12 w KPE 12 w KP
(n = 9) (n = 9) (n = 3)
Ercc1-negative cells
Ercc1-positive cells
2,000 µm 1,000 µm 100 µm
Anti-Ercc1, 12 weeks post Ad-Cre
1112 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.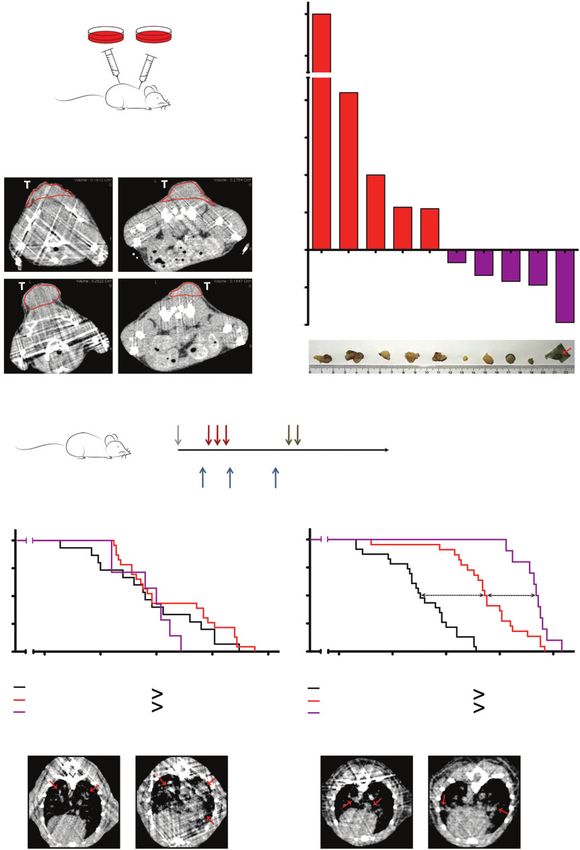
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
Immunohistochemical staining and quantification of protein Luminata Crescendo Western HRP Substrate (Millipore,
expression in murine FFPE tissue WBLUR0100) on a Bio-Rad ChemiDoc MP System.
Immunohistochemical staining against cleaved caspase-3
(CC3) and Ercc1 was conducted using the Ventana Discovery Clonogenic survival assay
XT automated staining system (Ventana Medical Systems) on A total of 2 103 cells per condition were seeded in 6-cm
FFPE mice lung samples. In brief, slides were incubated at room plates and incubated for 24 hours. Cells were either treated with
temperature with the primary antibody according to the man- mock or were exposed to 5 mmol/L cisplatin for 24 hours, and
ufacturers' instructions (dilutions, clones, manufacturer): anti- then the medium (RPMI) was replaced. Cells were incubated
CC3 rabbit polyclonal (1:100, #9661, Cell Signaling Technol- for 10 days with medium exchange, fixed, and stained with
ogy) and anti-Ercc1 rabbit monoclonal (1:100, EPR7277, 0.5% crystal violet (Sigma, HT90132).
Abcam). Antibody dilution was conducted using a Ventana
diluent. Signal detection was done using the UltraMap anti-Rb Immunostaining and measurement of Pt-(GpG) adducts in
HRP Detection Kit (Ventana Medical Systems). Finally, slides DNA
were counterstained with hematoxylin and bluing reagent, Plated cells were treated with cisplatin (20 mg/mL) for 4 hours
dehydrated, and mounted. Immunohistochemical stainings and were subsequently maintained in drug-free media for up to 48
were analyzed independently by two experienced observers hours. At various time points, cell aliquots were harvested, washed
(S. Perner and N. Kl€ umper). in PBS, resuspended, and placed onto microscopic slides (Super-
Quantification of staining intensity by CC3 and Ercc1 anti- frost Plus Gold Adhesion Slides, Thermo Scientific). Lungs of
bodies was performed using a semiquantitative image analysis tumor-bearing mice were perfused 24 hours after cisplatin treat-
program (Definiens Inc.) as described earlier (24). In short, the ment (7.5 mg/kg) with Tissue-Tek OTC/PBS solution (1:1;
slides were scanned and areas containing lung adenocarcinoma Sakura), resected, and placed in liquid N2. Frozen tissue sections
within the lung specimens were marked manually to exclude (12 mm) were prepared on adhesion slides. Pt-(GpG) intrastrand
normal and stromal areas. Afterward, the software analyzed the cross-links in the nuclear DNA of single cells were visualized and
staining intensity of tumor cells in the selected regions, and the measured by an immunocytologic assay using the Pt-(GpG)–
cells were defined as positive through a predefined intensity specific antibody "R-C18" essentially as described previously
threshold. The "positive index" was calculated as the ratio of (25). Briefly, cells were fixed in methanol (20 C), denatured
positive cells divided by all cells in the region of interest. by alkaline treatment (60% seventy mmol/L NaOH/140 mmol/L
NaCl; 40% methanol; 5 minutes, 0 C), and digested successively
with pepsin (100 mg/mL; 10 minutes; 37 C) and proteinase K
Cell culture
(200 mg/mL; 10 minutes; 37 C). After blocking (5% skim milk in
Murine lung adenocarcinoma tumor cells were isolated and
PBS), slides were immunostained with "R-C18" rat anti-Pt-(GpG)
cultured in RPMI containing 10% FBS. Retained Ercc1fl allele in
antibody (20 ng/mL in PBS/BSA; 12 hours; 4 C) and with Cy3-
KPE post-cisplatin cells was knocked out (recombined) by treat-
labeled rabbit anti-(rat Ig) antibody (Dianova) for 1 hour at 37 C.
ment with 2.5 107 PFU of Ad-CMV-Cre in three rounds.
Nuclear DNA was counterstained with DAPI (1 mg/mL in PBS).
DAPI- and Cy3-derived signals were integrated and measured
PCR separately for individual cell nuclei using a microscope-coupled
For genotyping purposes, Ercc1flox and Ercc1wt alleles were digital image analysis system (Zeiss Axioplan; ACAS 6.0 Image
detected using ERCC1_F (50 -TAC TTG CCA GGG CAT TTG Analysis System, Ahrens Electronics). Antibody-derived fluores-
TGT-30 ) and ERCC1_R (50 -GTA TTG AGT GTT TTG CCT GCA cence signals were normalized to the corresponding DNA content
T-30 ) primers. A 350-bp band indicated the Ercc1flox allele, while a of the same nucleus and expressed as arbitrary fluorescence units
500-bp band indicated the presence of the Ercc1wt allele. Recom- (AFU). Values were calculated as means of >100 measured cells
bined Ercc1flox allele was detected with primers F25731 and per sample, and error bars represent 95% confidence intervals. For
F25732 as described before (23). lung sections, randomly selected areas (100 100 mm) of tumor
cells were measured and calculated for AFU values.
