Pharmacological Inhibition of Centrosome Clustering by Slingshot-Mediated Cofilin Activation and Actin Cortex Destabilization
←
→
Page content transcription
If your browser does not render page correctly, please read the page content below
Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cancer
Therapeutics, Targets, and Chemical Biology Research
Pharmacological Inhibition of Centrosome
Clustering by Slingshot-Mediated Cofilin
Activation and Actin Cortex Destabilization
Gleb Konotop1, Elena Bausch1, Tomoaki Nagai2, Andrey Turchinovich3, Natalia Becker4,
€ mer6, and Marc Steffen Raab1
Axel Benner4, Michael Boutros5, Kensaku Mizuno2, Alwin Kra
Abstract
Centrosome amplification is a hallmark of virtually all types that these compounds induce mitotic spindle multipolarity by
of cancers, including solid tumors and hematologic malignan- activation of the actin-severing protein cofilin, leading to
cies. Cancer cells with extra centrosomes use centrosome clus- destabilization of the cortical actin network, and provide evi-
tering (CC) to allow for successful division. Because normal dence that this activation is dependent on slingshot phospha-
cells do not rely on this mechanism, CC is regarded as a tases 1 and 2 but unrelated to PDGFR-b inhibition. More
promising target to selectively eradicate cells harboring super- specifically, we found that although both compounds attenu-
numerary centrosomes. To identify novel inhibitors of CC, we ated PDGF-BB–induced signaling, they significantly enhanced
developed a cell-based high-throughput screen that reports the phosphorylation of PDGFR-b downstream effectors, Akt
differential drug cytotoxicity for isogenic cell populations with and MEK, in almost all tested cancer cell lines under physio-
different centrosome contents. We identified CP-673451 and logic conditions. In summary, our data reveal a novel mech-
crenolanib, two chemically related compounds originally anism of CC inhibition depending on cofilin-mediated corti-
developed for the inhibition of platelet-derived growth factor cal actin destabilization and identify two clinically relevant
receptor b (PDGFR-b), as robust inhibitors of CC with selective compounds interfering with this tumor cell–specific target.
cytotoxicity for cells with extra centrosomes. We demonstrate Cancer Res; 76(22); 1–11. 2016 AACR.
Introduction Centrosome amplification (CA) is found in most types of
cancers. Although it is still not clear whether CA is a cause or a
Centrosomes are cytoplasmic organelles composed of a pair
consequence of tumor initiation and progression, extra centro-
of centrioles, which nucleate and anchor microtubules. Cen-
somes strongly correlate with chromosomal instability, clinical
trosomes act as microtubule-organizing centers in animal cells
aggressiveness, and adverse clinical outcome in several tumor
and play a key role in mitotic fidelity by securing bipolar
types (4–10). Cancer cells carrying supernumerary centrosomes
mitotic spindle formation and equal chromosome segregation
escape detrimental multipolar divisions by coalescing multiple
(1, 2). The number of centrosomes is tightly regulated by
centrosomes into two functional spindle poles, a process
ensuring that centrosomes are duplicated exactly once per cell
known as centrosome clustering (CC; ref. 11). CC contributes
cycle (3).
to chromosome segregation errors by generating merotelic
microtubule–kinetochore attachment errors, leading to tolera-
ble levels of genomic instability (12). Because most healthy
1
Max-Eder Research Group "Experimental Therapies for Hematologic
tissues have normal centrosome content, they do not rely on
Malignancies", German Cancer Research Center (DKFZ) and Depart- CC for successful division, which makes this mechanism a
ment of Internal Medicine V, University of Heidelberg, Heidelberg, promising therapeutic target.
Germany. 2Department of Biomolecular Sciences, Graduate School
In addition to microtubule motor proteins, including dynein,
of Life Sciences, Tohoku University, Sendai, Miyagi, Japan. 3Molecular
Epidemiology Group, German Cancer Research Center, Heidelberg, Ncd/HSET, and Eg5 (11, 13–15), a role for cortical actin in CC was
Germany. 4Division of Biostatistics, German Cancer Research Center, initially suggested by a genome-wide RNAi screen in Drosophila S2
Heidelberg, Germany. 5Division of Signaling and Functional Genomics, cells, where depletion of several components of the actin cyto-
Medical Faculty Mannheim, German Cancer Research Center and
University of Heidelberg, Heidelberg, Germany. 6Clinical Cooperation skeleton led to CC inhibition (13). Also, depletion of the actin-
Unit Molecular Hematology/Oncology, German Cancer Research Cen- associated protein MISP destabilized attachments between astral
ter and Department of Internal Medicine V, University of Heidelberg, microtubules and the actin cortex, led to defects in spindle
Heidelberg, Germany.
orientation, and increased the incidence of multipolar spindles
Note: Supplementary data for this article are available at Cancer Research in cells with CA (16). Finally, CC requires a functional spindle
Online (http://cancerres.aacrjournals.org/).
assembly checkpoint (SAC) to provide the necessary time for
Corresponding Author: Alwin Kra€mer, German Cancer Research Center, Im effective centrosome coalescence (13, 14, 17).
Neuenheimer Feld 280, Heidelberg 69120, Germany. Phone: 4962-2142-1440; Cell-permeable small molecules that exclusively eradicate cells
Fax: 4962-2142-1444; E-mail: a.kraemer@dkfz.de
with extra centrosomes might be promising tools for targeted
doi: 10.1158/0008-5472.CAN-16-1144 cancer therapy. CC can be inhibited by molecules that interfere
2016 American Association for Cancer Research. with MT dynamics, such as taxanes, Vinca alkaloids, or the
www.aacrjournals.org OF1
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.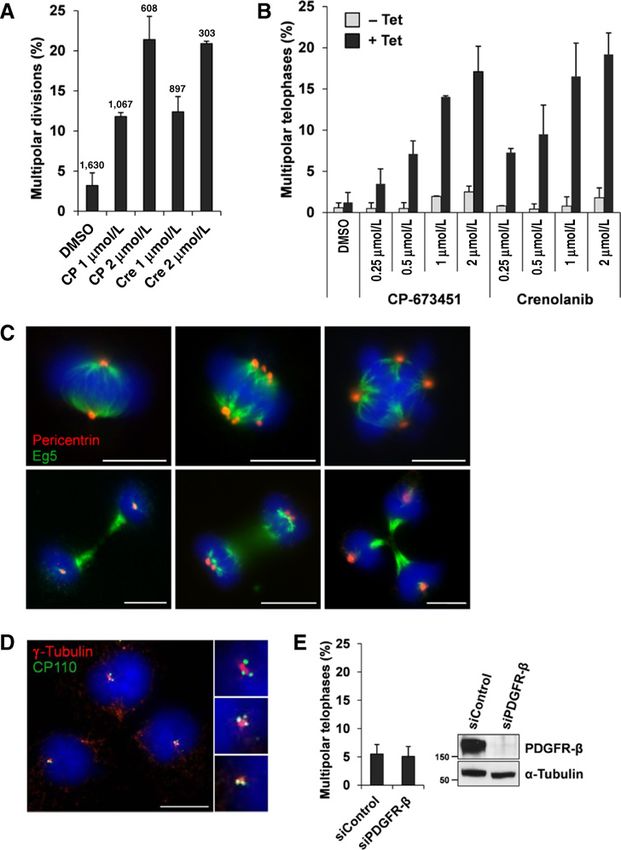
Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Konotop et al.
noscapinoid EM011 (18–20). However, these drugs are not HRP-conjugated secondary antibodies (Santa Cruz Biotechnology);
selective for cells with supernumerary centrosomes. Molecules a-tubulin (Sigma); Eg5 (BD), and phospho-Eg5 (Novus).
