Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Methamphetamine induces neuronal apoptosis via
cross-talks between endoplasmic reticulum and
mitochondria-dependent death cascades
SUBRAMANIAM JAYANTHI, XIAOLIN DENG, PIERRE-ANTOINE H. NOAILLES,
BRUCE LADENHEIM, AND JEAN LUD CADET1
Molecular Neuropsychiatry Branch, National Institute on Drug Abuse-Intramural Research Program,
National Institutes of Health, DHHS, Baltimore, Maryland, USA
ABSTRACT Methamphetamine (METH) is an illicit nondopaminergic regions of the brains of METH abus-
drug that causes neurodegenerative effects in humans. ers have been observed in various imaging studies (2, 3,
In rodents, METH induces apoptosis of striatal glu- 7–9). Some of the results of the imaging studies are
tamic acid decarboxylase (GAD) -containing neurons. supported by a postmortem study that showed marked
This paper provides evidence that METH-induced cell decreases in the levels of striatal dopamine, dopamine
death occurs consequent to interactions of ER stress transporters, and serotonin in the brains of METH-
and mitochondrial death pathways. Specifically, injec- addicted patients (1).
tions of METH are followed by an almost immediate In animals, METH causes substantial brain damage
activation of proteases calpain and caspase-12, events characterized by decreases in striatal dopamine and
consistent with drug-induced ER stress. Involvement of serotonin levels, decreased tyrosine hydroxylase activ-
ER stress was further supported by observations of ity, and loss of dopamine transporters (see reviews, refs
increases in the expression of GRP78/BiP and CHOP. 10 –12). Although studied less, incontrovertible evi-
Participation of the mitochondrial pathway was demon- dence has long existed that METH can cause cell death
strated by the transition of AIF, smac/DIABLO, and in the brain (13–16). Zalis et al. (13) injected dogs with
cytochrome c from mitochondrial into cytoplasmic frac- 2.5–5 mg/kg i.v. or 10 mg/kg p.o and reported the
tions. These changes occur before the apoptosome- occurrence of neuronal cell degeneration in the cere-
associated pro-caspase-9 cleavage. Effector caspases-3 bral cortex, basal ganglia, brainstem, and cerebellum of
and -6, but not -7, were cleaved with the initial time of these animals. Ellinwood and collaborators (14) had
caspase-3 activation occurring before caspase 9 cleav- reported chromatolysis in various regions of the brains
age; this suggests possible earlier cleavage of caspase-3 of cats treated with METH (2 wk of chronic intoxica-
by caspase-12. These events preceded proteolysis of the tion with increasing doses of METH starting at 15
caspase substrates DFF-45, lamin A, and PARP in mg/kg and reaching 28 mg·kg–1·day–1). Subsequently,
nuclear fractions. These findings indicate that METH Ellison and Switzer (16) demonstrated that METH (6
causes neuronal apoptosis in part via cross-talks be- mg/kg given every 2 h for 8 h) caused pronounced
tween ER- and mitochondria-generated processes, degeneration in the striatum and cerebellum of rats
which cause activation of both caspase-dependent and killed 36 h after drug administration. More recently,
-independent pathways.OJayanthi, S., Deng, X., No- Schmued and Bowyer (17) reported that neuronal cell
ailles, P.-A. H., Ladenheim, B., Cadet, J. L. Metham- death was apparent in the hippocampal remnants of
phetamine induces neuronal apoptosis via cross-talks rats within 5 days after injections of METH (15 mg/
between endoplasmic reticulum and mitochondria-de- kg ⫻ 4 administered at 2 h intervals). Eisch et al. (18,
pendent death cascades. FASEB J. 18, 238 –251 (2004) 19) had shown that METH (4 mg/kg ⫻ 4 given at 2 h
intervals) caused cell death that peaked at ⬃3 days after
Key Words: apoptosis-inducing factor 䡠 CHOP transcription drug administration. In a series of recent papers, we
factor 䡠 BiP/GRP78 䡠 DNA fragmentation have demonstrated that METH-induced cell death
showed characteristics of apoptosis (20 –26), findings
that have been replicated by others (27–29).
Because of its popularity and the neuropsychiatric Apoptosis is a highly regulated death process that
and neurodegenerative effects associated with its abuse occurs during development and is thought to be dys-
(1–3), it is essential to study the molecular and cellular
mechanisms of methamphetamine (METH) toxicity. 1
For example, acute METH intoxication can result in Correspondence: Molecular Neuropsychiatry Branch, Na-
tional Institute on Drug Abuse, Intramural Research Pro-
belligerent and psychotic behavior (4) as well as multi- gram, National Institute of Health, DHHS, 5500 Nathan
ple organ failure (5, 6) in humans. Myocardial infarc- Shock Dr., Baltimore, MD 21224, USA. E-mail: jcadet@intra.
tion, stroke, cerebral hemorrhages, and death have also nida.nih.gov
been reported (6). Dysfunctions in dopaminergic and doi: 10.1096/fj.03-0295com
238 0892-6638/04/0018-0238 © FASEB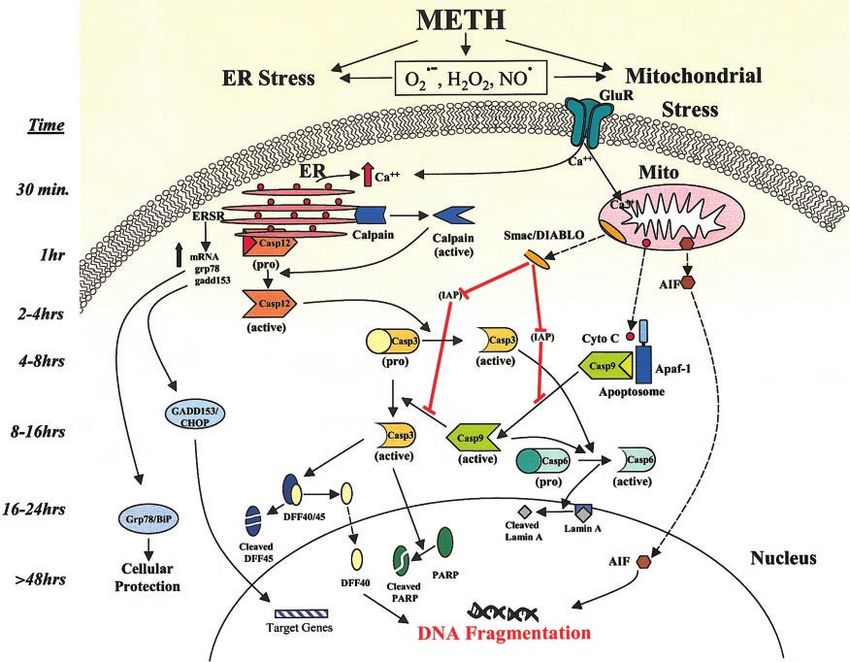
regulated in several neurodegenerative disorders in- MATERIALS AND METHODS
cluding Parkinson’s disease, Huntington’s chorea,
amyotrophic lateral sclerosis, and Alzheimer’s disease Animals and drug treatment
(30). Neuronal apoptosis can be induced by stimula-
tion of plasma membrane death receptors and by Male CD-1 mice (Charles River, Raleigh, NC, USA) weighing
perturbation of intracellular homeostasis via activation 30 –35 g were used. The mice tested were 9 to 11 wk old.