Western blot analysis
Murine lung adenocarcinoma cells were collected, lysed Immunofluorescence
[Invitrogen NP40-based lysis buffer (FNN0021) with protease For the staining of KPE and KPE post-cisplatin cells for g-H2AX,
inhibitors], and quantified using the BCA Protein Assay Kit we seeded 105 cells per 18 cm2 coverslip and incubated for
(Pierce). Protein extracts (30 mg) were subjected to 10% SDS- 24 hours. Cells were either mock treated or were exposed to
PAGE, immunoblotted for Ercc1 (Abcam, EPR7277, 1:1,000) and 10 mmol/L etoposide for 60 minutes, and then the medium
b-actin (Sigma-Aldrich, A5316, 1:5,000), and analyzed using the (RPMI) was replaced. Cells were collected at 6, 24, and 96 hours
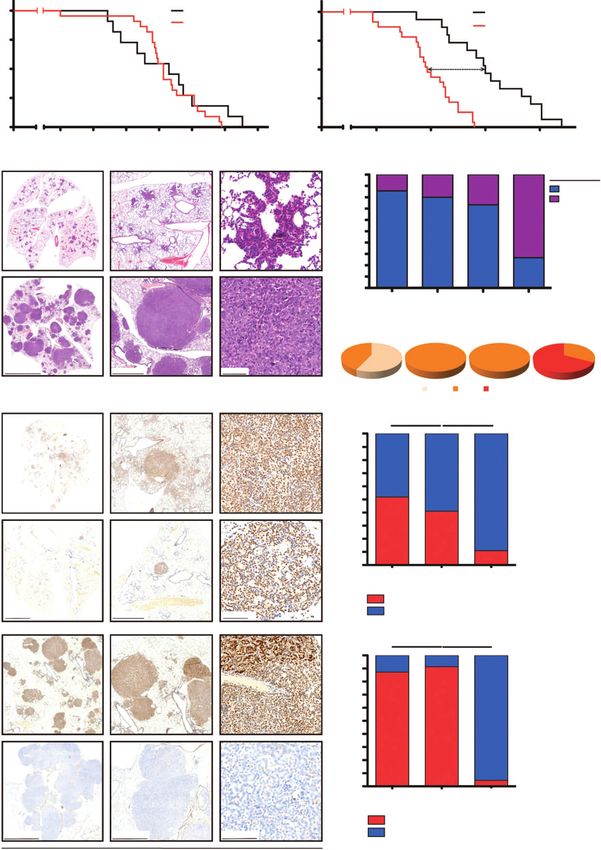
Figure 1.
Ercc1 deletion enhances more aggressive tumor phenotype in Tp53-deficient lung adenocarcinoma. A, survival of chemo-na€ve KrasLSL.G12D/wt (K) and
KrasLSL.G12D/wt;Ercc1fl/fl (KE; left) and KrasLSL.G12D/wt;Tp53fl/fl (KP) and KrasLSL.G12D/wt;Tp53fl/fl;Ercc1fl/fl (KPE) mice (right) inhaled with Ad-CMV-Cre. P
values were calculated using log-rank test. B, representative H&E staining of murine lung adenocarcinoma from KPE mice at 4 weeks (early-stage tumors)
and of KPE mice at 12 weeks after tumor induction (late-stage tumors). C, top, distribution of tumor versus normal lung tissue in K, KE, KP, and KPE
mice at 12 weeks after tumor induction; bottom, distribution of murine lung adenocarcinoma grades (G1, G2, G3) in corresponding K, KE, KP, and KPE mice at 12
weeks after tumor induction. D, left, anti-Ercc1 immunostaining of lung sections of K, KE, KP, and KPE mice at 12 weeks after tumor induction (three
magnifications for each genotype; right top, quantification of Ercc1-positive tumor cells in KE mice (n ¼ 4) at 4 weeks after tumor induction, KE mice (n ¼ 3) at
12 weeks after tumor induction, and of control K mice (n ¼ 3) at 12 weeks after tumor induction; right bottom, quantification of Ercc1-positive tumor
cells in KPE mice (n ¼ 9) at 4 weeks after tumor induction, KPE mice (n ¼ 9) at 12 weeks after tumor induction, and of control KP mice (n ¼ 3) at 12 weeks
after tumor induction. P values were calculated by comparing percentages using two-tailed t test. , P < 0.05.
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1113
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.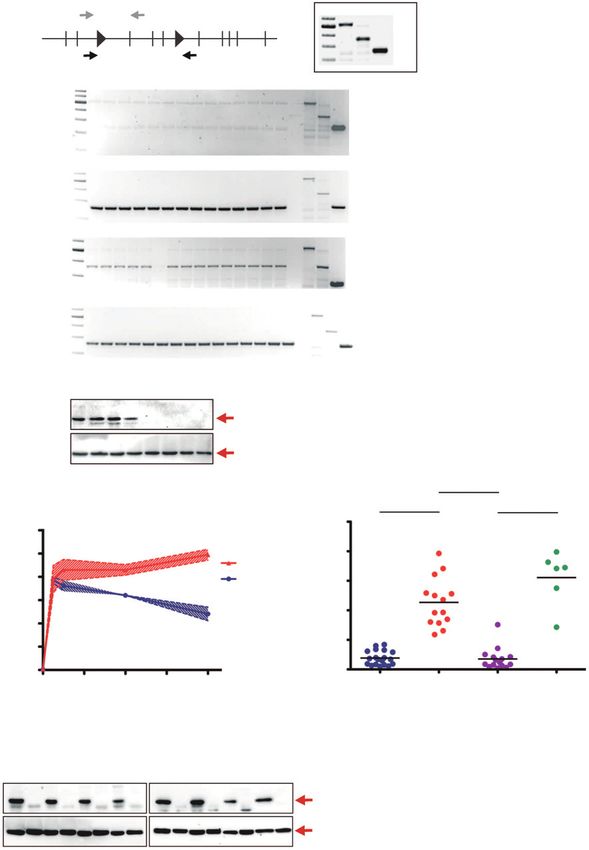
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
A KP KPE
**P = 0.0015 ***P = 0.0001
100
Pre-cisplatin
Volume of tumor/lung tissue (%)
80
60
40
Post-cisplatin
20
0
KP pre cis KP post cis KPE pre cis KPE post cis
B ***P < 0.0001
100
Fraction of apoptotic cells (%)
KP
80
60
40
KPE
20
0
5,000 mm 1,000 mm 100 mm KP KPE
Anti-CC3, 24 h post-cisplatin
C
100 KrasLSL.G12D/wt; Trp53fl/fl(KP)
KrasLSL.G12D/wt; Trp53fl/fl(KP) cis
Survival (%)
P = 0.356
75
50
25
0
75 100 125 150 175
Time (days)
100 KrasLSL.G12D/wt; p53fl/fl; Ercc1fl/fl(KPE)
KrasLSL.G12D/wt; p53fl/fl; Ercc1fl/fl(KPE) cis
Survival (%)
75 ***P < 0.0001
30.5 d
50
25
0
50 75 100 125 150
Time (days)
Figure 2.
Ercc1 deficiency sensitizes Tp53-deficient lung adenocarcinoma to cisplatin in vivo. A, left, mCT-based monitoring of tumor response in KP and KPE mice
prior to and after cisplatin treatment; right, quantification of tumor volume in KP and KPE mice prior to and after cisplatin treatment. P values were
calculated using two-tailed t test. B, left, representative anti–cleaved caspase-3 immunostaining of lung sections of KP and KPE mice after single
cisplatin treatment; right, quantification of the fraction of apoptotic cells in KP and KPE mice after single-dose cisplatin treatment. C, survival
comparison of KP chemo-na€ve and KP cisplatin-treated mice (top) and of KPE chemo-na€ve and KPE cisplatin-treated mice (bottom). P values were
calculated using log-rank test.