with increased selectivity include griseofulvin and its derivatives
and HSET inhibitors, which effectively decluster multiple centro- Time-lapse microscopy and image acquisition
somes, but lead at higher concentrations to the formation of Time-lapse microscopy was performed on a Zeiss Cell Observer.
multipolar spindles with acentriolar poles (13, 21–24). Z1 under controlled environmental conditions. The numbers of
Experimentally, cells with extra centrosomes can be obtained total mitotic cells counted are indicated over each bar. Fluorescence
by increasing the expression levels of key components of the microscopy was performed as described previously (16) using a
centriole replication machinery, such as Polo-like kinase 4 (PLK4) Zeiss Axiovert 200M. Antibodies used were as follows: Eg5 (BD);
or the scaffolding proteins HsSAS-6 and STIL (25–30). CP110 (Acris); g-tubulin (Exbio); pericentrin (Abcam); AlexaFluor
In this study, we employed a novel small-molecule screening 488 or 568-conjugated secondary antibodies (Molecular Probes).
strategy based on a differential viability readout between two
isogenic cell populations with different centrosome content to In vitro kinase assay
identify CP-673451 and crenolanib, two class III receptor tyrosine Kinase assay was performed as described previously (32).
kinase (RTK) inhibitors, as CC inhibitors. We demonstrate that
the inhibition of CC was attributed to activation of the actin- Results
severing protein, cofilin, which constitutes a novel mechanism of Establishment of a cell-based high-throughput screening assay
cortical actin-mediated CC inhibition. Furthermore, our work for the identification of small-molecule inhibitors of CC
sheds light on the mechanisms of CP-673451 and crenolanib- To identify novel inhibitors of CC, we developed a cell-based
induced cofilin activation mediated by the slingshot phospha- screening assay that reports on the differential effects of small
tases (SSH) SSH1 and SSH2. molecules on the viability of two isogenic cell populations
with different centrosome content. Specifically, we engineered
Materials and Methods a human osteosarcoma cell line (U2OS) to conditionally over-
Detailed experimental procedures are included in the Supple- express EGFP-tagged PLK4 (EGFP-PLK4) from a tetracycline-
mentary Data. inducible promoter. Under noninduced conditions, only 2%
to 3% of EGFP-PLK4-U2OS cells harbored aberrant centrosome
numbers (i.e., >2 g-tubulin signals), whereas 48 hours after
Cells and reagents
induction, over 80% of cells exhibited CA, which remained stable
To generate EGFP-PLK4-U2OS, human osteosarcoma cells
for several days despite tetracycline withdrawal (Fig. 1A–C).
carrying the regulatory plasmid pcDNA6/TR were transfected
Induced EGFP-PLK4-U2OS cells were CC proficient, as 98.8
with ToPuro-EGFP-PLK4. Plasmid generation is described in
0.7% of cells underwent bipolar cell division (n ¼ 1,783). To test
Supplementary Data. EGFP-PLK4-U2OS and H2B-mCherry-
the suitability of EGFP-PLK4-U2OS cells for viability-based high-
a-tubulin-EGFP-HeLa (31) cells were cultivated in DMEM þ
throughput screening, we treated control and induced cells with
GlutaMAX (Life Technologies) supplemented with 10% FCS
increasing concentrations of griseofulvin, an inhibitor of CC (21),
(Biochrom). All unmodified cancer cell lines were obtained from
for 5 days and subsequently measured the viabilities of both cell
ATCC and authenticated by MCA (2014). For PDGF-BB stimu-
populations using a luminescence reporter assay based on quan-
lation, starved cells (0% FCS, 24 hours) were pretreated with drug
tification of ATP. As expected, griseofulvin induced more cytotox-
or vehicle for 3 hours and stimulated with 500 mg/mL PDGF-BB
icity in EGFP-PLK4-U2OS cells with CA as compared with cells with
(Biotrend) for 15 minutes. Inhibitors included LIMKi3 (Merck),
normal centrosome content (Fig. 1D). Furthermore, live cell imag-
damnacanthal (Enzo), griseofulvin (Sigma), BYL719, CP-
ing demonstrated that treatment of induced EGFP-PLK4-U2OS
673451, and crenolanib (Selleckchem). Cells were synchronized
cells with 4 mmol/L griseofulvin (i.e., the concentration with the
with 100 ng/mL nocodazole (24 hours) or 2 mmol/L thymidine
largest viability difference between control and induced cells)
(16–18 hours; Sigma).
increased the rate of multipolar divisions by more than 5-fold in
comparison with DMSO (Fig. 1E). More than 80% of the progeny
Differential viability readout of multipolar divisions underwent cell death, in comparison with
EGFP-PLK4-U2OS cells were split into two populations and only 17% of the progeny of bipolar divisions (Fig. 1F).
incubated with 2 mg/mL tetracycline (Sigma) or vehicle. After 2
days, induced and noninduced cells were seeded in 384-well or Identification of CP-673451 and crenolanib as inhibitors of CC
96-well plates and rested (24 hours) prior to small-molecule To identify new cell-permeable molecules that target CC, we
addition. After 5-day exposure, cell viabilities were determined screened two small-molecule libraries consisting of 843 FDA-
with CellTiter-Glo (Promega). approved compounds and 273 kinase inhibitors (Fig. 2A).
The FDA-approved library was screened at a concentration of 10
Statistical analysis mmol/L, whereas the kinase inhibitor library was screened at three
Results are given as mean percentages SD. Significances were different concentrations (100 nmol/L, 1 mmol/L, and 10 mmol/L)
calculated by two-tailed t test or two-way ANOVA methods. because of the concentration-dependent target specificity of many
kinase inhibitors. Hits were ordered according to their CC inhi-
Immunoblotting bition (CCI) index, calculated as ratio of viabilities between
Cell lysis and immunoblotting was performed according to control and induced cells, and normalized to the viability ratio
standard protocols. Antibodies used were as follows: cofilin, phos- of vehicle-treated populations. Thus, a positive CCI index indi-
pho-cofilin, phospho-Akt, phospho-MEK1/2, phospho-LIMK1/ cated that a small molecule compromised the viability of induced
LIMK2, LIMK1, LIMK2, and SSH1 (CST); GFP, MCM7, PDGFR-b; cells with CA over that of noninduced controls. For further
OF2 Cancer Res; 76(22) November 15, 2016 Cancer Research
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.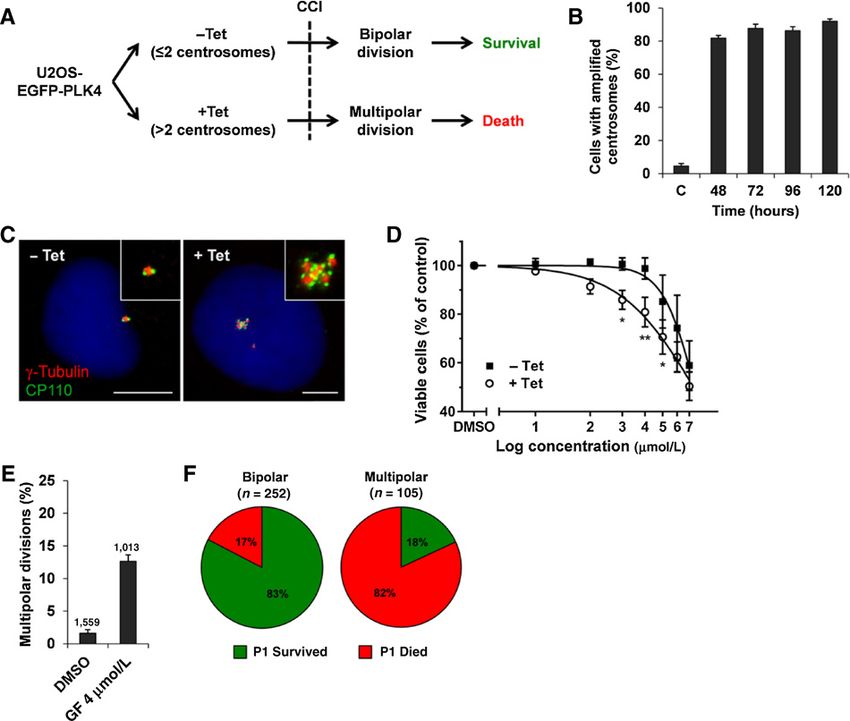
Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cofilin Activation Inhibits Centrosome Clustering
Figure 1.