of specific organelle-mediated death cascades (31). For Animals were housed two or three per cage with food and
water available ad libitum. Temperature (23⫾1°C) and hu-
example, damage to mitochondria and the endoplas- midity (53⫾15%) were controlled. Mice received a single
mic reticulum (ER) (32, 33) can cause release of intraperitoneal dose of 40.0 mg/kg METH or saline as
cytochrome c (cyto c) and subsequent activation of described previously by our laboratory (25, 26, 39, 42) and by
caspases that are major mediators of apoptotic signals. other investigators (43– 47). Animals showed no evidence of
These enzymes are broadly divided into two groups: seizures and all mice survived this dose regimen throughout
the duration of the study (7 days at most), during which they
initiator caspases whose main function is to activate
were killed at various times after drug treatment. As suggested
downstream caspases and effector/executor caspases by some investigators (45– 47), this approach might help to
responsible for dismantling cellular proteins. Activa- more specifically define the biochemical and molecular bases
tion of effector caspases leads to the proteolysis of of METH-induced neurotoxicity. The use of large doses of
several target proteins, including poly (ADP-ribose) METH constitutes an attempt to mimic the large doses taken
polymerase (PARP), lamins and DNA fragmentation by human METH abusers, which can amount to several grams
taken in a day (4, 48). Because there are significant species
factor 45 kDa subunit (DFF-45) (34). The importance differences in METH elimination half-lives (49) and metabo-
of proteolytic cleavage to the ensuing morphological lism (50), the often used binging patterns introduced in 1988
and molecular changes associated with apoptotic phe- by Sonsalla et al. (51) are only approximations of what is
nomena is being actively investigated. DFF-40/CAD, a actually encountered in clinical situations. This issue has been
subunit of the heterodimeric DFF, has been shown to discussed extensively in a recent review of METH neurotox-
mediate genomic DNA degradation during apoptosis icity (12).
Brain tissues were processed for use in immunohistochem-
(35) whereas PARP cleavage might cause dysfunctions istry, Western blot, and real-time RT-PCR as described below.
in DNA repair mechanisms (36). Cleavage of lamins All animal use procedures were according to the NIH Guide
may interfere with the integrity of the nuclear envelope for the Care and Use of Laboratory Animals and were
(37). Apoptosis can occur via caspase-independent approved by the local NIDA Animal Care Committee.
mechanisms after the mitochondrial release of the
apoptosis-inducing factor (AIF) (38). Immunocytochemistry and TUNEL histochemistry
Our efforts to characterize mechanisms involved in
METH-induced neuronal apoptosis have provided Procedures were performed according to previously reported
some novel clues on the cellular and molecular effects methods (24, 25). At designated times after METH adminis-
tration, the animals were perfused transcardially with 4%
of this illicit neurotoxin (12). We have found that paraformaldehyde under deep pentobarbital anesthesia. The
METH-induced cell death is associated with the activa- brain tissues were subsequently removed and postfixed over-
tion of the SAPK/JNK (stress-activated protein kinase/ night in the same fixative. On the next day, 30 m coronal
c-Jun amino-terminal kinase) pathway (39). Because sections were cut using a cryostat. Sections were mounted
the majority of TUNEL-positive cells express phosphor- onto Superfrost/Plus microscopy slides (Fisher, PA, USA)
ylated c-Jun and c-jun knockout mice show partial and stored at –20°C.
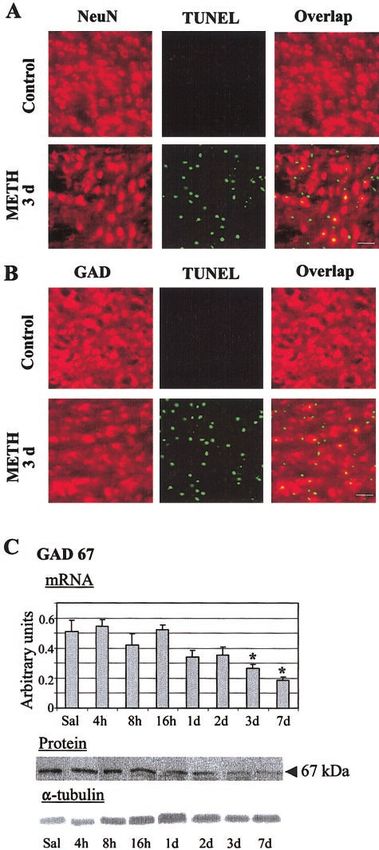
METH-induced striatal apoptotic cells and their content
protection against METH-mediated neuronal apoptosis were detected by using dual antigen staining of sections with
(23), it appears that the SAPK pathway plays a role in TUNEL histochemistry and neuronal nuclei (NeuN) or GAD
causing METH-induced cell death. These findings are immunohistochemistry as described before (23). Slide-
consistent with a wealth of information that implicates mounted sections from saline- or 3 day METH-treated ani-
c-Jun as a proapoptotic agent (40). We and others have mals were incubated with anti-NeuN or anti-GAD primary
antibody (both from Chemicon International Inc., Temecula,
found that METH-induced neurodegeneration is de-
CA, USA). They were subsequently incubated with biotinyl-
pendent on mitochondrial mechanisms in vivo (26) ated secondary antibody and Texas red-avidin-DCS (both
and in vitro (22, 27). Specifically, administration of from Vector Laboratories, Burlingame, CA, USA). After
METH to mice causes increases in pro-death (BAX, NeuN or GAD immunostaining, the sections were processed
BAD, and BID) but decreases in antiapoptotic Bcl-2- by TUNEL histochemical staining as reported earlier by this
related proteins (Bcl-2 and Bcl-XL) (26). These pro- laboratory (23, 25). The immunostained sections were rinsed
in 0.5% Triton X-100 in 0.01M phosphate-buffered saline for
teins are known to be involved in either activating or 20 min at 80°C to increase permeabilization of the cells. To
inhibiting the mitochondria-dependent cell death label damaged nuclei, 50 L of the TUNEL reaction mixture
pathway in the mammalian brain (41). We report here (Roche Diagnostics Corporation, IN, USA) were added onto
that, in addition to the mitochondrial death pathway, each sample in a humidified chamber, followed by a 60 min
METH can exert its neurodegenerative effects by caus- incubation at 37°C. Procedures for negative controls were
carried out as described in the manufacturer’s manual and
ing ER stress in the striatal neurons of mice treated with
consisted of not adding the label solution (terminal deoxy-
toxic doses of the drug and that the mitochondria and nucleotidyl transferase) to the TUNEL reaction mixture. No
ER pathways are activated within the same cells before TUNEL-positive cells were observed in the negative controls.