1114 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.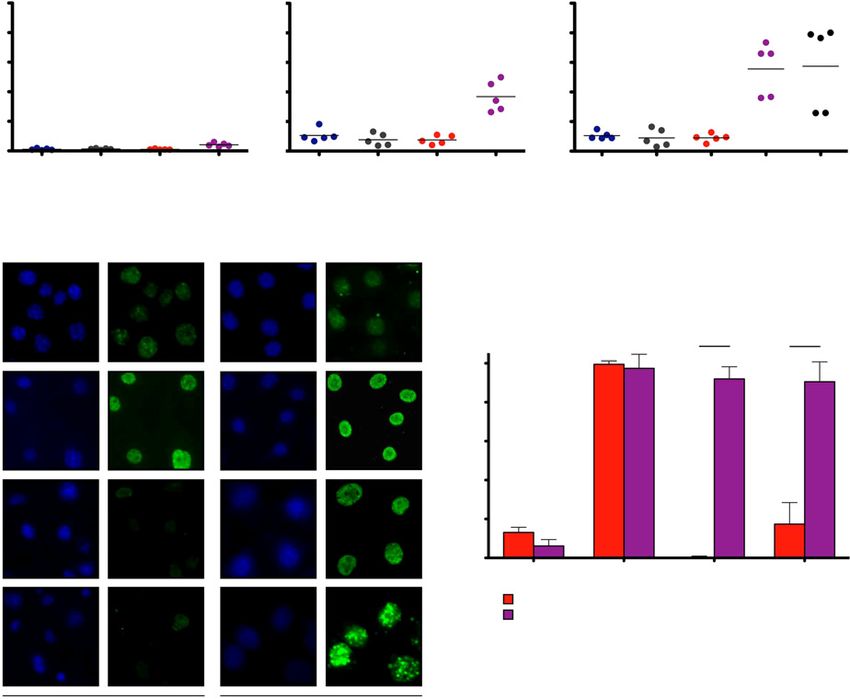
Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
A B
**P = 0.0080
KPE 4 w
100
Ercc1-positive tumor cells (%)
80
60
40
KPE 12 w
20
0
4 w KPE 12 w KPE KPE
chemo-naïve chemo-naïve cisplatin
(n = 9) (n = 9) (n = 10)
KPE cis
Ercc1-negative cells
Ercc1-positive cells
2,000 mm 1,000 mm 100 mm
Anti-Ercc1
Figure 3.
Cisplatin treatment selects for Ercc1 expression in lung adenocarcinoma. A, top, representative anti-Ercc1 immunostaining of lung sections of KPE mice
at 4 weeks after tumor induction shown in three magnifications; middle, representative anti-Ercc1 immunostaining of lung sections of KPE mice at
12 weeks after tumor induction shown in three magnifications; bottom, representative anti-Ercc1 immunostaining of lung sections of KPE mice after
three cycles of cisplatin treatment shown in three magnifications. B, quantification of Ercc1-positive tumor cells in KPE mice (n ¼ 9) at 4 weeks after
tumor induction, KPE mice (n ¼ 9) at 12 weeks after tumor induction, and of KPE mice (n ¼ 10) after cisplatin treatment. P value was calculated by
comparing percentages using a two-tailed t test.
after etoposide removal and fixed in 4% PFA for 15 minutes on minutes at 20 C. After several washing steps with PBS and
room temperature. Cells were washed in PBS, permeabilized in 0.25% Triton X-100 or PBS and 1% BSA, cells were incubated at
cytoskeleton buffer (10 mmol/L PIPES pH 6.8, 100 mmol/L NaCl, 4 C overnight with Purified Rabbit Anti-Active Caspase-3 primary
300 mmol/L sucrose, 3 mmol/L MgCl2, 1 mmol/L EGTA pH 8.0, antibody solution (BD Pharmingen, 559565, 1:200). Cells were
and 0.5% Triton X-100) for 10 minutes on ice and then counterstained with propidium iodide (PI), and after 15-minute
incubated in cytoskeleton stripping buffer (10 mmol/L Tris-HCl incubation in the dark, cells were assayed on Beckman Coulter
pH 7.4, 10 mmol/L NaCl, 3 mmol/L MgCl2, 2% Tween 20, and Gallios Flow Cytometer. Cell populations were analyzed using
0.4% sodium deoxycholate) for 10 minutes on ice. Cells were Kaluza software and 50,000 to 100,000 events were counted in
blocked in PBS supplemented with 5% BSA, 2% NGS, and each assay.
0.01% Triton X-100 for 1 hour at room temperature and incu-
bated with anti-g-H2AX (phospho S140) [3F2] primary
antibody solution (Abcam, ab22551, 1:250) at 4 C overnight. Results
Cell nuclei were counterstained with Prolong Gold Antifade Conditional Ercc1 deletion enhances tumorigenesis in
Reagent with DAPI (Molecular Probes, Life Technologies). Tp53-deficient lung adenocarcinomas
Cells were imaged on Axiovert 200M inverted fluorescence imag- To directly validate Ercc1 deficiency as a predictor for cisplatin
ing microscope equipped with a CCD camera (Zeiss) and with sensitivity in vivo, we aimed to assess the effect of Ercc1 deletion
AxioVision Rel. 4.8 software. A total of 400 to 500 cells per in a model of Kras-driven lung adenocarcinoma. For this purpose,
condition on several independent positions equally distributed we employed an established genetically engineered mouse
over the slides were evaluated for g-H2AX positivity by AxioVision model, in which expression of oncogenic KrasG12D is pre-
Rel. 4.8 software. vented through the insertion of a LoxP-flanked transcriptional
and translational STOP cassette in the endogenous locus
Quantification of apoptosis by flow cytometry (KrasLSL.G12D allele, referred to as K in the following; ref. 22).