Performance assessment of EGFP-PLK4-U2OS cells for high-throughput small-molecule screening. A, schematic overview of the screening concept. Induced (þTet)
and noninduced (Tet) EGFP-PLK4-U2OS cells are exposed to small molecules. Induction of spindle multipolarity by CC inhibitors (CCI) will selectively impair
survival of cells with CA. B, mean percentages SD of EGFP-PLK4-U2OS cells with more than two g-tubulin signals. Tetracycline (Tet) was removed 48 hours
after induction. C, noninduced cells (counts/sample 500, averaged from two independent experiments). C, representative images of noninduced (left) and induced
(right) EGFP-PLK4-U2OS cells. Cells were treated with vehicle (Tet) or tetracycline (þTet) for 48 hours and stained for g-tubulin (red), CP110 (green),
and DNA (blue) 96 hours postinduction. Scale bar, 10 mm. D, dose–response curves SD comparing relative viabilities of induced (þTet) and noninduced (Tet)
EGFP-PLK4-U2OS cells after 5 days of exposure to griseofulvin (1–7 mmol/L; , P < 0.02; , P < 0.01; n ¼ 3). E, time-lapse imaging over 48 hours showing average
percentages of multipolar divisions in induced EGFP-PLK4-U2OS cells after exposure to 4 mmol/L griseofuilvin (GF) from two independent experiments. F, fate of
progeny resulting from bipolar and multipolar divisions of induced EGFP-PLK4-U2OS cells after exposure to 4 mmol/L griseofulvin. Daughter cells were
tracked by time-lapse microscopy for up to 48 hours.
evaluation, we chose the kinase inhibitors, CP-673451 and CP- nolanib increased the percentage of multipolar divisions of
868596 (crenolanib), because they exhibited the highest CCI index induced EGFP-PLK4-U2OS cells by approximately 3-fold at 1
values and due to their structural homology (Fig. 2B and C; Sup- mmol/L and 5-fold at 2 mmol/L (Fig. 3A). To test whether
plementary Table S1). Both compounds are composed of amino- multipolar divisions were caused by centrosome declustering,
piperidine-, quinoline- and benzimidazole-ring systems and we treated control and induced EGFP-PLK4-U2OS cells with
termed quinolinobenzimidazoles. Detailed dose–response viabil- increasing concentrations of both compounds and quantified
ity analyses revealed that the presence of supernumerary centro- the percentage of multipolar telophases, resulting in more
somes reduced IC50 values of CP-673451 and crenolanib from 1.6 than two daughter cells. As expected, both drugs increased
to 0.6 mmol/L and 1.2 to 0.6 mmol/L, respectively (Fig. 2D). the rate of multipolar telophases in a dose-dependent manner,
reaching maxima of about 20% at 2 mmol/L. The percentage
CP-673451 and crenolanib inhibit CC of multipolar telophases in control cells remained less than 2%,
Consistent with the increased cytotoxicity seen in cells with indicating that only cells carrying supernumerary centrosomes
CA, live cell imaging demonstrated that CP-673451 and cre- were prone to multipolar cell division (Fig. 3B and C).
www.aacrjournals.org Cancer Res; 76(22) November 15, 2016 OF3
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Konotop et al.
Figure 2.
CP-673451 and crenolanib show selective lethality toward cells with CA. A, screening timeline. B, scatter plot showing the screening results of the 1 mmol/L
kinase inhibitor library screen. Positive hits with a CCI index >0.3 and adjusted P < 0.05 are highlighted in black. CP-673451 and crenolanib scored the highest
values. C, molecular structures of CP-673451 (1-(2-(5-(2-methoxyethoxy)-1H-benzo[d]imidazol-1-yl)quinolin-8-yl)piperidin-4-amine) and crenolanib
(1-(2-(5-((3-methyloxetan-3-yl)methoxy)-1H-benzo[d]imidazol-1-yl)quinolin-8-yl)piperidin-4-amine). D, dose–response curves SD comparing relative
viabilities of induced (þTet) and noninduced (Tet) EGFP-PLK4-U2OS cells after 5 days of exposure to increasing concentrations of CP-673451 and crenolanib
( , P < 0.01; , P < 0.001; n ¼ 3).
Importantly, virtually all multipolar telophases exhibited cen- 3Flag-STIL-HCT116, another cell line with inducible CA result-
trioles at each pole (100/101 for 1 mmol/L CP-673451, 91/91 ing from conditional STIL overexpression (Supplementary
for 1 mmol/L crenolanib), emphasizing the inhibition of CC by Table S2). Both compounds increased the rates of multipolar
both compounds (Fig. 3D). Neither CP-673451 nor crenolanib telophases by at least 2-fold in all cell lines with CA, including
caused centrosome amplification (Supplementary Fig. S1). nonmalignant MCF10A cells, which harbor about 10% CA. As
Because a SAC-mediated mitotic delay is required for CC expected, no significant multipolarity was observed in BJ fibro-
(13, 17), we addressed whether CP-673451 and crenolanib blasts, which do not contain extra centrosomes. Taken together,
affect the timing of mitosis. Fluorescence time-lapse microsco- these observations suggest that both compounds act as inhi-
py of dividing HeLa cells, stably expressing H2B-mCherry and bitors of CC in all cell lines tested and thereby preferentially
a-tubulin-EGFP, revealed that at 1 mmol/L, crenolanib affect cells that carry supernumerary centrosomes.
increased the duration of mitosis by about 2-fold, while CP-
673451 did not delay mitosis. These effects were more prom- Depletion of PDGFR-b has no effect on CC
inent at 2 mmol/L, leading to 2- and 3-fold mitosis prolonga- CP-673451 and crenolanib are potent inhibitors of platelet-
tion for CP-673451 and crenolanib, respectively (Supplemen- derived growth factor receptor b (PDGFR-b; refs. 33, 34). Because
tary Fig. S2). These data indicate that inhibition of CC was not both molecules share PDGFR-b as their main target, we next
caused by SAC inactivation. sought to analyze the effects of RNAi-mediated PDGFR-b deple-
Finally, we tested the effect of CP-673451 and crenolanib tion on CC. Surprisingly, downregulation of PDGFR-b did not
on CC in various cancer and nontransformed cell lines increase the percentage of multipolar divisions in EGFP-PLK4-
that harbor varying degrees of spontaneous CA as well as in U2OS cells with CA (Fig. 3E), indicating that inhibition of CC
OF4 Cancer Res; 76(22) November 15, 2016 Cancer Research
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cofilin Activation Inhibits Centrosome Clustering
Figure 3.