experiencing their ultimate demise. To detect the activation of caspase-3 in striatal neurons, we
METH, ER STRESS, AND APOPTOSIS 239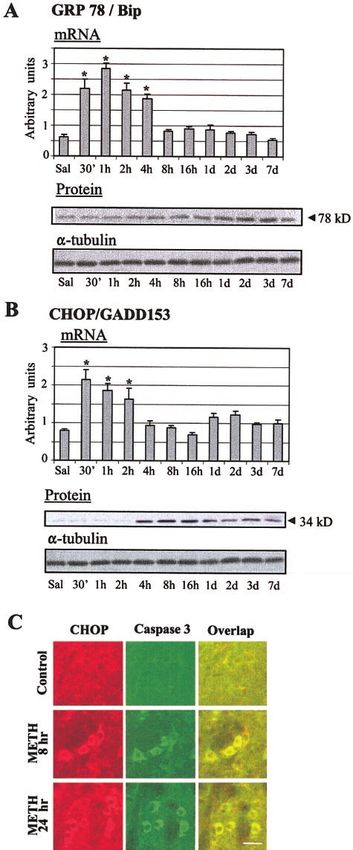
used costaining with antibodies against NeuN and activated caspase-3. Briefly, slide-mounted sections from saline- and METH-treated animals (8 and 24 h) were incubated with monoclonal anti-NeuN (Chemicon) and polyclonal anti- cleaved caspase-3 (Cell Signaling, Beverly, MA, USA) primary antibodies. This was followed by incubation with Texas red- conjugated anti-mouse antibody and biotinylated anti-rabbit antibody, then FITC-avidin-DCS immersion to detect the polyclonal antibody (caspase-3). Dual antigen immunostaining of CHOP/GADD153, a marker for ER stress, and cleaved caspase-3 antibodies was performed as described above. After staining, images were processed by using a Carl Zeiss Laser Scanning Confocal System with Axiovert 135 inverted microscopy. Excitation and emission wavelengths were selected according to the sug- gested index. To estimate the percentages of cell death of neuronal or GABAergic origin, striata of mice (n⫽10) were randomly viewed under 20⫻ objective lens. Total neurons dying (green) ⫽ a, total GAD neurons that are dying (yel- low) ⫽ b, and total number of GABA cells (red) ⫽ c were counted (see Fig. 1B for representative pictures). The per- centage of GABA cells that die was calculated using the equation, 100 ⫻ b/c. Western blot analysis Immunoblot analysis was carried out with the striatum dis- sected from six mice of METH- and saline-treated wild-type mice. Samples from six mice were pooled to form one group. Experiments were performed from three different groups for quantitation. The pooled mouse striatum was homogenized in buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 0.5% NP-40, 0.25% SDS, 5 g/mL leupeptin, and 5 g/mL aprotinin. Homogenates were cen- trifuged at 750 g for 10 min at 4°C and the precipitates that belong to nuclear fractions were resuspended in the lysis buffer. The supernatant fraction was subsequently centri- fuged at 10,000 g for 15 min at 4°C. The resulting pellet belongs to mitochondrial fraction and was resuspended in the lysis buffer. Supernatants were further centrifuged at 100,000 g for 1 h at 4°C. The pellet was discarded and the remaining supernatant is the cytosolic fraction. After deter- mining the protein concentration of lysates with a Bio-Rad assay system (Bio-Rad, San Francisco, CA, USA), the lysates were denatured with sample buffer (62.5 mM Tris-HCl, 10% glycerol, 2% SDS, 0.1% bromophenol blue, and 50 mM DTT) at 100°C for 5 min and subjected to SDS-PAGE. Proteins were electrophoretically transferred to Hybond-PTM membrane (Amersham Pharmacia Biotech. Piscataway, NJ, USA). Mem- brane blocking, primary and secondary antibody incubations, and chemiluminescence reactions were carried out according to the protocol described by individual antibody suppliers. Antibodies included monoclonal for Apaf-1, cyto c (BD Figure 1. METH causes apoptosis in striatal GABAergic neu- PharMingen, San Diego, CA, USA), rabbit polyclonal caspase rons. DNA fragmentation was analyzed using the TUNEL assay 3 for Western blot, DFF-40 and DFF-45 (BD PharMingen), as described in the method section. METH caused significant rabbit polyclonal for Smac/DIABLO, caspases 9, 6, cleaved increases in TUNEL-positive cells (green) in the mouse striatum caspase 3 for immunohistochemistry, cleaved PARP and (A) . Double labeling experiments showed that some TUNEL- cleaved lamin A (Cell Signaling), rabbit polyclonal GAD-67, positive cells were NeuN-positive (yellow in the overlap). Some mouse monoclonal CHOP/GADD153 (Santa Cruz Biotech- TUNEL-positive neurons (green) were GAD-positive (red), as nology, Inc., Santa Cruz, CA, USA), rabbit polyclonal calpain, shown in yellow in the overlap (B). Scale bars ⫽ 50 m. caspase 12 and AIF (Biovision Research Products, Mountain Quantitative PCR analyses revealed very early METH-induced View, CA, USA), and rabbit polyclonal BiP/GRP78 (Stressgen decreases in GAD67 mRNA levels (C). These were followed by Biotechnologies, Victoria, Canada). To confirm equal protein decreases in GAD67 protein levels 3–7 days after METH injec- loading, blots were reprobed with ␣-tubulin antibody (1:2000; tion. mRNA levels were measured as fluorescent intensities Sigma, 2 h at RT). Signal intensity was measured using using quantitative real-time PCR and normalized to light chain densitometric analysis (IS-1000 Digital Imaging System, Alpha clathrin mRNA levels. Values for the quantitative PCR represent InnoTech Corp., San Leandro, CA, USA) and quantitated means ⫾ se (6 –10 animals/time point). Statistical analysis was using FluorChem version 2.0 software (AlphaEaseFC analysis done by ANOVA, followed by Fisher’s protected least square software) (26). difference (PLSD). *P ⬍ 0.01 vs. saline control group. 240 Vol. 18 February 2004 The FASEB Journal JAYANTHI ET AL.
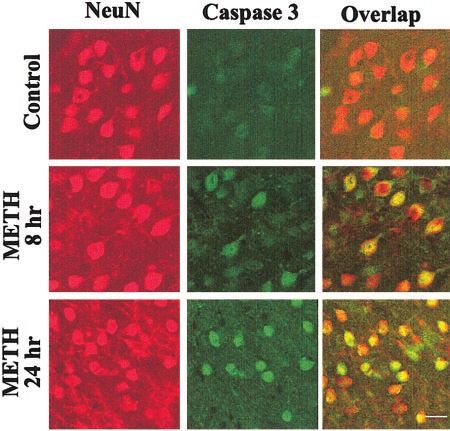
TABLE 1.
Gene Upstream primer Downstream primer
Grp78/Bip CAGAGACCCTTACTCG GTTTATGCCACGGGAT
chop/gadd153 GGAAGTGCATCTTCATACACCACC TGACTGGAATCTGGAGAGCGAGGGC
GAD67 AAACTCAGCGGCATAG CCCTGTATCGTAGGAGAC
Reverse transcription (RT) -PCR and detection of mRNA majority of the TUNEL-positive cells were NeuN posi-
expression using LightCycler technique tive (Fig. 1A). The striatum is a complex structure of
several subtypes of neurons, 95% of which are GABAer-
Real-time PCR to detect mRNA expression for GAD67 and the gic neurons that project to the outside of that structure
ER stress-related genes chop and Grp78/BiP was done using the (53). We thus wanted to ascertain whether the affected
LightCycler thermal cycler system (Roche Diagnostics) essen-
tially as described by us (52). For real-time PCR, unpooled neurons were GABAergic by running double-label ex-
total RNA (1 g) from 6 to 10 CD1 mice per group was periments using an antibody against glutamic acid
reverse-transcribed with oligo dT primer using Advantage RT decarboxylase (GAD), the enzyme involved in the
for PCR kit (BD Biosciences Clontech Laboratories, Palo Alto, GABA synthetic pathway (54), in conjunction with
CA, USA). PCR experiments were then performed using light TUNEL histochemistry. Figure 1B shows that many of
cycler technology and the LightCycler FastStart DNA Master the dying cells were GAD positive, indicating that
SYBR Green I kit (Roche Molecular Biochemicals). HPLC-
METH causes death of striatal GABAergic neurons. Cell
purified and gene-specific primers corresponding to PCR
targets were obtained from the Synthesis and Sequencing counting established that 17% of the GAD-positive cells
Facility of Johns Hopkins University (Baltimore, MD, USA) were TUNEL-positive. To test the effects of METH on
(Table 1). GAD expression, we performed quantitative RT-PCR
For PCR, 2 L of template was placed into 18 L reaction and Western blot analysis using primers and antibody
volume containing 0.5 L of each primer, 2.4 L 25 mM specific for GAD67. Figure 1C shows that METH caused
MgCl2 , 2 L FastStart DNA Master SYBR Green I, and 13.6 L a marked reduction in GAD67 transcript and protein
dH2O. Nucleotides, Taq DNA polymerase, and buffer were
included in the FastStart DNA Master SYBR Green I mix
levels measured several days after administration of the
(Roche Diagnostics). A typical protocol took ⬃60 min to drug.
complete and included denaturation at 95°C with a preincu-
bation time of 8 min, followed by 45 cycles with 95°C
denaturation for 15 s, 64°C annealing for 5 s, and 72°C
METH induces release of apoptogenic molecules
extension for 15 s. Extension periods varied with the specific from mitochondria
primers, depending on the length of the product. Negative
controls were run concomitantly to confirm the overall spec-
ificity and to verify that no primer/dimer was generated. The
Because release of cyto c from mitochondria into the
relative standard curve was established with serial dilution of cytoplasm has been documented in apoptosis caused by
a cDNA solution with an unknown concentration that corre- several toxic triggers (55), we wanted to know whether
sponds to a mix of five randomly picked samples. To confirm METH injections to mice would cause similar changes
the amplification specificity, the PCR products were subjected in the compartmentalization of cyto c. To test this, we
to a melting curve analysis. Amplification curves were gener- performed a detailed time course assessment of the
ated by the LightCycler Instrument Quantification program concentration of cyto c in cellular subfractions. There
and displayed the fluorescence values vs. cycle number.