For quantification of apoptosis, 106 cells were seeded in 10-cm Intratracheal administration of adenoviral Cre recombinase
dishes 24 hours prior to treatment. Cells were treated for 24 hours (Ad-CMV-Cre) leads to the expression of oncogenic KrasG12D
with mock, 5 mmol/L cisplatin, 10 mmol/L etoposide, 0.1 mmol/L from its endogenous locus. In addition to this simple Kras-
gemcitabine, or 10 mmol/L taxol. Next, cells were collected, driven model, we also used KrasLSL.G12D/wt;Tp53fl/fl mice (KP
washed with ice-cold PBS, and fixed in methanol for at least 30 in the following), in which both Tp53 alleles are flanked by
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1115
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
WT MT REC
A -500 bp
1 2 IoxP 3 4 5 IoxP 6 78 9 10
Ercc1fl -350 bp
-250 bp
b1 b2 b3 b4 b5 b6 b7 b8 b9 b10 b11 b12 b13 b14 E WTc MTc Recc
-500 bp
350 bp
-250 bp
b1 b2 b3 b4 b5 b6 b7 b8 b9 b10 b11 b12 b13 b14 E WTc MTc Recc
-500 bp
350 bp
-250 bp
c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11 c12 c13 c14 c15 E WTc MTc Recc
-500 bp
350 bp
-250 bp
c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11 c12 c13 c14 c15 E WTc MTc Recc
-500 bp
350 bp
-250 bp
1 2 3 4 5 6 7 8
Ercc1
(33 kDa)
b-Actin
(42 kDa)
***P < 0.0001
B C ***P < 0.0001 ***P < 0.0001
50
Fraction of apoptotic cells (%)
1.2
Pt-(GpG) in DNA (AFU)
1.0 40
KPE Cell lines
0.8 KP Cell lines 30
0.6
20
0.4 c6
10
0.2
0.0 0
0 12 24 36 48 KP KPE KPE KPE
post cis post cis + Ad-Cre
Time after cisplatin (h)
D
3 + Ad-Cre
4 + Ad-Cre
5 + Ad-Cre
7 + Ad-Cre
8 + Ad-Cre
9 + Ad-Cre
1
2
3
4
5
1
6
7
8
9
Ercc1
(33 kDa)
b-Actin
(42 kDa)
1116 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
LoxP sites (22). Here, Ad-CMV-Cre drives expression of KrasG12D (59% 17.39 of Ercc1-positive tumor cells, n ¼ 3) after tumor
and simultaneous deletion of both Tp53 alleles (22). The induction (Fig. 1D and Supplementary Fig. S1B). In stark
impact of Ercc1 deletion was assessed by crossing conditional contrast, the Ercc1 deletion in Tp53-deficient KPE animals was
LoxP-flanked Ercc1 alleles (23) homozygously into our K and almost complete [12.68 4.24% and 8.65 3.23% of Ercc1-
KP models, yielding KE and KPE mice, respectively. The effect of positive tumors cells at 4 (n ¼ 9) and 12 weeks (n ¼ 9) after
Ercc1 deletion was gauged through assessment of the overall tumor induction, respectively; Fig. 1D and Supplementary
survival of K, KE, KP, and KPE animals that inhaled Ad-CMV- Fig. S1C]. These data suggest that Ercc1 deletion is selected
Cre at 8 weeks of age. As shown in Fig. 1A, the survival of K (n ¼ against in p53-proficient settings, while it is tolerated in the
11) and KE (n ¼ 22) animals was not statistically different. In absence of functional p53. Furthermore, concomitant Tp53 and
contrast, KPE animals (n ¼ 23) displayed a significantly Ercc1 deletion appears to promote the development of more
reduced overall survival, compared with their Ercc1-proficient aggressive lung adenocarcinomas (Fig. 1A).
KP counterparts (n ¼ 15). To further dissect this observation,
we next performed histologic tumor volume assessments at 4 Ercc1 deficiency increases cisplatin sensitivity in Tp53-deficient
and 12 weeks after Ad-CMV-Cre (Fig. 1B and C and Supple- lung adenocarcinomas in vivo
mentary Fig. S1A). These experiments revealed that lungs of K The conflicting clinical data on the role of ERCC1 expression
and KE animals were infiltrated with similar tumor volumes at as a predictive biomarker for cisplatin response and overall
both 4 and 12 weeks following Ad-CMV-Cre inhalation (Sup- survival (11–18) have led to a hesitance in pursuing ERCC1
plementary Fig. S1A and S1C). Similarly, no difference between expression as a potential stratifier for the selection of chemo-
KP and KPE lungs was observed at 4 weeks (Supplementary Fig. therapeutic regimens in the treatment of NSCLC. To formally
S1A). However, at 12 weeks, lungs derived from KPE animals address the potential impact of low-level ERCC1 expression in a
displayed a substantially higher degree of tumor infiltration, relevant autochthonous mouse model of NSCLC in vivo, we
compared with KP mice (73.34 13.92% vs. 26.66 next assessed the cisplatin response in tumor-bearing K, KE, KP,
7.64%; Fig. 1C). Consistent with the enhanced aggressiveness and KPE animals (Fig. 2 and Supplementary Fig. S2). CT-based
of KPE tumors compared with KP tumors (Fig. 1A), we found imaging was used prior to initiation of treatment to confirm the
that KPE tumors were of substantially higher grade (G3) com- presence of neoplastic lesions formed 5 weeks following Ad-
pared with their KP counterparts (Fig. 1C). No substantial CMV-Cre inhalation in KP and KPE mice and 12 weeks fol-
difference was observed between K and KE tumors, although lowing Ad-CMV-Cre inhalation in K and KE mice (Fig. 2A and
a trend toward a shift from grade 1 to 2 was detected in KE Supplementary Fig. S2A). Upon tumor formation, we admin-
tumors. Both, K and KE tumors were entirely of low tumor istered three courses of cisplatin (7.5 mg/kg, i.p., once weekly).
grade (G1 and G2; Fig. 1C). Therapeutic response was evaluated 7 days following the last
Acute Ercc1 deletion leads to apoptotic cell death in murine cisplatin dose by mCT-based restaging. As shown in Supple-
hepatocytes (26). Furthermore, p53 protein levels were found to mentary Fig. S2A, cisplatin treatment induced significant tumor
be elevated in Ercc1-deficient murine liver-, kidney-, and brain regression in both K and KE animals (P ¼ 0.0051 and 0.0012,
tissue (27). These observations suggest that Ercc1 deletion pro- respectively). KP lung adenocarcinomas were entirely resistant
motes the induction of a p53 response. Given the proapoptotic and displayed significant (P ¼ 0.0015) tumor growth following
cellular outcome of p53 signaling (28), we hypothesized that three cycles of cisplatin exposure (Fig. 2A). This observation
Ercc1 deletion is tolerable in a p53-defective background, while it is in agreement with previous experiments that indicated cis-
is detrimental in a p53-proficient context. platin resistance of the KP model (29). In stark contrast, KPE
To directly assess Ercc1 deletion in tumors, we next per- tumors displayed a significant (P ¼ 0.0001) volume reduction
formed immunohistochemical staining of lungs isolated from at follow-up mCT. We note that this result reflects the average
tumor-bearing KE and KPE animals 4 and 12 weeks following of multiple tumors, which may have discordant responses
Ad-CMV-Cre inhalation with the adequate controls (K and with some tumors responding and others showing stable, or
KP tumors 12 weeks after tumor induction, respectively). In even progressive disease. These CT-morphologic cisplatin
KE tumors, we observed incomplete Ercc1 deletion at 4 (48.17 responses were mirrored by a highly significant (P < 0.0001)
6.82% of Ercc1-positive tumor cells, n ¼ 4) and 12 weeks increase in cleaved caspase-3–positive apoptotic tumor cells in
Figure 4.
Ercc1 deficiency sensitizes Tp53-deficient lung adenocarcinoma to cisplatin, in vitro. A, schematic representation of the Ercc1fl allele showing primer
positions for detection of the floxed allele (top gray arrows) and of the recombined allele [bottom black arrows; left; model partially taken from original
publication (23)]. Schematic representation showing the positions of Ercc1 wild-type (WT), Ercc1fl (MT), and Ercc1fl recombined (REC) bands (right).