CP-673451 and crenolanib inhibit CC in
induced EGFP-PLK4-U2OS cells. A,
average percentage of multipolar
divisions SD of induced EGFP-PLK4-
U2OS cells within the first 24 hours after
exposure to DMSO, CP-673451 (CP), or
crenolanib (Cre) by time-lapse imaging
(n ¼ 2). B, average percentages of
multipolar telophases in control (Tet)
and induced EGFP-PLK4-U2OS cells
(þTet) from two independent
experiments, treated with increasing
drug concentrations for 24 hours
(counts/sample 200). C,
representative images of normal bipolar
(left), clustered bipolar (middle), and
multipolar (right) metaphases and
telophases in EGFP-PLK4-U2OS cells
stained for Eg5 (green), pericentrin
(red), and DNA (blue). Scale bars, 10 mm.
D, multipolar telophase of an induced
EGFP-PLK4-U2OS cell treated with 1
mmol/L CP-673451 (24 hours) and
stained for CP110 (green), g-tubulin
(red), and DNA (blue). Note that part of
the green signal could be due to residual
EGFP-PLK4. Scale bar, 10 mm. E, average
percentage of multipolar telophases in
induced EGFP-PLK4-U2OS cells
after PDGFR-b knockdown (72 hours)
from two independent experiments
(counts/sample 1,000). Immunoblot
showing PDGFR-b depletion; a-tubulin
indicates equal loading.
caused by both quinolinobenzimidazoles was not mediated by both drugs led to a complete disorganization of stress fibers and
impaired PDGFR-b signaling. the appearance of aberrant F-actin arrangements. Similar results
were obtained in other cell lines, including MDA-MB-231,
CP-673451 and crenolanib affect the organization of the actin LOVO, and HCT116 (data not shown).
cytoskeleton
U2OS cells treated with 1 to 4 mmol/L CP-673451 or creno- CP-673451 and crenolanib activate cofilin
lanib showed a ruffled cell surface as a sign for alterations of the The observed rearrangements of the actin cytoskeleton indi-
cortical actin cytoskeleton. Phalloidin-FITC staining of the actin cated that the compounds might affect the regulation of actin
cytoskeleton revealed that both compounds markedly affect dynamics. Rapid actin remodeling in response to extracellular
the morphology of stress fibers and overall actin organization stimuli is elicited by the activation of cofilin, which is regulated
(Fig. 4A). Drug concentrations (1 mmol/L) led to the appear- by an inhibitory Ser3 phosphorylation (35, 36). To analyze
ance of bundled actin networks instead of characteristic stress changes in cofilin activity, we treated unsynchronized U2OS
fibers. Strikingly, treatment of U2OS cells with 4 mmol/L of cells with increasing quinolinobenzimidazole concentrations
www.aacrjournals.org Cancer Res; 76(22) November 15, 2016 OF5
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Konotop et al.
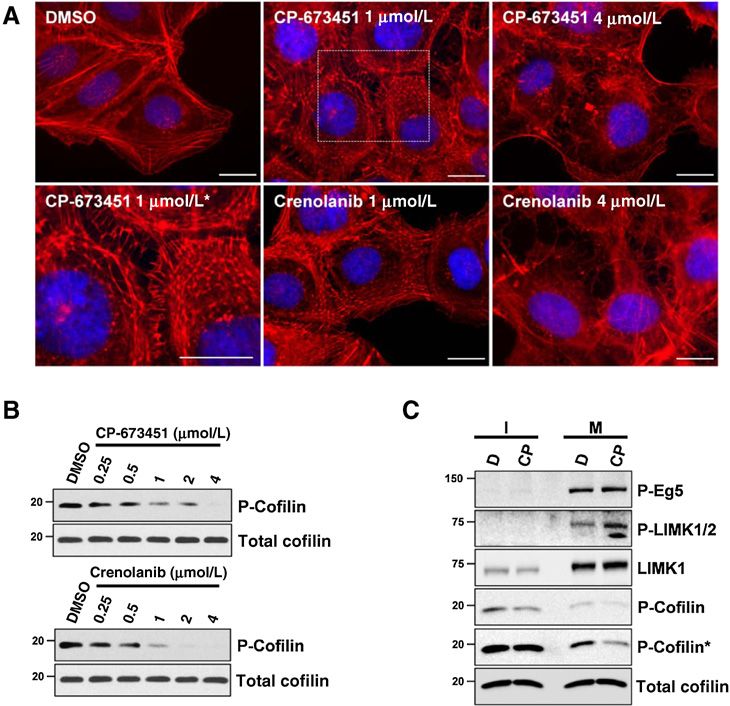
Figure 4.
CP-673451 and crenolanib disturb actin
organization associated with cofilin
activation both in interphase and
mitotic cells. A, representative images
of compound-induced disorganization
of the actin network in phalloidin-
TRITC–stained U2OS cells after
compound addition (3 hours).
, magnified view of a rectangular inset.
Scale bar, 20 mm. B, immunoblot
showing a decrease of phospho-Ser3-
cofilin levels in unsynchronized U2OS
cells after exposure to CP-673451 or
crenolanib (3 hours), in comparison
with DMSO. C, CP-673451 decreases
phospho-Ser3-cofilin levels in both
interphase (I) and mitotic (M) cells.
U2OS cells were synchronized with
nocodazole in the presence of 1 mmol/L
CP-673451 (CP) or DMSO (D). Phospho-
Thr927-Eg5 positivity characterizes the
mitotic fraction. , longer exposure.
and assessed the levels of phosphorylated (inactive) cofilin with 2 mmol/L CP-673451 increased the average spindle
using an antibody against phospho-Ser3-cofilin. Both com- rotation from 23 degrees to 59 degrees and the average
pounds induced a concentration-dependent reduction of phos- oscillation distance from 7 to 17 mm (Fig. 5A; Supplementary
pho-Ser3-cofilin levels, whereas overall cofilin levels remained Movies S1–S3).
unchanged in both noninduced (Fig. 4B) and induced EGFP- Because CP-673451 and crenolanib led to cofilin activation and
PLK4-U2OS cells carrying CA (data not shown). In addition, inhibition of CC, we next addressed whether increased cofilin
we treated several other cancer cell lines with increasing con- activity causes CC inhibition. We increased the levels of active
centrations of CP-673451. Immunoblot analysis of phospho- cofilin in dividing EGFP-PLK4-U2OS cells with CA by (i) inhibi-
Ser3-cofilin clearly showed that cofilin was activated in a dose- tion of cofilin phosphorylation and (ii) increasing overall cofilin
dependent manner in all cell lines examined (Supplementary levels. To inhibit cofilin phosphorylation, we suppressed the
Fig. S3). Next, we analyzed whether cofilin becomes activated activity of LIM kinases (LIMK) using two highly selective, cell-
in drug-exposed mitotic cells as well. Mitotic U2OS cells permeable LIMK inhibitors, LIMKi3 (38) and damnacanthal (32).
arrested in metaphase by nocodazole in the presence of CP- Time-lapse microscopy analysis of induced EGFP-PLK4-U2OS
673451 were separated from interphase cells by intensive cells after exposure to LIMKi3 or damnacanthal revealed a
shaking. As expected, the levels of phospho-Ser3-cofilin were concentration-dependent increase of multipolar divisions
reduced in both CP-673451–treated interphase and mitotic (Fig. 5B and C), suggesting that cofilin activation disturbs CC.
cells as compared with controls (Fig. 4C). Next, we examined the effect of cofilin overexpression on
inhibition of CC by transiently transfecting induced EGFP-
Accumulation of active cofilin during mitosis inhibits CC PLK4-U2OS cells with wild-type cofilin (Cof-WT), non-phos-
A previous study has shown that the accumulation of active phorylatable cofilin (Cof-S3A), or cofilin containing a phos-
cofilin during mitosis strongly affects the orientation of the phomimetic mutation (Cof-S3E). To increase the number of
mitotic spindle in HeLa cells due to decreased stability of mitotic events, cells were synchronized in G1–S-phase by a
the cortical actin meshwork (37). Accordingly, time-lapse single thymidine block and released before transfection. Time-
fluorescence microscopy analysis of spindle dynamics in HeLa lapse microscopy revealed that overexpression of wild-type and
cells stably expressing H2B-mCherry and a-tubulin-EGFP constitutively active but not inactive cofilin significantly
revealed that CP-673451 markedly affected spindle orienta- increased the frequency of multipolar divisions in comparison
tion and caused spindle oscillation. Specifically, treatment with cells transfected with empty vector (Fig. 5D). These results
OF6 Cancer Res; 76(22) November 15, 2016 Cancer Research
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cofilin Activation Inhibits Centrosome Clustering
Figure 5.