Template concentrations using the relative standard curve was only a negligible amount of cyto c in the cytosolic
were given arbitrary values. The mean concentration of fractions obtained from control mice. Administration
clathrin light chain was used as control for input RNA of METH caused a gradual appearance of cyto c in the
because it is considered a stable housekeeping gene. The cytosol of striatal cells, which began at ⬃30 min to 1 h,
mean clathrin concentration was determined once for each peaked at 4 – 8 h, then tapered off 2 days after drug
cDNA sample and used to normalize all other genes tested injection (Fig. 2A). These changes were contrasted by
from the same cDNA sample. The relative change in gene
marked decreases in cyto c in mitochondrial fractions,
expression was recorded as the ratio of normalized data over
saline. One-way ANOVA was followed by Fisher’s PLSD test with its almost total disappearance from the mitochon-
for testing differences between the different time points and dria from 2 to 7 days postdrug. Because cyto c is known
the saline-treated animals. All analyses were done using the to bind to dATP and apoptotic protease-activating
program Statview 4.02 (SAS Institute, Cary, NC, USA). The factor-1 (Apaf-1) in order to form the apoptosome that
null hypothesis was rejected at P ⬍ 0.05. activates caspase-9 (56), we determined whether METH
would affect Apaf-1 and caspase-9 protein levels in the
striatum. As is observed in Fig. 3A, A’, Apaf-1 showed
RESULTS initial increases at ⬃4 h after METH administration,
with maximal changes occurring during the 8 –24 h
METH kills striatal GABAergic neurons interval after the drug injection. These were accompa-
nied by initial caspase-9 cleavage at ⬃4 h, with the
Figure 1A shows the effects of METH on NeuN-positive cleaved protein reaching a peak concentration between
striatal cells after 3 days of METH administration. The 8 –24 h postdrug (Fig. 3A, A’).
METH, ER STRESS, AND APOPTOSIS 241Figure 2. METH administration induced the release of apoptogenic proteins from the mitochondria. Nuclear, mitochondrial, and cytosolic fractions were separated by ultra-centrifugation and queried for cyto c (A), Smac/DIABLO (B), and AIF (C). The fractions were obtained from pooled stria- tal samples of 6 animals for each time point. Quantitative data are given in panels A’, B’, C’ (n⫽3/time point, where ‘n’ represents one group of pooled striatal samples) for each protein, respectively. Membranes were reprobed with ␣-tubulin antibody to con- firm equal protein loading in each lane. The immunoblots were visualized using ECL detection agents from Amersham. Besides cyto c, several other apoptogenic substances after treatment. In addition, METH caused AIF, which can be released from the mitochondria during the has been reported to cause caspase-independent apo- apoptotic process. These include Smac/DIABLO (57) ptosis (59), to be released from the mitochondria (30 and AIF (58). We measured these two substances in min–1 h) and to be translocated to the nucleus (8 –72 order to test the possibility that METH administration h) via transit through the cytosol (Fig. 2C, C’). These can release proteins that might be involved in caspase- findings are consistent with a recent paper in which independent effects in the brain. Figure 2B, B’ shows similar AIF transit was observed during apoptosis that METH induced transit of Smac/DIABLO from the caused by N-methyl-d-aspartate (NMDA) glutamate re- mitochondria to the cytosol as early as 30 min to 1 h ceptor-mediated toxicity in cortical neurons (60). Figure 3. METH induces activation of the caspase-dependent mitochondrial apopto- tic pathway. Administration of METH was associated with time-dependent increases in Apaf-1 protein levels in the cytosol of striatal neurons, which gradually returned to normal levels ⬃7 days after the METH injection (A, A’). The initiator, caspase-9, was activated via its cleavage, as demon- strated by loss in its pro-enzyme form and increases in its cleaved product, which peaked at ⬃8 h to 1 day after drug treat- ment (A, A’). METH injections caused cleavage of caspase–3 in mice striata (B, B’). Caspase-6 cleavage occurred in the striata of METH-treated mice (C, C’). The quantitative data represent means ⫾ se (n⫽3). 242 Vol. 18 February 2004 The FASEB Journal JAYANTHI ET AL.
include caspase-12, calpain (65), GRP78/BiP (glucose-
regulated protein/immunoglobulin heavy chain bind-
ing protein) (66), and CHOP/GADD153 (C/EBP ho-
mology protein/growth arrest and DNA damage) (67).
As shown in Fig. 5A, METH administration caused
marked changes in the pattern of caspase-12 expres-
sion. Specifically, 1 h after METH injection there was
an initial cytosolic appearance of the smaller cleaved 40
kDa fragment that peaked at 3– 4 h, then gradually
decreased toward control levels 7 days after drug treat-
ment (Fig. 5A’). Because caspase-12 is cleaved by cal-
pain during ER stress (65), we assessed the effects of
METH on calpain expression by using an antibody that
detects the original and activated forms of this pro-
tease. We found that METH administration was associ-
ated with activation of calpain as early as 30 min after
drug injection (Fig. 5B, B’). These changes lasted
throughout the time course of the experiments (7
days), suggesting the possible occurrence of METH-
Figure 4. METH caused cleavage of caspase-3 (green) in induced prolonged changes in calcium homeostasis
striatal neurons (red). The overlap of the two stains is shown since calpain is activated by increased intracellular
as yellow-orange stained. Scale bar ⫽ 30 m. calcium (68).
The ER stress-generated UPR is characterized by
Neuronal apoptosis is accompanied by cleavage of increased transcription of ER chaperones, such as
caspases-3 and -6 but not of caspase-7 BiP/GRP78, which are meant to increase the ER capac-
ity to cope (66). To confirm that METH was indeed
Because activation of the mitochondrial death pathway causing ER stress, we decided to measure BiP mRNA
is known to lead to cleavage of terminal caspases and protein expression after drug treatment. Figure 6A
involved in the proteolysis of many proteins (34), we (upper panel) shows that METH indeed caused very
sought to determine whether this was the case with early increases (2- to 2.5-fold) in BiP/Grp78 mRNA at 30
METH treatment. There was indeed cleavage of the min–2 h after its injection. Western blot analysis re-
caspase-3 pro-enzyme (32 kDa), with the cleaved (17 vealed a gradual increase in BiP protein starting at 8 h
kDa) product appearing and increasing from 3 to 48 h and reaching a maximal peak by 2–3 days after the
after drug administration (Fig. 3B, B’). To determine METH injection (Fig. 6A, lower panel).
whether caspase-3 cleavage was occurring in striatal In addition to increasing its coping mechanisms,
neurons, we performed double-label immunostaining organisms can trigger apoptosis by increasing the tran-
experiments using antibodies against active caspase-3 scription of genes that are involved in causing cellular
and NeuN. As shown in Fig. 4, METH-induced changes demise if the ER stress is overwhelming (reviewed in ref
in the activation of caspase-3 occurred in NeuN-positive 69). One such gene is chop/gadd153 (67), which inhibits
cells in the mouse striatum. Figure 3C shows that some (63) but induces other genes (70). We found that
METH administration caused cleavage of pro-caspase-6, METH-treated mice showed rapid increases in chop
beginning at ⬃16 h and almost completely disappear- mRNA that lasted from 30 min to 2 h (Fig. 6B, upper
ing at 7 days post-METH treatment. In contrast, METH panel). Figure 6B (lower panel) shows that METH
caused no changes in the expression of caspase-7 (data treatment caused sustained increases in CHOP protein
not shown). from 4 h for up to 7 days after drug treatment. To
examine whether ER stress was occurring in the same
neurons that showed expression of activated caspase-3,
METH administration causes ER stress in the
we performed double-label experiments with antibod-
mouse striatum
ies against CHOP and active caspase-3. Figure 6C shows
that these two proteins colocalize in all affected cells.