Absence of Ercc1fl allele in KPE tumor cells (b1-b14; top gel), presence of recombined Ercc1fl allele in KPE tumor cells (b1-b14; second gel), presence of Ercc1fl
allele in KPE post-cisplatin (cis) cells (c1-c5 and c7-c15, except clone c6; third gel), and presence of recombined Ercc1fl allele in KPE post-cisplatin cells (c1-c15;
bottom gel). Anti-Ercc1 immunoblotting in KP cells (n ¼ 4, lanes 1–4) and KPE cells (n ¼ 4, lanes 5–8; top blot). Control b-actin immunoblotting in
corresponding tumor cell lines (bottom blot). B, measurement of Pt-(GpG) adduct levels (AFU) in KP (n ¼ 2) and KPE tumor cell lines (n ¼ 3) at
different time points after exposure to cisplatin. C, determination of the fraction of apoptotic cells (cleaved caspase-3 positive) using flow cytometry in
KP cell lines (n ¼ 19), KPE cell lines (n ¼ 14), KPE post-cisplatin cell lines (n ¼ 15), and KPE post-cisplatin pretreated with Ad-CMV-Cre (n ¼ 6), after
cisplatin exposure in vitro. c6, clone 6. P values were calculated using a two-tailed t test. D, anti-Ercc1 immunoblotting in control KP cell line (lane 1),
control KPE cell line (lane 2), KPE post-cisplatin, and corresponding KPE post-cisplatin cell lines treated with Ad-CMV-Cre (lanes 3 and 3 þ Ad-Cre, 4
and 4 þ Ad-Cre, 5 and 5 þ Ad-Cre; top left blot). Control b-actin immunoblotting in corresponding tumor cell lines (bottom left blot). Anti-Ercc1
immunoblotting in control KP cell line (lane 1), KPE post-cisplatin cell line clone 6 (lane 6), KPE post-cisplatin, and corresponding KPE post-cisplatin cell
lines treated with Ad-CMV-Cre (lanes 7 and 7 þ Ad-Cre, 8 and 8 þ Ad-Cre, 9 and 9 þ Ad-Cre; top right blot). Control b-actin immunoblotting in
corresponding tumor cell lines (bottom right blot).
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1117
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
KPE animals (n ¼ 4) compared with their KP counterparts (n ¼ zygous (Ercc1fl/null) on the Ercc1 locus, as indicated by the presence
4; Fig. 2B). In line with this, exposure to cisplatin (single dose, of both Ercc1 floxed and Ercc1 recombined bands (c1–c5 and
i.p., 7.5 mg/kg) leads to significant increase of DNA Pt-(GpG) c7–c15). Only one of these 15 clones (clone c6), was homozy-
adduct levels in KPE tumors (n ¼ 4) when compared with Ercc1- gously Ercc1-deficient, as only the recombined allele was detect-
proficient KP tumors (n ¼ 3), in vivo (Supplementary Fig. S2B). able on the PCR. These observations were confirmed by Ercc1
Furthermore, we monitored the survival of cisplatin-treated K immunoblotting (Fig. 4A and D).
(n ¼ 11), KE (n ¼ 17), KP (n ¼ 23), and KPE (n ¼ 23) animals. We next asked whether the retained Ercc1 allele is necessary
As shown in Supplementary Fig. S2C, median survival and sufficient to mediate cisplatin resistance. For this purpose,
of chemo-na€ve K and KE animals was 159 and 154 days, we initially compared the cisplatin response of cell lines iso-
respectively. Cisplatin treatment substantially enhanced overall lated from KP tumors (KP cells) with those derived from
survival by 51 and 76 days in the K and KE animals, respec- chemotherapy-na€ve KPE tumors (KPE cells). The ability of the
tively. The cisplatin-induced survival gains did not significantly different cell lines to clear cisplatin-induced DNA adducts was
(P ¼ 0.165) differ between K and KE animals, consistent with assessed using a Pt-(GpG) intrastrand cross-link–specific mAb
the incomplete Ercc1 deletion that we had observed histolog- that detects the most frequently occurring adduct formed by
ically in the adenocarcinoma-bearing KE animals (Fig. 1D and cisplatin, which is associated with its cytotoxicity and antican-
Supplementary Fig. S1B). A different picture emerged when we cer activity (25, 30). Exposure of both KP (n ¼ 5) and KPE
analyzed the overall survival of animals bearing Tp53-deficient (n ¼ 6) cell lines to cisplatin (20 mg/mL, 4 hours) led to the
tumors. In agreement with previously published results, cis- occurrence of Pt-(GpG) adducts that remained present for up
platin failed to significantly (P ¼ 0.356) enhance the survival of to 48 hours after the initial insult in KPE cells (Supplemen-
tumor-bearing KP animals, indicating that the KrasLSL-G12D/wt; tary Fig. S4A). To assess the kinetics of Pt-(GpG) adduct
Tp53fl/fl-driven lung adenocarcinoma model mimics a chemo- removal, we next performed a time course experiment. As
therapy-resistant high-risk clinical scenario (Fig. 2C; ref. 29). In shown in Fig. 4B, cisplatin treatment (20 mg/mL, 4 hours) in-
marked contrast, cisplatin treatment significantly prolonged duced the rapid occurrence of Pt-(GpG) adducts, in both KP
the overall survival in KPE mice, compared with chemothera- (n ¼ 2) and KPE (n ¼ 3) cells, as early as 4 hours following drug
py-na€ve (P < 0.0001) controls (Fig. 2C). Together, these data exposure. Ercc1-proficient KP cells continuously cleared these
indicate that Ercc1 deficiency significantly enhances cisplatin adducts over time, and 48 hours following cisplatin treatment,
sensitivity in aggressive Kras-driven and Tp53-deficient lung adduct level was reduced by 52% in these cells. In marked
adenocarcinomas, in vivo. However, cisplatin alone does not contrast, KPE cells completely failed to remove cisplatin
appear to have curative potential in this model. Thus, we next adducts at the end of the 48-hour observation period (Fig. 4B).
aimed to understand the mechanisms of acquired cisplatin We next asked whether KPE post-cisplatin cell lines isolated
resistance in KPE animals. from KPE mice treated with cisplatin in vivo display cisplatin
resistance, as one might predict based on their Ercc1 proficiency
Ercc1 expression is selected in cisplatin-resistant lung (Fig. 4A and D). We exposed KPE post-cisplatin cells (n ¼ 15),
adenocarcinomas as well as cells isolated from chemotherapy-na€ve KP (n ¼ 19)
To assess the biological mechanisms of cisplatin resistance in and KPE (n ¼ 14) tumors to 5 mmol/L cisplatin for 24 hours.