Cofilin activation inhibits CC. A, spindle
rotation (left, angle variation between
initial metaphase plate and anaphase)
and oscillation (right, cumulative travel
distance of the metaphase plate center)
induced by 2 mmol/L CP-673451 as
quantified by fluorescence time-lapse
imaging of H2B-mCherry-a-tubulin-
EGFP-HeLa cells. Bars, averages. Cells
were synchronized in G1–S-phase
(thymidine, 18 hours), drug or DMSO were
added 3 hours after release, and imaging
started 6 hours after release (nangle ¼ 100;
nmovement ¼ 70; , P < 0.0001). B and C,
average percentages SD of multipolar
divisions from two independent
experiments in induced EGFP-PLK4-
U2OS cells after exposure to the indicated
concentrations of LIMKi3 (B) or
damnacanthal (C), determined by time-
lapse microscopy over the first
24 hours after drug addition.
Immunoblots showing the respective
phospho-Ser3-cofilin levels are shown in
the corresponding Supplementary Fig.
S4A and S4B. D, average percentages of
multipolar divisions in induced EGFP-
PLK4-U2OS cells, transiently transfected
with the indicated constructs,
determined by time-lapse microscopy
over the first 12 hours after transfection
( , P < 0.05; , P < 0.01; n ¼ 5;
Supplementary Fig. S5A and S5B).
demonstrate that increased amounts of active cofilin in U2OS revealed that exposure to CP-673451 had no effect on LIMK1
cells with amplified centrosomes perturb CC. activity (Fig. 6B), indicating that impaired kinase activity is not
responsible for the decrease in cofilin phosphorylation.
CP-673451- and crenolanib-induced cofilin activation is In contrast, cofilin activation might be triggered by increased
mediated by SSHs SSH activity. Indeed, we observed that transient overexpression of
The putative mechanisms of cofilin activation upon treatment GFP-tagged SSH1 in U2OS cells decreased phospho-cofilin to
with CP-673451 or crenolanib include drug-induced inhibition similar levels as exposure to CP-673451 or crenolanib (Fig. 6C).
of LIMK and/or the activation of SSHs (35). Because insufficient To examine the involvement of SSH in drug-induced cofilin
activity of LIMK leads to the accumulation of active cofilin (39, activation, we depleted SSH isoforms 1, 2, or 3 from U2OS cells
40), we first analyzed the phosphorylation status of LIMK1 and and monitored cofilin activation after exposure to both quinoli-
LIMK2 in U2OS cells after exposure to increasing concentrations nobenzimidazoles. RNAi-mediated depletion of SSH1 and SSH2
of CP-673451 or crenolanib. Immunoblot analysis using a phos- partially rescued drug-induced inhibition of cofilin phosphory-
pho-LIMK1/2 antibody revealed that the levels of phosphorylated lation (Fig. 6D). Importantly, SSH2 depletion had the most
LIMK did not decrease, suggesting that both compounds do not pronounced effect, while knockdown of SSH3 failed to rescue
inhibit kinase activity. In fact, LIMK phosphorylation appeared to cofilin activation. Next, we investigated whether SSH depletion
increase after compound addition both in interphase (Fig. 6A) can also rescue drug-induced CC inhibition. We depleted each
and mitotic cells (Fig. 4C). To exclude direct inhibition of LIMK, SSH isoform in induced EGFP-PLK4-U2OS cells with CA and
independent from its phosphorylation status, LIMK1 expressed assessed CC by time-lapse microscopy during the first day fol-
in kidney HEK293T cells was immunoprecipitated and subjected lowing addition of the compounds. Despite relatively low knock-
to an in vitro kinase assay in the presence of CP-673451, using down efficiencies (Supplementary Fig. S6), silencing of SSH1 and
His6-cofilin as a substrate. Autoradiography of incorporated 32P SSH2 partially rescued induction of multipolar cell divisions by
www.aacrjournals.org Cancer Res; 76(22) November 15, 2016 OF7
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Konotop et al.
Figure 6.
CP-673451- and crenolanib-induced cofilin activation is mediated by SSH1 and SSH2. A, analysis of LIMK1/LIMK2 phosphorylation (Thr508/Thr505) in U2OS cells
exposed to indicated drug concentrations or DMSO for 3 hours. B, LIMK1 kinase activity assay. Ectopically expressed Myc-hLIMK1 was immunoprecipitated from
HEK293T cells and kinase activity was analyzed in vitro, by comparing the amounts of incorporated 32P into His6-cofilin. CP-673451 was added to the assay
buffer at the indicated concentrations. LIMK1-D460A–inactive mutant (DA) and damnacanthal were used as controls. Whole-cell lysates show overexpression of
Myc-hLIMK1 variants. C, phospho-Ser3-cofilin levels in U2OS cells transiently transfected with empty vector (GFP) or GFP-SSH1L (24 hours) and treated with DMSO
(D), 2 mmol/L CP-673451 (CP), or crenolanib (Cre; 3 hours). D, partial rescue of drug-induced cofilin activation by knockdown of SSH1 and SSH2. U2OS cells
were transfected with RNAi pools against SSH1, SSH2, SSH3, or control (72 hours) and exposed to 2 mmol/L compound (3 hours). Silencing was validated by
qPCR (Supplementary Fig. S6) and for SSH1L by immunoblotting as well (top). Relative levels of phospho-Ser3-cofilin are indicated for each sample. E, average
percentages SD of multipolar divisions in induced EGFP-PLK4-U2OS cells, depleted for the indicated SSH isoform (72 hours) and treated with DMSO or
1 mmol/L compound. Quantification was done by time-lapse microscopy during the first 24 hours after drug addition ( , P < 0.05; , P < 0.01; , P ¼ 0.001;
, P < 0.001).
both drugs (Fig. 6E). Again, SSH2 knockdown had the strongest suggest that SSH-mediated cofilin activation by quinolinoben-
effect and decreased the percentage of multipolar divisions zimidazoles may be mediated by PI3K. However, both com-
induced by CP-673451 and crenolanib by 35% and 43%, respec- pounds are known to potently inhibit PDGFR-b, and several
tively, whereas depletion of SSH3 had no effect. These results studies have demonstrated their inhibitory effects on PDGFR-b
correlate with cofilin activation observed in U2OS cells under downstream signaling (33, 34, 43, 44). To assess the effects of
similar conditions (Fig. 6D), emphasizing the negative effect of selective PDGFR-b inhibition on cofilin activation, we depleted
cofilin activation on CC. It can be concluded that CP-673451 and PDGFR-b by RNAi and found levels of phospho-cofilin to be
crenolanib-induced cofilin activation is mediated by slingshot unaltered (Fig. 7C). In addition to our finding that PDGFR-b
phosphatases 1 and 2. depletion had no effect on CC (Fig. 3E), we conclude that CP-
673451- and crenolanib-induced cofilin activation is indepen-
CP-673451 and crenolanib activate PI3K/Akt and MEK/ERK dent of PDGFR-b.