The ER is an important organelle that participates in
cellular homeostasis by regulating calcium signaling
and protein folding (61). However, dysregulation of METH administration causes cleavage of various
intracellular homeostasis (62) and oxidative stress (63) caspase target proteins
can cause ER stress and ER-induced apoptosis (62).
Because METH is known to cause oxidative stress (10, Because activation of terminal caspases leads to the
64), we wanted to know whether ER-mediated events cleavage of several nuclear and cytoplasmic proteins,
might participate in METH-induced cellular demise. we investigated the effects of METH on the status of
We measured a number of substances that have been DFF45, DFF40, PARP, and lamin A, known caspase
reported to participate in ER-induced apoptosis and targets (34). Figure 7A shows the effects of METH on
the unfolded protein response (UPR) (62). These the status of DFF45. In samples from control mice,
METH, ER STRESS, AND APOPTOSIS 243Figure 5. Injection of METH
causes almost immediate activa-
tion of caspase-12 (A, A’) and
calpain (B, B’) in the mouse stri-
atum. Quantitative data repre-
sent means ⫾ se (n⫽3).
there were two protein bands: one of 45 kDa and one of cause cell death in the brains of various animal species
34 kDa. METH injection caused decreases in the cyto- (see review by Davidson et al., ref 73). Results from
solic DFF45 kDa band but increases in the 34 kDa band. several laboratories have now confirmed that METH
There was also the appearance of METH-induced 32 can indeed cause neuronal death in vitro (20, 22,
and 11 kDa bands first observed at 8 h after drug 27–29) and in vivo (17, 18, 24 –26). This is the first
treatment and present almost concurrently (Fig. 7A). demonstration, however, that METH can kill GAD-
The concentration of the carboxyl-terminal 11 kDa positive striatal neurons in vivo.
fragment (p11), essential for activation of the DFF GAD is the rate-limiting enzyme in GABA biosynthe-
activity (71), peaked after 2 days and returned toward sis (54) and has been used to characterize GABAergic
control levels by 7 days (Fig. 7A’). Almost simulta- neurons in various regions of the brain (74). GAD
neously, DFF40, the active component of DFF that exists as two major isoforms, GAD65 and GAD67, which
triggers DNA fragmentation (72), showed increases in are products of two different genes (75). GAD65 is
the cytosolic fractions after drug administration (Fig. membrane-associated whereas GAD67 is cytoplasmic
7A, A’). Another caspase-3 target, PARP, was cleaved to (75); GAD65 is thought to be involved in short-term
its 89 kDa product in nuclear fractions (Fig. 7B). regulation of GAD activity whereas GAD67 is thought to
Because caspase-dependent degradation of lamins is participate in its long-term regulation (76). GAD67 is
thought to facilitate nuclear dissolution during nuclear responsible for the majority of GABA synthesis within
apoptosis (37), we measured its status after METH the brain because lack of GAD65, as observed in GAD65
treatment. We found that METH administration re- knockout mice, does not change brain GABA content
sulted in a time-dependent cleavage of lamin A in (77) whereas significant decreases in GAD activity and
nuclear fractions (Fig. 7C). The 46 and 28 kDa lamin A GABA content are observed in the brains of GAD67
fragments, which are indicative of proteolytic digestion, knockout mice (78). In the present study, we thus focus
were quite visible 8 h after METH administration. on studies of GAD67 and have found significant METH-
These changes in lamin A peaked at ⬃24 h after induced decreases in GAD67 mRNA and protein levels.
METH, then tapered off until 7 days after drug treat- These results are consistent with the double label
ment (Fig. 7C’). The time (24 h after drug) of greatest experiments where we found a substantial number of
changes in lamin A status corresponds to the time of TUNEL-positive cells that are GAD-positive cells. Taken
peaked caspase-6 cleavage (see Fig. 3C). Unexpectedly, together, these observations suggest that many of the
there were no changes in the pattern of lamin B dying/dead cells are indeed GABAergic. Although
expression in spite of our running these experiments these observations are not consistent with those of
with three different antibodies (data not shown). others who had reported that toxic doses of METH did
not affect GABA systems in the brain (79, 80), there are
some differences between these studies and ours.
DISCUSSION Hotchkiss et al. (79) injected 15 mg/kg ⫻ 4 of METH
given 6 h apart for 24 h and measured GAD activity in
Most studies of the mechanisms of METH-induced rats killed from 6 h to 30 days after the last injection. In
neurotoxicity have until recently been limited to inves- another study (80), GABA uptake was measured only
tigating its damaging effects on dopaminergic and 1 h after the 1 day binge of four injections of 10 mg/kg.
serotonergic terminals (11) although, as discussed ear- In contrast, our observations revealed that decreases in
lier, there was substantial evidence that METH can GAD67 did not occur until 3 to 7 days after injection of
244 Vol. 18 February 2004 The FASEB Journal JAYANTHI ET AL.a single large toxic dose of the drug (see Fig. 1); these
observations are consistent with our previous demon-
stration that the number of METH-induced TUNEL-
positive cells peaked 3 days postdrug (24, 25). This
peak is followed by a decrease in the number of
TUNEL-positive cells, suggesting that these cells had
been removed by scavenger cells (81). Thus, the de-
creases in GAD67 protein levels 3–7 days after METH
appear to correspond to the death of striatal GABAer-
gic neurons caused by METH. It is thus likely that the
discrepancies between our results and those of others
(79, 80) are due to the doses of METH, the schedule of
its administration, the time of animal death, and the
different assays used in the studies. In what follows, we
discuss the possible scenarios that might lead to the
demise of these cells.
The mechanisms by which these GABAergic cells die
subsequent to METH might include excitotoxic dam-
age (for a review, see ref 82) since toxic doses of METH
are known to cause glutamate release in the striatum
(83), production of free radicals (10, 64, 84, 85); or
perturbations of DNA repair mechanisms (86), and the
activation of mitochondrial and ER death pathways (see
below). The cytotoxic effects of glutamate are known to
occur through receptor-mediated and receptor-inde-
pendent events (87). Receptor-mediated mechanisms
could involve glutamate-induced NO synthesis with
subsequent production of reactive oxygen and nitrogen
species that could lead to a shift in mitochondrial
membrane potential with subsequent production of
metabolic stress and the occurrence of cell death (82).
Another major player in glutamate-mediated excitotox-
icity leading to neuronal apoptosis involves NMDAR-
induced excessive Ca2⫹ influx, which activates Ca2⫹/
calmodulin-regulated protein phosphatase and causes
the release of cytochrome c from mitochondria, subse-
quent activation of the caspase cascade, and cytoskel-
etal breakdown (88). The idea for the possible involve-
ment of glutamate in METH-induced GABAergic cell
death is supported by the presence of glutamate recep-
tors on these neurons (89, 90). Nevertheless, studies
using specific glutamate receptor blockers need to be
conducted to test this idea further.