KPE animals, we performed histologic and immunohistochem- The fraction of apoptotic cells was assessed by flow cytometry,
ical analysis of KPE tumors that had relapsed after cisplatin using an antibody detecting cleaved caspase-3. As shown
treatment (Fig. 3 and Supplementary Fig. S3). We specifically in Fig. 4C and Supplementary Fig. S4C, chemotherapy-na€ve
stained tumor sections with an antibody detecting Ercc1, as we KPE cells were significantly more sensitive to cisplatin than
hypothesized that rare tumor cells that have undergone incom- their Ercc1-proficient KP counterparts. In contrast, cells isolated
plete recombination of the Ercc1fl/fl alleles might have escaped from KPE tumors that had relapsed after cisplatin treatment
cisplatin treatment, similar to the KP tumors, which are entirely in vivo (KPE post-cisplatin cells) were almost entirely resistant
resistant against cisplatin. Consistent with this hypothesis, tumors against cisplatin, with the exception of clone c6, which we
that had relapsed after cisplatin exposure in KPE animals, stained had previously shown to be Ercc1 deficient (Fig. 4A and D).
strongly positive for Ercc1 (Fig. 3 and Supplementary Fig. S3). We Similar results were obtained when we assessed the long-term
next aimed to verify that incomplete recombination of the Ercc1fl/fl effect of cisplatin treatment using clonogenic survival assays
alleles in early tumors of KPE mice is the molecular explanation (Supplementary Fig. S4B).
for Ercc1 expression in relapsed tumors. We further aimed to To directly test whether the retained Ercc1fl allele in KPE post-
validate that resistance in cisplatin-treated KPE tumors is indeed cisplatin cells that were isolated from cisplatin-treated KPE
dependent on Ercc1 expression. For this purpose, we isolated tumors was responsible for the cisplatin resistance that we had
cell lines from KP tumors (n ¼ 19; KP cells), as well as chemo- observed in vitro, we next deleted the retained Ercc1fl allele,
therapy-na€ve (n ¼ 14; KPE cells) and cisplatin-treated (n ¼ 15) using Ad-CMV-Cre, in vitro. Immunoblotting was used to verify
tumors (KPE post-cisplatin cells) from KPE animals. To directly Cre-mediated deletion of Ercc1fl (Fig. 4D). These cisplatin-
assess recombination efficiency, we checked for the presence of experienced Ercc1null/null cells (KPE post-cisplatin þ Ad-Cre
floxed and recombined Ercc1 alleles in chemotherapy-na€ve KPE cells) displayed a cisplatin-induced apoptotic response in vitro
and in cisplatin-treated KPE post-cisplatin cell lines. As shown in that was indistinguishable from that observed in chemother-
Fig. 4A, all 14 chemotherapy-na€ve KPE cell lines (b1–b14) apy-na€ve KPE cells (Fig. 4C). Altogether, these data strongly
displayed complete Ercc1 recombination, indicated by the suggest that the retained Ercc1 allele in cell lines isolated from
absence of the Ercc1 floxed (350 bp) and by the presence of KPE tumors that relapsed after cisplatin treatment was neces-
Ercc1 recombined (250 bp) bands. In marked contrast, cisplatin- sary and sufficient for the cisplatin resistance observed both
treated KPE post-cisplatin cell lines were almost entirely hetero- in vitro and in vivo.
1118 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
Cisplatin-relapsed lung adenocarcinomas display increased this effect was even more apparent when the cells were expos-
etoposide sensitivity ed to etoposide (10 mmol/L, 24 hours; Fig. 5A). To determine
As NSCLC patients typically receive multiple lines of whether this etoposide sensitivity in the cisplatin-relapsed
genotoxic chemotherapy, we next examined whether KPE KPE post-cisplatin cells is dependent on the Ercc1 status, we
post-cisplatin cells that were isolated from tumors that had exposed KPE post-cisplatin cells pretreated with Ad-CMV-Cre
relapsed after three cycles of cisplatin displayed differential in vitro (KPE post-cisplatin þ Ad-Cre; described in Fig. 4C)
sensitivity to other chemotherapeutic agents. We specifically to etoposide (10 mmol/L, 24 hours; Fig. 5A, far right). We
evaluated the cytotoxic effects of the spindle poison taxol, the observed no difference in the fraction of apoptotic cells
nucleoside analogue gemcitabine, and the topoisomerase-II when compared with their Ercc1-proficient counterparts (KPE
inhibitor etoposide using flow cytometry–based quantifica- post-cisplatin cells), indicating that the observed sensitivity
tion of apoptosis. As shown in Fig. 5, chemotherapy-na€ve and to etoposide is Ercc1 independent and therefore exclusively
cisplatin-experienced KP and KPE cell lines did not display a cisplatin induced.
differential taxol (10 mmol/L, 24 hours) sensitivity. In con- To further assess the differential cytotoxicity inflicted by
trast, cisplatin-experienced KPE post-cisplatin cell lines show- etoposide on chemotherapy-na€ve and cisplatin-experienced
ed sensitivity to gemcitabine (0,1 mmol/L, 24 hours), and KPE cells, we next quantified etoposide-induced genotoxic
A
Taxol, 10 mmol/L, 24 h Gemcitabine, 0.1 mmol/L, 24 h Etoposide, 10 mmol/L, 24 h
Fraction of apoptotic cells (%)
Fraction of apoptotic cells (%)
Fraction of apoptotic cells (%)
50 50 50
40 40 40
30 30 30
20 20 20
10 10 10
0 0 0
KP KP KPE KPE KP KP KPE KPE KP KP KPE KPE KPE
post cis post cis post cis post cis post cis post cis post cis
+ Ad-Cre
DAPI g-H2AX DAPI gH2AX
B
Mock
***P < 0.0001 ***P < 0.0001
Fraction of gH2AX-positive cells (%)
100
80
6h
60
40
20
24 h
0
Mock 6h 24 h 96 h
KPE cells
KPE post cis cells
96 h
KPE cells KPE post cis cells
Figure 5.
Cisplatin-relapsed tumor cells display etoposide sensitivity, in vitro. A, determination of fraction of apoptotic cells (cleaved caspase-3 positive) using flow
cytometry in KP, KP post-cisplatin (cis), KPE, and KPE post-cisplatin cell lines after exposure to taxol (left plot) and gemcitabine (middle plot). Determination
of fraction of apoptotic cells (cleaved caspase-3 positive) using flow cytometry in KP, KP post-cisplatin, KPE, KPE post-cisplatin cell lines, and
corresponding KPE post-cisplatin cell lines pretreated with Ad-CMV-Cre in vitro after exposure to etoposide (right plot). B, Immunofluorescence staining
for g-H2AX in chemo-na€ve KPE and KPE post cisplatin cells. Left, foci were evaluated in mock, 6, 24 and 96 hours after etoposide exposure. Right,
quantification of fraction of g-H2AX-positive cells. P value was calculated by comparing percentages using two-tailed t-test.
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1119
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
A C
KPE post cis cells KPE cells 5.0
4.5
2.0
Tumor growth (fold change)
1.5
B
KPE KPE post cis 1
1.0
Pre Eto
0.5
0.0
–0.5
Post Eto
–1.0 KPE tumors KPE post cis tumors
D
Ad-Cre Cisplatin 3x Etoposide 2x
0 5 6 7 Weeks
KP/KPE
mCT mCT mCT
Tumor Tumor Tumor
presence reduction relapse
E
100 100
75 75
Survival (%)
Survival (%)
30.5 d 24 d
50 50
25 25
0 0
75 100 125 150 175 50 75 100 125 150
Time (days) Time (days)
KrasLSL.G12D/wt; Trp53fl/fl(KP) KrasLSL.G12D/wt; p53fl/fl;Ercc1fl/fl(KPE)
P = 0.356 ***P < 0.0001
KrasLSL.G12D/wt; Trp53fl/fl(KP) cis KrasLSL.G12D/wt; p53fl/fl;Erccfl/fl(KPE) cis
P = 0.154 **P = 0.0026
KrasLSL.G12D/wt; Trp53fl/fl(KP) 1st cis/2ndeto KrasLSL.G12D/wt; p53fl/fl;Erccfl/fl(KPE) 1st cis/2ndeto
Pre cis Pre eto Pre cis Pre eto
KPE
KP
1120 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Ercc1 Loss Sensitizes Lung Adenocarcinoma to Cisplatin
lesions, using indirect immunofluorescence. We specifically cisplatin treatment of Ercc1-deficient Kras-driven lung adeno-
treated cells with etoposide (10 mmol/L, 60 minutes). Zero, carcinomas induces a hard-wired etoposide sensitivity that can
6, 24, and 96 hours following removal of etoposide, cells were be therapeutically exploited upon relapse following first-line
fixed and stained with antibody detecting g-H2AX. Both che- cisplatin-based chemotherapy.