signaling under physiologic conditions To gain further insights into signaling alterations caused by
Earlier studies have demonstrated that isoforms of PI3K play both quinolinobenzimidazoles, we next analyzed the impact of
an important role in mediating extracellular signals leading to these compounds on Akt and MEK, the main signaling branches
the activation of SSH1 and SSH2, resulting in cofilin activation downstream of several RTKs, in different cancer cell lines. Under
and actin cytoskeleton rearrangement (41, 42). As direct SSH normal growth conditions, CP-673451 unexpectedly elevated the
activation by CP-673451 was not observed (Supplementary Fig. levels of phospho-Akt and phospho-MEK in almost all cell lines
S7), we next examined whether PI3K signaling is required for within 3 hours of exposure (Supplementary Fig. S8A). Although
CP-673451–induced cofilin activation by SSHs. We preincu- Akt phosphorylation was not increased in U2OS cells at that time
bated U2OS cells with the PI3Ka inhibitor BYL719 before the point, CP-673451 treatment led to a significant increase in phos-
addition of CP-673451. Immunoblot analysis showed that pho-Akt levels at 24 hours in a dose-dependent manner (Supple-
cofilin phosphorylation was partially rescued by BYL719 (Fig. mentary Fig. S8B). In conclusion, both compounds stimulate Akt
7A). Similar results were obtained by preincubating cells with and MEK in cultured cells.
the pan-PI3K inhibitor wortmannin, but not the PI3Kd-specific Because crenolanib acts as a type I tyrosine kinase inhibitor
inhibitor CAL-101 (data not shown). Importantly, pretreat- (TKI) and binds preferentially to phosphorylated RTKs (45,
ment of induced EGFP-PLK4-U2OS cells with BYL719 also 46), we reasoned that CP-673451 and crenolanib would inhibit
partially rescued CP-673451–induced multipolar divisions in RTK signaling only when receptors are in their active confor-
time-lapse microscopy experiments (Fig. 7B). These results mation. To validate this hypothesis, we assessed Akt and MEK
OF8 Cancer Res; 76(22) November 15, 2016 Cancer Research
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cofilin Activation Inhibits Centrosome Clustering
Figure 7.
CP-673451 and crenolanib stimulate Akt and MEK signaling under physiologic conditions. A, immunoblot analysis of phospho-Ser3-cofilin levels in U2OS cells
pretreated with BYL719 for 2 hours, followed by addition of 2 mmol/L CP-673451 (CP) or DMSO (D) for 3 hours. Phospho-Ser473-Akt levels indicate PI3K inhibition.
MCM7 shows equal loading. B, average percentage SD of multipolar cell divisions of induced EGFP-PLK4-U2OS cells, pretreated with BYL719 (2 hours) and
exposed to 1 mmol/L CP-673451 in the continuous presence of BYL719. Quantification was done by time-lapse microscopy during the first 24 hours after the addition
of CP-673451 ( , P < 0.001; n ¼ 3). C, immunoblot showing the effects of RNAi-mediated PDGFR-b silencing (72 hours) in U2OS cells on phospho-Ser3-cofilin levels.
Akt and MEK phosphorylation was detected using phospho-Ser473-Akt and phospho-Ser217/221-MEK1/2 antibodies. D, immunoblot comparing downstream MEK1/
2-Ser217/221-, Akt-Ser473-, and cofilin-Ser3-phosphorylation between PDGF-BB–stimulated and nonstimulated U2OS cells in the presence or absence of
compounds. Starved U2OS cells were preincubated with DMSO (D), 2 mmol/L CP-673451 (CP), or crenolanib (Cre) for 2 hours before PDGF-BB addition.
phosphorylation (as indicators of PDGFR-b downstream sig- of this mechanism, the discovery of new druggable target
naling) in PDGF-BB–stimulated and nonstimulated U2OS proteins, and the identification of small-molecule inhibitors.
cells pretreated with either CP-673451 or crenolanib. As To date, most cell-based assays have utilized high-content
expected, stimulation of PDGFR-b strongly enhanced Akt microscopic imaging (13, 14, 21, 47). However, these screens
and MEK phosphorylation and activated cofilin (Fig. 7D). delivered little information on direct cellular cytotoxicity
Preincubation with CP-563451 and crenolanib attenuated and thus therapeutic potential because their readouts were
PDGFR-BB–induced Akt and MEK activation, demonstrating confined to metaphase multipolarity induction. In this study,
their inhibitory role on PDGFR-b signaling. In contrast, expo- we employed a screening concept to identify small-molecule
sure of nonstimulated, serum-starved U2OS cells to CP-673451 or inhibitors of CC based on differential viabilities of induced
crenolanib increased Akt and MEK phosphorylation. This con- versus noninduced isogenic EGFP-PLK4-U2OS cells. High
firms that the inhibitory capability of both molecules depends on levels of CA and robust CC in these cells allowed for the
the RTK activation state. Finally, we tested whether PDGFR-b was identification of small molecules, which selectively interfere
required for drug-induced activation of downstream signaling in with the mechanisms of CC.
nonstimulated cells. Exposure of PDGFR-b–depleted U2OS cells With this screening approach, we identified CP-673451 and
to CP-673451 and crenolanib still resulted in Akt and MEK crenolanib (CP-868596), two molecules with similar chemical
phosphorylation (Supplementary Fig. S8C), indicating that other structures and proven antitumor activity, as inhibitors of CC. At
kinases are involved in this signaling. clinically relevant concentrations (48), both compounds effec-
tively induced multipolar cell divisions and consequent cell death
in EGFP-PLK4-U2OS cells with CA as well as in a variety of cancer
Discussion cell lines harboring different degrees of spontaneous CA. Impor-
Because CC is regarded as a promising target for cancer tantly, drug-induced multipolarity was restricted to cells with
treatment, several studies have focused on the characterization supernumerary centrosomes and did not lead to the formation
www.aacrjournals.org Cancer Res; 76(22) November 15, 2016 OF9
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Konotop et al.
of acentrosomal spindle poles as seen with the other inhibitors of Our data suggest that the downstream inhibitory effect of these
CC (21–23, 47). compounds is dependent on the activation state of PDGFR-b.
Previous studies have shown that CC is inhibited upon inter- Although CP-673451 and crenolanib attenuated PDGF-BB–
ference with spindle pole integrity, microtubule–kinetochore induced Akt and MEK activation, in the absence PDGF-BB stim-
attachment, SAC activation, or cortical actin cytoskeleton ulation, they enhanced downstream Akt and MEK pathway sig-
(13, 14, 17). The absence of acentriolar spindle poles in cells naling in almost all cell lines tested. These observations may be
treated with CP-673451 and crenolanib suggests that spindle pole explained by the fact that crenolanib behaves as a type I TKI and
integrity is not affected by these compounds. As both CP-673451 therefore preferentially binds to RTKs in their active conformation
and crenolanib prolonged the average duration of mitosis but did (45). Its affinity toward active FLT3 is more than 10-fold higher
not induce mitotic arrest, SAC inactivation and interference with than toward inactive FLT3, and for ABL1, phosphorylation
microtubule–kinetochore attachment is unlikely. increases drug affinity by 7-fold (46). To the best of our knowl-
We show here that CP-673451 and crenolanib affect the orga- edge, no data concerning this matter are available for CP-673451.
nization of cortical actin filaments by the activation of cofilin. In summary, we present a novel high-throughput screening
Cofilin is one of the key regulators of actin remodeling in response concept for the identification of small molecules that inhibit CC.
to external stimuli; it promotes severing and dissociation of F- By applying this strategy, we have identified CP-673451 and
actin filaments and increases the cellular pool of G-actin for new crenolanib as inhibitors of CC with increased cytotoxicity for
filament growth (36). Cofilin activity is negatively regulated by cells with CA. Both compounds induce multipolar cell division
Ser3 phosphorylation, mediated by LIM-domain kinases (LIMK1 and subsequent cell death by cofilin-mediated disruption of the
and LIMK2) and related testicular kinases TESK1 and TESK2. cortical actin cytoskeleton, reemphasizing the importance of
Cofilin dephosphorylation is mainly regulated by slingshot phos- cortical actin for CC.
phatases SSH1, SSH2, and SSH3 (35).