Our repeated attempts to further characterize the
molecular and cellular bases of METH-induced apopto-
sis have now revealed that ER and mitochondria-asso-
ciated events are involved in causing the demise of
these GABAergic neurons in a fashion similar to what
occurs in other models of cell death (31, 91). Specifi-
Figure 6. METH caused rapid increases in Grp78/BiP mRNA cally, we have shown that METH injections can pro-
levels (A). Western analyses showed increases in GRP78/BiP mote a shift in the balance of pro-death/anti-death
protein levels that reached a maximum 2–3 days postdrug proteins of the BAX/Bcl-2-related family (26). These
(A). Values represent means ⫾ se (6 –10 animals/time point). changes are known to trigger programmed cell death
Statistical analysis was done by ANOVA, followed by Fisher’s in various models of apoptosis (92). These earlier
protected least square difference (PLSD). *P ⬍ 0.001 vs. with observations had led us to conclude that mitochondria-
saline control group. METH caused increases in chop/gadd153
mRNA levels (B). Changes in protein levels occurred ⬃4 h mediated caspase-dependent events might play impor-
after injection. METH caused increases in CHOP (red) tant roles in METH-induced deleterious effects (22,
protein expression and of cleaved caspase-3 (green) within 26). Nevertheless, several other apoptogenic substances
the same cells (C). Scale bar ⫽ 30 m. can be released from the mitochondria during the
apoptotic process, one of which is AIF, which promotes
METH, ER STRESS, AND APOPTOSIS 245Figure 7. METH induced the cleavage of caspase target proteins. METH causes changes in the pattern of DFF-45 and DFF-40 expression in the mouse striatum (A). Representative Western blots of DFF-45 and DFF-40 proteins in the cytoso- lic fractions in saline-treated control and in METH-treated animals show cleavage of DFF45 with the appearance of three bands at 45, 34, and 11 kDa, respectively. METH- induced PARP cleavage was observed in the nuclear fractions (B). The band at 110 kDa represents intact PARP and the one at 89 kDa is the cleaved product. METH administration caused cleavage of lamin A in the nucleus (C) . Three bands were detected at 70, 45, and 28 kDa. The 45 and 28 kDa bands represent the cleaved bands. The quantitative data for these METH- induced changes are shown in panels A’, B’, C’ (n⫽3/time point). cellular suicide in a caspase-independent manner (58). exposure of cells to H2O2 (95), both of which have To test the role of AIF, we measured its exit from the been shown to be involved in METH-induced toxicity mitochondria after METH and have provided the first (10, 12, 87). Cyto c release is followed by the formation detailed evidence for the concurrent involvement of of the apoptosome and subsequent cleavage of multiple mitochondrial pathways that appear to inter- caspase-9 and caspase-3 (56). Nevertheless, in our act to cause METH-induced neuronal death. Several model of METH-induced cell death, the fact that lines of evidence had in fact suggested a role for caspase-3 cleavage anteceded caspase-9 activation sug- mitochondrial dysfunction in METH-induced neuro- gests that its initial cleavage might be secondary to toxicity. Gluck et al. (84) had shown that METH can mechanisms other than the formation of the apopto- disrupt the electron transport chain by inhibiting com- some/caspase-9 complex. Such possibilities include ER- plex I activity, an event associated with decreased ATP dependent events associated with the METH-induced production. Burrows et al. (93) had reported rapid activation of caspase-12. METH-mediated decreases in cytochrome oxidase As stated, damage to mitochondria is known to cause (complex IV) activity and striatal depletion of ATP the release of another proapoptotic factor, AIF, which stores in the rat brain, observations that are consistent can activate caspase-independent neuronal apoptosis with our recent observations of complex IV inhibition (38). Normally localized to the intermembrane com- by METH using an in vitro system consisting of immor- partment of mitochondria (96), AIF is released into the talized striatal cells (22). Further evidence that mito- cytoplasm after induction of permeability transition chondria may be involved in METH toxicity was pro- pores (96). The early transit of AIF out of the mito- vided by our previous report that BAX, a known chondria after METH may be due to a similar mecha- proapoptotic agent that acts via cyto c release (33), is nism. In the present study, the release of AIF from the induced according to a time point that suggested a mitochondria was detected 30 min after METH and in similar role of BAX in METH-induced cyto c release the nuclear fraction 4 h thereafter. Rapid AIF release (22, 26). These observations are consistent with data and its translocation to the nucleus, which preceded from other models of neuronal apoptosis in which cyto c release from mitochondria, have been reported mitochondrial mechanisms are involved. These include by others (58, 59, 97). In contrast, Cregan et al. (38) neuronal apoptosis associated with NO toxicity (94) or reported that the release of AIF occurs later than cyto c 246 Vol. 18 February 2004 The FASEB Journal JAYANTHI ET AL.
in a model of p53-dependent cell death. In the present have occurred secondary to METH-mediated oxidative model, the peak of AIF release (1–2 h post-METH) stress (10, 64, 103). When taken together, these obser- appears to occur before peaked cyto c release (4 – 8 h vations suggest that calpain might be acting to cleave postdrug). Thus, our data are more consistent with caspase-12 in the METH neurodegeneration model in a those of the former investigators (58, 59, 97). The fashion similar to that observed in other models of cell reasons for these discrepant reports regarding the death (65). ER stress is associated with increased tran- timing of AIF release are not clear, but suggest inter- scription of the ER resident chaperone GRP78/BiP esting questions that need to be addressed when trying (104) and the nuclear protein CHOP (102). To assess to identify and explain the mechanisms involved in the whether METH-induced changes in these two proteins efflux of proapoptotic substances from the mitochon- were occurring at transcriptional and translational lev- dria. In any case, after its transit into the nucleus, AIF els in the METH toxicity model, we measured their might function as an endonuclease that causes DNA mRNA and protein levels. We observed almost imme- fragmentation (59, 98), even though the possibility that diate increases in BiP mRNA levels. These changes AIF might promote the activity of endonucleases (such might be related to METH-induced oxidative stress (10, as endonuclease G) needs to be considered since these 64, 103) because BiP mRNA is induced in cultures of two appear to work in concert to cause DNA fragmen- neurons exposed to oxidative insults (104). The in- tation in C. elegans (99). Because AIF entry into the creases in BiP protein levels might serve a protective nucleus precedes DFF45 cleavage in the METH apopto- function because BiP overexpression protects cells sis model, it is not farfetched to suggest that the earliest against apoptotic insults (105). In contrast, chop induc- appearance of DNA damage after METH injection tion might play a role in promoting METH-induced might occur in a caspase-independent fashion, with the apoptosis, as it has been reported that the brains of destruction of the nucleus being dependent on the chop/gadd153 null mice exhibit a fourfold reduction in involvement on caspase-dependent and -independent apoptosis that resulted from ER stress (67). This view is pathways. supported by our observation that cells that demon- In addition to cyto c and AIF, METH injection caused strated METH-induced increased CHOP protein the release of smac/DIABLO from the mitochondria, showed active caspase-3 expression. The link between with the appearance of small amounts of the protein as CHOP and apoptosis is thought to occur via down- early as 30 min postdrug injection. Smac/DIABLO is a regulation of Bcl-2 expression and exaggerated produc- proapoptotic molecule recently identified by two differ- tion of reactive oxygen species (63). This supposition ent groups (57, 100). Smac/DIABLO functions as an might provide a partial explanation for the METH- indirect activator of caspases by inhibiting the inhibitor induced down-regulation of Bcl-2 (26) and increased of apoptosis proteins (IAPs) (57, 100). It has been lipid peroxidation (64) we observed in mice striata. suggested that the release of smac/DIABLO from the Activation of downstream or executioner caspases, mitochondria during apoptosis is dependent on active including caspase-3, -6, -7, is the reported penultimate caspases and occurs downstream of cyto c release (101). step in the induction of apoptosis. Their participation In contrast, we found early and potent METH-induced in neuronal cell death is thought to be influenced by mitochondrial release of smac/DIABLO, which pre- cell types and specific apoptotic signals (106). The cedes caspases-3 and 9 activation. Moreover, Smac/ present observation of METH-induced activation of DIABLO release was more coincident with AIF release caspase-6, but not of caspase-7, indicates there might be in the METH model. These observations might impli- selective activation of specific downstream caspases cate