motherapy-na€ve KPE and cisplatin-experienced KPE cells
stained strongly positive for g-H2AX after 6 hours of etoposide
exposure (Fig. 5B). While chemotherapy-na€ve KPE cells were Discussion
largely devoid of nuclear g-H2AX foci 24 and 96 hours follow- ERCC1 deficiency in humans is rarely recognized and has been
ing etoposide exposure (levels similar to KPE mock), cisplatin- implicated in severe developmental and neurodegenerative dis-
experienced KPE post-cisplatin cells remained g-H2AX positive orders emphasizing the importance of ERCC1 that extends
even 96 hours after the initial etoposide-induced insult. These beyond NER (31). In line with this, constitutive Ercc1 knockout
observations suggest that in vivo cisplatin exposure leads to a mice show growth retardation and die several weeks after birth
preserved impairment of the capacity to repair etoposide- due to severe liver damage and hepatocyte polyploidy (27, 32).
induced DNA double-strand breaks in cisplatin-relapsed KPE These findings would suggest the importance of Ercc1 in the
post-cisplatin cells. development, at least in certain tissues. However, the role of
To validate the enhanced etoposide sensitivity that we ERCC1 in the tumor initiation and progression has been poorly
observed specifically in cisplatin-experienced KPE cells, we next investigated. By employing a murine model resembling KRAS-
performed a series of in vivo experiments. First, we transplanted driven human lung adenocarcinoma, we aimed to investigate
a control chemotherapy-na€ve KPE cell line (n ¼ 1) and cis- whether lung cells that have undergone oncogenic transformation
platin-experienced KPE post-cisplatin cells (n ¼ 5 distinct can tolerate loss of Ercc1. Our experiments show that complete
cell lines) subcutaneously into syngeneic KPE recipient animals Ercc1 loss is only tolerated, when coupled with Tp53 deficiency
(n ¼ 5) that had not been exposed to Ad-CMV-Cre and allowed and that this combined loss induces a more aggressive tumor
tumors to form (Fig. 6A). Tumor onset was monitored by phenotype, leading to reduced overall survival of the affected
palpation, and tumor volumes were quantified using mCT, prior animals. This finding corresponds to earlier studies showing
to initiation of etoposide treatment (20 mg/kg, 3 consecutive that the genotoxic lesions, which are otherwise repaired through
days per week, for 2 weeks, i.p.). Tumors that formed from an Ercc1-dependent mechanism, accumulate in liver and kidneys
chemotherapy-na€ve KPE cells were entirely resistant and con- of Ercc1-deficient mice (33). These lesions trigger the activation
tinued to grow under etoposide exposure (Fig. 6B and C and of p53, resulting in early cell-cycle arrest and cellular senes-
Supplementary Fig. S5). In sharp contrast, tumors that formed cence (33). Therefore, our results suggest that Kras-mutant and
from cisplatin-experienced KPE post-cisplatin cells were exqui- Ercc1-deficient murine lung adenocarcinoma might escape
sitely sensitive to etoposide and shrunk under therapy (Fig. 6B these tumor-suppressive mechanisms in the absence of Tp53.
and C and Supplementary Fig. S5). These allograft tumors thus Furthermore, the DNA repair deficiency associated with Ercc1
fully recapitulated our in vitro observations. loss might lead to a mutator phenotype that promotes the
We next aimed to validate the enhanced etoposide sensitivity acquisition of additional mutations that ultimately cumulate in
of KPE tumors that had relapsed following cisplatin treatment a more aggressive phenotype of the resulting tumors.
in an autochthonous mouse model. For this purpose, we Acquired resistance to platinum-based chemotherapy is fre-
generated KP (n ¼ 7) and KPE (n ¼ 10) animals, induced quently observed in human NSCLC, and only a small fraction
lung adenocarcinomas through Ad-CMV-Cre inhalation, and of patients that were initially diagnosed with advanced
administered three cycles of cisplatin (7.5 mg/kg, once a week stage disease survive 5 years beyond diagnosis (34). The
for 3 weeks, i.p.), once mCT-confirmed tumors had formed implication of the role of ERCC1 expression as a predictor
(Fig. 6D and E). Upon mCT-confirmed relapse, animals for a cisplatin sensitivity in human lung adenocarcinoma is
received two courses of etoposide (20 mg/kg, 2 consecutive still not clear due to conflicting clinical data, and thus far to our
days per week, for 2 weeks, i.p.). While this etoposide exposure knowledge, no in vivo lung adenocarcinoma study has
did not lead to a significant gain in overall survival in KP addressed this problem thoroughly. Moreover, human lung
animals (P ¼ 0.154), KPE mice derived a substantial median adenocarcinomas harboring inactivating TP53 mutations are
survival gain (24 days) and displayed a significantly (P ¼ frequent (35) and are present in approximately half of the
0.0026) increased overall survival in response to sequen- KRAS-mutated lung adenocarcinoma tumors (36, 19). How-
tial cisplatin/etoposide treatment, compared with cisplatin ever, their correlation with resistance to platinum-based che-
treatment alone (Fig. 6E). Together, these data indicate that motherapy was not always straightforward (37–42). Here, we
Figure 6.
Cisplatin-relapsed tumors are sensitive to etoposide (Eto) in allograft and in autochthonous model. A, schematic representation of allograft experiment.
Control KPE cell line (n ¼ 1) was injected into the upper back of each mouse (n ¼ 5). KPE post-cisplatin (cis) cell line (n ¼ 5) was injected into the
lower back of a mouse (n ¼ 5). B, mCT-based monitoring of KPE and KPE post-cisplatin tumor response in allograft mice. Representation of resistant
KPE tumor and sensitive KPE post-cisplatin tumor after exposure to etoposide (mouse 1 shown). C, top, quantification of response of KPE and KPE
post-cisplatin tumors to etoposide based on mCT imaging; bottom, macroscopic images of corresponding tumors (KPE tumors, left; KPE post-cisplatin
tumors, right). D, schematic representation of autochthonous model treatment schedule. E, left, survival comparison of chemo-na€ve KP mice (black
line), cisplatin-only treated KP mice (red line), and KP mice treated with cisplatin and after relapse with etoposide (purple line); right, survival comparison
of chemo-na€ve KPE mice (black line), cisplatin-only treated KPE mice (red line), and KPE mice treated with cisplatin and after relapse with etoposide
(purple line); bottom, mCT-based confirmation of tumor presence before cisplatin and before etoposide treatment in KP mice (bottom left) and in KPE
mice (bottom right). P values were calculated using log-rank test.
www.aacrjournals.org Mol Cancer Res; 14(11) November 2016 1121
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.Published OnlineFirst August 11, 2016; DOI: 10.1158/1541-7786.MCR-16-0094
Jokic et al.