We demonstrate that cofilin activation in EGFP-PLK4-U2OS
cells with CA inhibits CC. A previous study showed that elevated Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
levels of active cofilin strongly affect spindle orientation and
positioning in dividing HeLa cells with regular centrosome con-
tent (37). We observed similar effects upon exposure of HeLa cells Authors' Contributions
to CP-673451 and crenolanib. In cells with extra centrosomes, Conception and design: G. Konotop, A. Kr€amer, M.S. Raab
normal actin and actin-based contractility has been shown to Development of methodology: G. Konotop, M. Boutros, A. Kr€amer, M.S. Raab
promote bipolar spindle assembly and suppress spindle multi- Acquisition of data (provided animals, acquired and managed patients,
provided facilities, etc.): G. Konotop, E. Bausch, T. Nagai, A. Turchinovich,
polarity (13). In accordance with our results, an independent
K. Mizuno, A. Kr€amer
study identified LIMK2 and TESK1 as important regulators of CC Analysis and interpretation of data (e.g., statistical analysis, biostatistics,
(15). computational analysis): G. Konotop, E. Bausch, N. Becker, A. Benner,
Our results indicate that CP-673451 and crenolanib stimulate K. Mizuno, A. Kr€amer, M.S. Raab
phosphatase activity of SSH1 and SSH2 to decrease cofilin phos- Writing, review, and/or revision of the manuscript: G. Konotop, N. Becker,
phorylation. SSH1 and SSH2 are known to be activated by A. Benner, A. Kr€amer, M.S. Raab
Administrative, technical, or material support (i.e., reporting or organizing
external factors that involve production of PI(3,4,5)P3 (41, 42).
data, constructing databases): A. Kr€amer
Accordingly, we found PI3K inhibition by BYL719 or wortmannin Study supervision: A. Kr€amer, M.S. Raab
to partially rescue CP-673451/crenolanib-induced cofilin activa-
tion, suggesting that both drugs activate cofilin, at least in part,
through PI3Ka stimulation. Acknowledgments
It is important to note that CP-673451 has been described to be We thank Barbara Schmitt and Thilo Miersch for excellent technical assis-
a highly selective ATP-competitive inhibitor of PDGFR-b (33). tance and advice during the small-molecule screen. We acknowledge Bianca
Kraft for the 3Flag-STIL-HCT116 cells and Marion Schmidt-Zachmann for the
Similarly, crenolanib is a potent TKI with strongest affinity for
NO66 antibody.
PDGF-a and -b receptors and FLT3 (34). Stimulation of several
different RTKs and G protein–coupled receptors, for example,
insulin receptor, formyl peptide receptor 1, and PDGFR-b, pro- Grant Support
motes cofilin activity via activation of SSH1/2 to generate rapid This study was supported by the Max-Eder program of the German Cancer
turnover of actin filaments in different cell types (42, 49–51). Aid (Deutsche Krebshilfe; awarded to M.S. Raab) and a German Research
Foundation (DFG) grant (KR 1981/4-1 to A. Kr€amer).
Accordingly, although PDGFR-b depletion did not affect cofilin
The costs of publication of this article were defrayed in part by the payment of
regulation, we corroborate that stimulation of U2OS cells with page charges. This article must therefore be hereby marked advertisement in
PDGF-BB decreases overall cofilin phosphorylation. As CP- accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
673451 and crenolanib stimulated cofilin activation in all tested
cell lines in a concentration-dependent manner, we conclude that Received May 8, 2016; revised July 15, 2016; accepted August 21, 2016;
this effect is not mediated by RTK/PDGFR-b inhibition. published OnlineFirst September 13, 2016.
References
1. Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: 3. Brito DA, Gouveia SM. Deconstructing the centriole: structure and number
centrosomics brings new understanding. Nat Rev Mol Cell Biol 2007;8: control. Curr Opin Cell Biol 2012;152:8–11.
451–63. 4. Lingle WL, Salisbury JL. Altered centrosome structure is associated
2. Bornens M. The centrosome in cells and organisms. Science 2012; with abnormal mitoses in human breast tumors. Am J Pathol
335:422–6. 1999;155:1941–51.
OF10 Cancer Res; 76(22) November 15, 2016 Cancer Research
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Cofilin Activation Inhibits Centrosome Clustering
5. Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, et al. 30. Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof Y-D, Nigg
Centrosome defects and genetic instability in malignant tumors. Cancer EA. Plk4-induced centriole biogenesis in human cells. Dev Cell
Res 1998;58:3974–85. 2007;13:190–202.
6. Nigg EA. Centrosome aberrations: cause or consequence of cancer 31. Schmitz MHA, Held M, Janssens V, Hutchins JRA, Hudecz O, Ivanova E,
progression? Nat Rev Cancer 2002;2:815–25. et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and impor-
7. Koutsami MK, Tsantoulis PK, Kouloukoussa M, Apostolopoulou K, Pateras tin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol
IS, Spartinou Z, et al. Centrosome abnormalities are frequently observed in 2010;12:886–93.
non-small-cell lung cancer and are associated with aneuploidy and cyclin E 32. Ohashi K, Sampei K, Nakagawa M, Uchiumi N, Amanuma T, Aiba S, et al.
overexpression. J Pathol 2006;209:512–21. Damnacanthal, an effective inhibitor of LIM-kinase, inhibits cell migration
8. Chan JY. A clinical overview of centrosome amplification in human and invasion. Mol Biol Cell 2014;25:828–40.
cancers. Int J Biol Sci 2011;7:1122–44. 33. Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang H,
9. G€onczy P. Centrosomes and cancer: revisiting a long-standing relationship. et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine
Nat Rev Cancer 2015;15:639–52. kinase inhibitor, CP-673,451 antiangiogenic and antitumor activity of a
10. Denu RA, Zasadil LM, Kanugh C, Laffin J, Weaver BA, Burkard ME. selective PDGFR. Cancer Res 2005;65:957–66.
Centrosome amplification induces high grade features and is prognostic 34. Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachan-
of worse outcomes in breast cancer. BMC Cancer 2016;16:47. dran A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V
11. Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle mutation associated with imatinib-resistant gastrointestinal stromal
multipolarity is prevented by centrosomal clustering. Science 2005;307: tumors. Clin Cancer Res 2012;18:4375–84.
127–9. 35. Mizuno K. Signaling mechanisms and functional roles of cofilin phos-
12. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centro- phorylation and dephosphorylation. Cell Signal 2013;25:457–69.
somes to chromosomal instability. Nature 2009;460:278–82. 36. Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin
13. Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, et al. dynamics. Annu Rev Cell Dev Biol 1999;15:185–230.