similar mechanisms for their release, mechanisms during METH-mediated neuronal apoptosis. These that may differ from cyto c release. Furthermore, the findings are consistent with the report of increased temporal sequence of smac/DIABLO release is consis- caspase-6 activity but not of caspase-7 in human neu- tent with its known action on the IAPs and supports its rons undergoing apoptosis after serum deprivation involvement in promoting the activation of various (106). Caspase-3 is the caspase most frequently re- caspases in the present neurodegeneration model. ported to participate in various models of neuronal This is the first evidence that injections of METH can apoptosis, including that caused by oxidative stress (96, trigger ER stress in the mouse brain. The ER is an 107). Thus, our present data extend a role for this organelle actively involved in the synthesis and proper enzyme to another model of neuronal apoptosis in vivo. folding of proteins as well as in their transport via the Our results show that a panoply of proteins known to be Golgi apparatus to their ultimate destinations (91). targeted for proteolytic cleavage by these enzymes is Dysfunctions of calcium homeostasis, protein misfold- affected after METH treatment. We find proteolysis of ing, or oxidative stress can cause ER stress and cell the caspase-6 substrate lamin A, whose cleavage has death (102). ER-induced apoptosis is associated with been reported to be necessary for complete condensa- early calpain-dependent activation of caspase-12 (65), tion of DNA during apoptosis (108). The caspase-3 which can lead to caspase-3 cleavage. Calpain, a Ca2⫹- substrates ICAD/DFF 45 and PARP (34) are cleaved responsive cytosolic cysteine protease (65) that is an during METH-induced apoptosis. DFF is comprised of important early mediator of ER-dependent cell death, DFF45/ICAD and DFF40/CAD subunits. Its cleavage by is activated by METH, suggesting that METH might caspase-3 results in the liberation of the active DFF40, cause dysregulation of calcium mechanisms. This could the major nuclease implicated in caspase-dependent METH, ER STRESS, AND APOPTOSIS 247
Figure 8. Schematic representation of METH-induced activation of ER- and mitochondria-dependent events during drug-
mediated progressive paths to neuronal apoptosis in the mouse striatum. METH itself or via the generation of superoxide,
hydrogen peroxide, or nitric oxide radicals might have caused stress to the ER and mitochondria. The prolonged
METH-induced activation of calpain observed for up to 7 days after drug injection suggests possible METH-glutamate or reactive
species-mediated dysregulation of calcium homeostasis after the METH injection, as calpain activation is a known calcium-
dependent event. METH-induced CHOP overexpression suggests there may be thus-far unknown gene products involved in
causing METH-mediated neurodegeneration in the mouse striatum. Thus, METH is able to trigger multiple pathways that
interact to lead to the ultimate demise of striatal GABAergic neurons.
DNA fragmentation (72). As stated earlier, our findings culprits in METH-induced neurodegeneration in the
of a substantial increase in DFF-40 and in AIF suggest mouse brain. Finally, further investigations are needed
for the first time that METH-induced DNA fragmenta- to characterize the role, if any, of dopamine, glutamate,
tion in the mouse brain (25) might be the result of and/or temperature regulation in METH-induced apo-
concerted efforts of various nucleases. ptosis in the striatum because these have all been
In summary, our observations indicate that the ad- implicated in METH-mediated toxic effects on mono-
ministration of METH to mice causes activation of aminergic systems (12, 73, 109).
several apoptotic pathways that have documented roles
in neuronal apoptosis (31). Our results indicate that
METH activates these death cascades in a sequential
manner. We have summarized our observations in a
theoretical schema that seeks to detail the temporal REFERENCES
sequence of these molecular events (Fig. 8). Early
1. Wilson, J. M., Kalasinsky, K. S., Levey, A. I., Bergeron, C.,
activation of calpain and caspase-12 indicates that Reiber, G., Anthony, R. M., Schmunk, G. A., Shannak, K.,
METH-induced ER stress might be the earliest contrib- Haycock, J. W., and Kish, S. J. (1996) Striatal dopamine nerve
utor to the appearance of apoptosis in the mouse brain terminal markers in human, chronic methamphetamine users.
after administration of the drug. This is followed by Nat. Med. 2, 699 –703
2. Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Franceschi,
activation of mitochondria-mediated events that trigger D., Sedler, M., Gatley, S. J., Miller, E., Hitzemann, R., Ding,
both caspase-dependent and independent pathways. Y. S., et al. (2001) Loss of dopamine transporters in metham-
Our observations are beginning to shed more light on phetamine abusers recovers with protracted abstinence. J. Neu-
rosci. 21, 9414 –9418
the diverse pathways that are involved in METH-in- 3. Ernst, T., Chang, L., Leonido-Yee, M., and Speck, O. (2000)
duced neuronal apoptosis and to identify ER- and Evidence for long-term neurotoxicity associated with metham-
mitochondria-mediated events as important causative phetamine abuse: A 1H MRS study. Neurology 54, 1344 –1349
248 Vol. 18 February 2004 The FASEB Journal JAYANTHI ET AL.4. Kramer, J. C., Fischman, V. S., and Littlefield, D. C. (1967) evidence from using an improved TUNEL histochemical
Amphetamine abuse. Pattern and effects of high doses taken method. Mol. Brain Res. 93, 64 – 69
intravenously. J. Am. Med. Assoc. 201, 305–309 26. Jayanthi, S., Deng, X., Bordelon, M., McCoy, M. T., and Cadet,
5. Lan, K. C., Lin, Y. F., Yu, F. C., Lin, C. S., and Chu, P. (1998) J. L. (2001) Methamphetamine causes differential regulation
Clinical manifestations and prognostic features of acute meth- of pro-death and anti-death Bcl-2 genes in the mouse neocor-
amphetamine intoxication. J. Formos. Med. Assoc. 97, 528 –533 tex. FASEB J. 15, 1745–1752
6. Perez, J. A., Jr., Arsura, E. L., and Strategos, S. (1999) Meth- 27. Stumm, G., Schlegel, J., Schafer, T., Wurz, C., Mennel, H. D.,
amphetamine-related stroke: four cases. J. Emerg. Med. 17, Krieg, J. C., and Vedder, H. (1999) Amphetamines induce
469 – 471 apoptosis and regulation of bcl-x splice variants in neocortical
7. Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Ding, Y. S., neurons. FASEB J. 13, 1065–1072
Sedler, M., Logan, J., Franceschi, D., Gatley, J., Hitzemann, R., 28. Choi, H. J., Yoo, T. M., Chung, S. Y., Yang, J. S., Kim, J. I., Ha,
et al. (2001) Low level of brain dopamine D2 receptors in E. S., and Hwang, O. (2002) Methamphetamine-induced apo-
methamphetamine abusers: association with metabolism in the ptosis in a CNS-derived catecholaminergic cell line. Mol. Cells
orbitofrontal cortex. Am. J. Psychiatry 158, 2015–2021 13, 221–227
8. McCann, U. D., Wong, D. F., Yokoi, F., Villemagne, V., Dan- 29. Genc, K., Genc, S., Kizildag, S., Sonmez, U., Yilmaz, O.,
nals, R. F., and Ricaurte, G. A. (1998) Reduced striatal dopa- Tugyan, K., Ergur, B., Sonmez, A., and Buldan, Z. (2003)
mine transporter density in abstinent methamphetamine and Methamphetamine induces oligodendroglial cell death in
methcathinone users: evidence from positron emission tomog- vitro. Brain Res. 982, 125–130
raphy studies with [11C] WIN-35, 428. J. Neurosci. 18, 8417– 30. Mattson, M. P. (2000) Apoptosis in neurodegenerative disor-
8422 ders. Nat. Rev. Mol. Cell Biol. 1, 120 –129
9. Sekine, Y., Iyo, M., Ouchi, Y., Matsunaga, T., Tsukada, H., 31. Putcha, G. V., Harris, C. A., Moulder, K. L., Easton, R. M.,
Okada, H., Yoshikawa, E., Futatsubashi, M., Takei, N., and Thompson, C. B., and Johnson, E. M., Jr. (2002) Intrinsic and
Mori, N. (2001) Methamphetamine-related psychiatric symp- extrinsic pathway signaling during neuronal apoptosis: lessons
toms and reduced brain dopamine transporters studied with from the analysis of mutant mice. J. Cell Biol. 157, 441– 453
PET. Am. J. Psychiatry 158, 1206 –1214 32. Hacki, J., Egger, L., Monney, L., Conus, S., Rosse, T., Fellay, I.,
10. Cadet, J. L., and Brannock, C. (1998) Free radicals and the and Borner, C. (2000) Apoptotic crosstalk between the endo-
pathobiology of brain dopamine systems. Neurochem. Int. 32, plasmic reticulum and mitochondria controlled by Bcl-2. On-
117–131 cogene 19, 2286 –2295
11. Seiden, L. S., and Sabol, K. E. (1996) Methamphetamine and 33. Nutt, L. K., Pataer, A., Pahler, J., Fang, B., Roth, J., McConkey,
methylenedioxymethamphetamine neurotoxicity: possible mech- D. J., and Swisher, S. G. (2002) Bax and Bak promote apoptosis
anisms of cell destruction. NIDA Res. Monogr. 163, 251–276 by modulating endoplasmic reticular and mitochondrial Ca2⫹
12. Cadet, J. L., Jayanthi, S., and Deng, X. (2003) Speed kills: stores. J. Biol. Chem. 277, 9219 –9225
cellular and molecular bases of methamphetamine-induced 34. Earnshaw, W. C., Martins, L. M., and Kaufmann, S. H. (1999)
nerve terminal degeneration and neuronal apoptosis. FASEB J. Mammalian caspases: structure, activation, substrates, and
17, 1775–1788 functions during apoptosis. Annu. Rev. Biochem. 68, 383– 424
13. Zalis, E. G., Lundberg, G. D., and Knutson, R. A. (1967) The 35. Tang, D., and Kidd, V. J. (1998) Cleavage of DFF-45/ICAD by
pathophysiology of acute amphetamine poisoning with patho- multiple caspases is essential for its function during apoptosis.