showed that cisplatin treatment specifically sensitizes aggres- tion therapy following tumor resection, there was no significant
sive Ercc1-deficient Kras-mutant and p53-deficient adenocarci- difference in overall survival, depending on the combination
noma, while their Ercc1-proficient counterparts remain resis- partner (etoposide, vinblastine, and vinorelbine; ref. 20). These
tant, indicating that Ercc1 is a potent predictor of the outcome data may suggest that the effects that we observed may indeed
to the cisplatin-based chemotherapy in lung adenocarcinoma, be selected by prior cisplatin treatment. Future experiments
in vivo. These findings were further supported in vitro where we should address this question.
showed that cisplatin-relapsed tumor cells expressed Ercc1 Altogether, our murine model faithfully resembling human
through retained Ercc1 allele. In vitro deletion of this allele lung adenocarcinoma with differential ERCC1 expression pro-
led to Ercc1 loss and resensitization against cisplatin. Our vides the basis for the investigation of the role of ERCC1 in the
results are in line with studies performed on human tumor prediction to cisplatin sensitivity. We conclude that acquired
cell lines, where cisplatin sensitivity was associated with low cisplatin resistance commonly observed in lung adenocarcino-
total ERCC1 expression (43, 44), although the correlation to ma patients might be challenged by employing etoposide as a
TP53 status is unclear. second line of chemotherapy to patients with low functional
KRAS-mutant lung adenocarcinomas, which account for ERCC1 and TP53-mutated lung adenocarcinoma once cisplat-
approximately 20% of all lung adenocarcinomas, are difficult in-based chemotherapy fails.
to treat, and targeted therapy has been proven to be clinically
challenging (45). Chemotherapy regimens for metastasized
Disclosure of Potential Conflicts of Interest
NSCLC are largely based on cisplatin, usually applied in the
No potential conflicts of interest were disclosed.
combination with another compound. However, chemothera-
py resistance almost inevitably occurs in these patients. Here,
we aimed to explore the possibility of sequential instead of Authors' Contributions
combined chemotherapy and therefore target relapsed murine Conception and design: M. Jokic, I. Vlasic, M. Rinneburger, J. Wolf,
lung adenocarcinomas that emerge after cisplatin treatment. B. Schumacher, H.C. Reinhardt
Development of methodology: M. Jokic, I. Vlasic, M. Rinneburger,
First, we exposed relapsed tumor cell lines to three mechanis- N. Kl€umper, A. Riabinska, D. Welcker, M. Nowak, J. Thomale, T. Persigehl,
tically different DNA-damaging agents in vitro, and we observed D. Maintz, S. Perner
that these tumor cells show pronounced sensitivity to etopo- Acquisition of data (provided animals, acquired and managed patients,
side by a mechanism not dependent on the Ercc1 status. This provided facilities, etc.): M. Jokic, I. Vlasic, M. Rinneburger, N. Kl€
umper,
sensitivity was further corroborated by their inability to effi- J. Spiro, C. Fritz, A. Schmitt, A. Riabinska, S. Michels, M.D. Akyuz, M. Nowak,
ciently clear etoposide-induced double-strand breaks. Etopo- M. Erkel, J. Wolf, R. B€ uttner, J. Thomale, T. Persigehl, S. Perner
Analysis and interpretation of data (e.g., statistical analysis, biostatistics,
side is occasionally used for the treatment of NSCLC as an computational analysis): M. Jokic, I. Vlasic, M. Rinneburger, J. Spiro,
additive drug to cisplatin (46), but not as a main drug choice A. Offermann, C. K€ umpers, M. Wittersheim, L. Ozretic, M. Erkel, J. Wolf,
once the cisplatin chemotherapy fails. Therefore, we next aimed R. B€uttner, J. Thomale, T. Persigehl, S. Perner, H.C. Reinhardt
to validate our observations in vivo by creating an in vivo Writing, review, and/or revision of the manuscript: M. Jokic, I. Vlasic,
syngeneic model, in which we showed sensitization of trans- M. Rinneburger, N. Kl€ umper, J. Spiro, M. Wittersheim, J. Wolf, R. B€ uttner,
planted cisplatin-relapsed cells to etoposide exposure. In line T. Persigehl, D. Maintz, S. Perner, H.C. Reinhardt
Administrative, technical, or material support (i.e., reporting or organiz-
with this, we were able to target cisplatin-relapsed tumors in an ing data, constructing databases): M. Rinneburger, A. Florin, T. Persigehl,
autochthonous in vivo lung adenocarcinoma model where the D. Maintz, S. Perner, H.C. Reinhardt
survival of KPE mice was significantly prolonged after etopo- Study supervision: B. Schumacher, H.C. Reinhardt
side treatment as a second line of therapy when compared with Other (IHC, HE staining, etc.): W. Vogel
KPE mice exposed only to a single line of cisplatin chemother-
apy. Conversely, KP mice that developed initially cisplatin-
Grant Support
resistant tumors did not experience tumor sensitivity to sub- This work was supported by the Volkswagenstiftung (Lichtenberg Pro-
sequent double-strand break insult. These findings may indi- gramA107062, to H.C. Reinhardt), the Deutsche Forschungsgemeinschaft
cate a requirement of initial Ercc1-dependent cisplatin sensi- (RE 2246/7-1, RE 2246/2-1 to H.C. Reinhardt), the Bundesministerium f€ ur
tivity for the occurrence of subsequent sensitivity to etoposide Bildung und Forschung (BMBF 01ZX1303A, to H.C. Reinhardt), the Boeh-
that is Ercc1 independent. However, we note that we did not ringer-Ingelheim Stiftung (Exploration Grants-Program, to H.C. Reinhardt),
the Helmholtz-Gemeinschaft (PCCC, to H.C. Reinhardt), the Else Kr€ oner-
directly examine the effect of combined cisplatin/etoposide
Fresenius Stiftung (EKFS-2014-A06, to H.C. Reinhardt), and the Deutsche
treatment in our models. Thus, we cannot formally exclude Krebshilfe (222521, to H.C. Reinhardt).
the possibility that a sequential approach is preferable to The costs of publication of this article were defrayed in part by the payment of
concurrent cisplatin/etoposide regimens. In fact, the interaction page charges. This article must therefore be hereby marked advertisement in
between cisplatin-induced DNA damage and etoposide sensi- accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
tivity may also be immediately synergistic rather than a trait
that is selected upon cisplatin exposure. However, at least in Received March 18, 2016; revised June 18, 2016; accepted July 6, 2016;
NSCLC patients receiving adjuvant cisplatin-based combina- published OnlineFirst August 11, 2016.
References
1. Bowden NA. Nucleotide excision repair: why is it not used to predict 3. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009;
response to platinum-based chemotherapy? Cancer Lett 2014;346:163–71. 361:1475–85.
2. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. 4. Friedberg EC. How nucleotide excision repair protects against cancer.
Nature 2001;411:366–74. Nat Rev Cancer 2001;1:22–33.
1122 Mol Cancer Res; 14(11) November 2016 Molecular Cancer Research
Downloaded from mcr.aacrjournals.org on February 23, 2021. © 2016 American Association for Cancer Research.You can also read

















