Mechanisms to suppress multipolar divisions in cancer cells with extra 37. Kaji N, Muramoto A, Mizuno K. LIM kinase-mediated cofilin phosphor-
centrosomes. Genes Dev 2008;22:2189–203. ylation during mitosis is required for precise spindle positioning. J Biol
14. Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, et al. Proteins required Chem 2008;283:4983–92.
for centrosome clustering in cancer cells. Sci Transl Med 2010;2:33ra38. 38. Ross-Macdonald P, de Silva H, Guo Q, Xiao H, Hung C-Y, Penhallow B,
15. Drosopoulos K, Tang C, Chao WCH, Linardopoulos S. APC/C is an et al. Identification of a nonkinase target mediating cytotoxicity of novel
essential regulator of centrosome clustering. Nat Commun 2014;5:3686. kinase inhibitors. Mol Cancer Ther 2008;7:3490–8.
16. Maier B, Kirsch M, Anderhub S, Zentgraf H, Kr€amer A. The novel actin/focal 39. Arber S, Barbayannis FA, Hanser H. Regulation of actin dynamics through
adhesion-associated protein MISP is involved in mitotic spindle position- phosphorylation of cofilin by LIM-kinase. Nature 1998;393:805–9.
ing in human cells. Cell Cycle 2013;12:1457–71. 40. Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, et al. Cofilin
17. Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, et al. phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin
Centrosome amplification can initiate tumorigenesis in flies. Cell 2008; reorganization. Nature 1998;393:809–12.
133:1032–42. 41. Nishita M, Wang Y, Tomizawa C, Suzuki A, Niwa R, Uemura T, et al.
18. Sertel S, Fu Y, Zu Y, Rebacz B, Konkimalla B, Plinkert PK, et al. Molecular Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase
docking and pharmacogenomics of vinca alkaloids and their monomeric Slingshot and its role for insulin-induced membrane protrusion. J Biol
precursors, vindoline and catharanthine. Biochem Pharmacol 2011;81: Chem 2004;279:7193–8.
723–35. 42. Tang W, Zhang Y, Xu W, Harden TK, Sondek J, Sun L, et al. A PLCb/PI3Kg-
19. Karna P, Rida PCG, Pannu V, Gupta KK, Dalton WB, Joshi H, et al. A novel GSK3 signaling pathway regulates cofilin phosphatase slingshot2 and
microtubule-modulating noscapinoid triggers apoptosis by inducing spin- neutrophil polarization and chemotaxis. Dev Cell 2011;21:1038–50.
dle multipolarity via centrosome amplification and declustering. Cell 43. Dai J, Kong Y, Si L, Chi Z, Cui C, Sheng X, et al. Large-scale analysis of
Death Differ 2011;18:632–44. PDGFRA mutations in melanomas and evaluation of their sensitivity to
20. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev tyrosine kinase inhibitors imatinib and crenolanib. Clin Cancer Res
Cancer 2004;4:253–65. 2013;19:6935–42.
21. Rebacz B, Larsen TO, Clausen MH, Rønnest MH, L€ offler H, Ho AD, et al. 44. Ehnman M, Missiaglia E, Folestad E, Selfe J, Strell C, Thway K, et al. Distinct
Identification of griseofulvin as an inhibitor of centrosomal clustering in a effects of ligand-induced PDGFRa and PDGFRb signaling in the human
phenotype-based screen. Cancer Res 2007;67:6342–50. rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res
22. Raab MS, Breitkreutz I, Anderhub S, Ronnest MH, Leber B, Larsen TO, et al. 2013;73:2139–49.
GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell 45. Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS,
growth in vitro and in vivo. Cancer Res 2012;72:5374–85. et al. Crenolanib is active against models of drug-resistant FLT3-ITD-
23. Watts CA, Richards FM, Bender A, Bond PJ, Korb O, Kern O, et al. Design, positive acute myeloid leukemia. Blood 2013;122:3607–15.
synthesis, and biological evaluation of an allosteric inhibitor of HSET that 46. Ramachandran A, Marshall H, Jain VK. Abstract 3683: Crenolanib, a novel
targets cancer cells with supernumerary centrosomes. Chem Biol 2013; Type I, mutant-specific inhibitor of Class III receptor tyrosine kinases,
20:1399–410. preferentially binds to phosphorylated kinases. Cancer Res 2012;72:3683.
24. Kleylein-Sohn J, P€ ollinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings 47. Kawamura E, Fielding AB, Kannan N, Balgi A, Eaves J, Roberge M, et al.
BA, et al. Acentrosomal spindle organization renders cancer cells depen- Identification of novel small molecule inhibitors of centrosome clustering
dent on the kinesin HSET. J Cell Sci 2012;125:5391–402. in cancer cells. Oncotarget 2013;4:1763–76.
25. Habedanck R, Stierhof Y-D, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 48. Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, et al. Phase I study
functions in centriole duplication. Nat Cell Biol 2005;7:1140–6. of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a
26. Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, Liu F, et al. STIL is highly specific platelet-derived growth factor receptor tyrosine kinase
required for centriole duplication in human cells. J Cell Sci 2012;125: inhibitor in patients with advanced cancers. J Clin Oncol 2009;27:5262–9.
1353–62. 49. Nebl G, Fischer S, Penzel R, Samstag Y. Dephosphorylation of cofilin is
27. Arquint C, Sonnen KF, Stierhof Y, Nigg EA. Cell-cycle-regulated expression regulated through Ras and requires the combined activities of the Ras-
of STIL controls centriole number in human cells. J Cell Sci 2012;125: effectors MEK and PI3K. Cell Signal 2004;16:235–43.
1342–52. 50. San Martín A, Lee MY, Williams HC, Mizuno K, Lassegue B, Griendling KK.
28. Tang CC, Lin S, Hsu W, Lin Y, Wu C, Lin Y, et al. The human microcephaly Dual regulation of cofilin activity by LIM kinase and Slingshot-1L phos-
protein STIL interacts with CPAP and is required for procentriole forma- phatase controls platelet-derived growth factor-induced migration of
tion. EMBO J 2011;30:4790–804. human aortic smooth muscle cells. Circ Res 2008;102:432–8.
29. Leidel S, Delattre M, Cerutti L, Baumer K, G€ onczy P. SAS-6 defines a protein 51. Homma Y, Kanno S, Sasaki K, Nishita M, Yasui A, Asano T, et al. Insulin
family required for centrosome duplication in C.elegans and in human receptor substrate-4 binds to Slingshot-1 phosphatase and promotes
cells. Nat Cell Biol 2005;7:115–25. cofilin dephosphorylation. J Biol Chem 2014;289:26302–13.
www.aacrjournals.org Cancer Res; 76(22) November 15, 2016 OF11
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.Published OnlineFirst September 13, 2016; DOI: 10.1158/0008-5472.CAN-16-1144
Pharmacological Inhibition of Centrosome Clustering by
Slingshot-Mediated Cofilin Activation and Actin Cortex
Destabilization
Gleb Konotop, Elena Bausch, Tomoaki Nagai, et al.
Cancer Res Published OnlineFirst September 13, 2016.
Updated version Access the most recent version of this article at:
doi:10.1158/0008-5472.CAN-16-1144
Supplementary Access the most recent supplemental material at:
Material http://cancerres.aacrjournals.org/content/suppl/2020/12/19/0008-5472.CAN-16-1144.DC1
E-mail alerts Sign up to receive free email-alerts related to this article or journal.
Reprints and To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Subscriptions Department at pubs@aacr.org.
Permissions To request permission to re-use all or part of this article, use this link
http://cancerres.aacrjournals.org/content/early/2016/11/03/0008-5472.CAN-16-1144.
Click on "Request Permissions" which will take you to the Copyright Clearance Center's
(CCC)
Rightslink site.
Downloaded from cancerres.aacrjournals.org on February 19, 2021. © 2016 American Association for Cancer
Research.You can also read

















