logic correlation. J. Pharmacol. Exp. Ther. 158, 115–127 J. Biol. Chem. 273, 28549 –28552
14. Escalante, O. D., and Ellinwood, E. H., Jr. (1970) Central 36. D'Amours, D., Sallmann, F. R., Dixit, V. M., and Poirier, G. G.
nervous system cytopathological changes in cats with chronic (2001) Gain-of-function of poly(ADP-ribose) polymerase-1
Methedrine intoxication. Brain Res. 21, 151–155 upon cleavage by apoptotic proteases: implications for apopto-
15. Ellinwood, E. H., Jr., and Escalante, O. (1970) Behavior and sis. J. Cell Sci. 114, 3771–3778
histopathological findings during chronic Methedrine intoxi- 37. Lazebnik, Y. A., Takahashi, A., Moir, R. D., Goldman, R. D.,
cation. Biol. Psychiatry 2, 27–39 Poirier, G. G., Kaufmann, S. H., and Earnshaw, W. C. (1995)
16. Ellison, G., and Switzer, R. C., III (1993) Dissimilar patterns of Studies of the lamin proteinase reveal multiple parallel bio-
degeneration in brain following four different addictive stim- chemical pathways during apoptotic execution. Proc. Natl.
ulants. NeuroReport 5, 17–20 Acad. Sci. USA 92, 9042–9046
17. Schmued, L. C., and Bowyer, J. F. (1997) Methamphetamine 38. Cregan, S. P., Fortin, A., MacLaurin, J. G., Callaghan, S. M.,
exposure can produce neuronal degeneration in mouse hip- Cecconi, F., Yu, S. W., Dawson, T. M., Dawson, V. L., Park, D. S.,
pocampal remnants. Brain Res. 759, 135–140 Kroemer, G., et al. (2002) Apoptosis-inducing factor is involved
18. Eisch, A. J., Schmued, L. C., and Marshall, J. F. (1998) in the regulation of caspase-independent neuronal cell death.
Characterizing cortical neuron injury with Fluoro-Jade labeling J. Cell Biol. 158, 507–517
after a neurotoxic regimen of methamphetamine. Synapse 30, 39. Jayanthi, S., McCoy, M. T., Ladenheim, B., and Cadet, J. L.
329 –333 (2002) Methamphetamine causes coordinate regulation of src,
19. Eisch, A. J., and Marshall, J. F. (1998) Methamphetamine cas, crk, and the jun N-terminal kinase-jun pathway. Mol.
neurotoxicity: dissociation of striatal dopamine terminal dam- Pharmacol. 61, 1124 –1131
age from parietal cortical cell body injury. Synapse 30, 433– 445 40. Mielke, K., and Herdegen, T. (2000) JNK and p38
20. Cadet, J. L., Ordonez, S. V., and Ordonez, J. V. (1997) stresskinasesOdegenerative effectors of signal-transduction-
Methamphetamine induces apoptosis in immortalized neural cascades in the nervous system. Prog. Neurobiol. 61, 45– 60
cells: protection by the proto-oncogene, bcl-2. Synapse 25, 41. Korsmeyer, S. J., Shutter, J. R., Veis, D. J., Merry, D. E., and
176 –184 Oltvai, Z. N. (1993) Bcl-2/Bax: a rheostat that regulates an
21. Deng, X., and Cadet, J. L. (2000) Methamphetamine-induced anti-oxidant pathway and cell death. Semin. Cancer Biol. 4,
apoptosis is attenuated in the striata of copper-zinc superoxide 327–332
dismutase transgenic mice. Mol. Brain Res. 83, 121–124 42. Cadet, J. L., Jayanthi, S., McCoy, M. T., Vawter, M., and
22. Deng, X., Cai, N. S., McCoy, M. T., Chen, W., Trush, M. A., and Ladenheim, B. (2001) Temporal profiling of methamphet-
Cadet, J. L. (2002) Methamphetamine induces apoptosis in an amine-induced changes in gene expression in the mouse
immortalized rat striatal cell line by activating the mitochon- brain: evidence from cDNA array. Synapse 41, 40 – 48
drial cell death pathway. Neuropharmacology 42, 837– 845 43. Xie, T., Tong, L., Barrett, T., Yuan, J., Hatzidimitriou, G.,
23. Deng, X., Jayanthi, S., Ladenheim, B., Krasnova, I. N., and McCann, U. D., Becker, K. G., Donovan, D. M., and Ricaurte,
Cadet, J. L. (2002) Mice with partial deficiency of c-Jun show G. A. (2002) Changes in gene expression linked to metham-
attenuation of methamphetamine-induced neuronal apopto- phetamine-induced dopaminergic neurotoxicity. J. Neurosci.
sis. Mol. Pharmacol. 62, 993–1000 22, 274 –283
24. Deng, X., Ladenheim, B., Tsao, L. I., and Cadet, J. L. (1999) 44. Barrett, T., Xie, T., Piao, Y., Dillon-Carter, O., Kargul, G. J.,
Null mutation of c-fos causes exacerbation of methamphet- Lim, M. K., Chrest, F. J., Wersto, R., Rowley, D. L., Juhaszova,
amine-induced neurotoxicity. J. Neurosci. 19, 10107–10115 M., et al. (2001) A murine dopamine neuron-specific cDNA
25. Deng, X., Wang, Y., Chou, J., and Cadet, J. L. (2001) Metham- library and microarray: increased COX1 expression during
phetamine causes widespread apoptosis in the mouse brain: methamphetamine neurotoxicity. Neurobiol. Dis. 8, 822– 833
METH, ER STRESS, AND APOPTOSIS 249You can also read

















































